
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceApplication of dissolvable Mg/Al layered double hydroxides as an adsorbent for the dispersive solid phase extraction of gold nanoparticles prior to their determination by atomic absorption spectrometry†
Tatiana G.
Choleva
and
Dimosthenis L.
Giokas
 *
*
Department of Chemistry, University of Ioannina, 45110, Ioannina, Greece. E-mail: dgiokas@uoi.gr
First published on 27th November 2019
Abstract
In this work, dissolvable layered double hydroxides (LDH) were used for the first time for the extraction and preconcentration of metallic nanoparticles from water samples. Magnesium–aluminum LDHs were prepared in situ in a sample solution by alkaline co-precipitation and separated by centrifugation. It was found that gold nanoparticles (AuNPs) and gold ions could be simultaneously extracted by the LDH by electrostatic interactions and anion exchange reactions, respectively. Separation of AuNPs from gold ions could be achieved by ultracentrifugation enabling the selective determination of AuNPs in the sample. Two analytical techniques based on molecular and atomic spectrometry (spectrophotometry and electrothermal atomic absorption spectrometry) were used to optimize the method and determine the concentration of AuNPs in spiked environmental water samples at femtomolar concentration levels, respectively. Under the optimized experimental conditions the method enables the determination and speciation of AuNPs at concentration levels as low as 16 femtomole L−1 with satisfactory recoveries (71–91%) and good reproducibility (<9%).
1. Introduction
Over the past decade the use of nanomaterials in a broad spectrum of applications and technologies has grown rapidly and is expected to increase even further in the near future.1,2 It is therefore reasonable that great concern has been raised regarding their potential release into the environment and their effects on ecosystems and living organisms.3,4 For this reason several regulation authorities have classified nanoparticles as emerging environmental pollutants and try to formulate a basis for potential preservation measures.5,6The first step is assessing the fate, transportation and potential risks of nanoparticles in the environment and their determination in various environmental compartments such as wastewater and natural waters. This task, however, is not facile for several reasons: (a) the concentrations of nanoparticles are expected within the pg L−1 to the low ng L−1 levels and current instrumental techniques can hardly accomplish such sensitivity, (b) matrix effects may interfere with the analysis and affect the accuracy of the results, and (c) dissolved ions of the same nature may be misinterpreted as nanoparticles.7,8 Therefore, the analysis of nanoparticles in environmental samples requires a combination of both appropriate analytical methods for their extraction and preconcentration and sensitive analytical techniques for their detection which almost invariably rely on atomic spectroscopy due to its high sensitivity and selectivity.9
The extraction and preconcentration of nanoparticles from environmental samples has been successfully accomplished by a variety of methods that use either liquid or solid extraction templates such as micelles and other surfactant aggregates (e.g. cloud point extraction),10–13 binary or ternary solvent mixtures (e.g. LPME and its modifications),14–16 solid phase extraction and microextraction (e.g. resins and magnetic nanoparticles)17–20etc. Most of these methods provide information regarding the chemical footprint of nanoparticles (such as composition, mass and concentration) while some methods may also provide physical information (e.g. size, shape, and aggregation). Importantly, almost all of these methods enable the selective determination of nanoparticles over their precursor metal ions thus avoiding misinterpretations that could lead to an overestimation of the nanoparticle concentration in the environment. Not all of these methods, however, can extract and preconcentrate both metal nanoparticles and their corresponding metal ions simultaneously which is necessary when investigating the environmental fate of nanoparticles that may involve several transformation pathways (dissolution, reformation, etc.).21
Although rapid progress has been made in the adaptation of conventional sample preparation methods to the extraction of nanoparticles, the use of layered double hydroxides (LDHs) for the extraction and preconcentration of nanoparticles has not been reported. LDHs are ionic lamellar solids made from the stacking of positively charged (brucite-like) layers of mixed metal hydroxides.22,23 These layers are bound together by electrostatic interactions and hydrogen bonds with interlayer counter-anions and solvation (i.e. water) molecules. LDHs are typically made of divalent and trivalent metal cations, (such as Mg2+, Zn2+, Al3+ or Fe3+) and interlayer charge-compensating anions (such as NO3−, Cl−, CO32−, etc.). Due to their high anion exchange capacity, increased porosity, and high specific surface area, LDHs have been considered as a promising sorbent for enriching inorganic and organic anions.24–26 Moreover, advanced treatment of the surface and structure of LDHs has extended their potential as extraction sorbents for a variety of other organic compounds.27–30
In this work, we examined the analytical utility of LDHs as sorbents for the extraction of metallic nanoparticles from water samples for the first time. Using AuNPs as model nanoparticle species, the experimental parameters affecting the extraction efficiency of AuNPs of variable size and surface coating were examined and optimized. We also developed a simple procedure for separating Au ions from AuNPs and demonstrate, as a proof-of-concept application, the utility of LDHs in the determination of AuNPs at ultra-trace levels in spiked environmental water samples by electrothermal atomic absorption spectrometry.
2. Experimental part
2.1. Reagents
Hydrogen tetrachloroaurate trihydrate (≥99.9% trace metals basis), aluminum nitrate nonahydrate (≥98%), magnesium nitrate hexahydrate (≥99%), sodium borohydride, tri-sodium citrate, and high purity nitric acid for inorganic trace analysis (TraceSELECT® Ultra) were purchased from Sigma-Aldrich (Steinheim, Germany). HPLC-grade methanol was obtained from Fisher Scientific (Loughborough, UK).2.2. Equipment
UV-Vis absorption measurements were performed with matched quartz cells of a 1 cm path length using a Jenway (Essex, UK) 6405 UV/Vis spectrophotometer. A PerkinElmer Spectrum Two attenuated total reflectance-infra red (ATR-IR) spectrometer was used for determining the vibrational relaxations of the collected LDH. Scanning electron microscopy (SEM) experiments were performed with a JEOL JSM-6300 instrument. A Shimadzu AA-6800 electrothermal atomic absorption spectrophotometer (ETAAS) equipped with an ASC-6100 autosampler was used for the determination of gold at 242.8 nm using a self-reversal hollow cathode lamp (Heraeus, Hanau, Germany) operating at 10 mA.2.3. Synthesis of gold nanoparticles
The synthesis of AuNPs of variable sizes and with different surface coatings was performed using the experimental procedures described in our previous studies without modifications.12,13,31 The average size distribution and the concentration of the synthesized AuNPs were calculated using the method of Haiss et al.32 by determining the ratio of the absorbance of the AuNP suspensions at the surface plasma resonance peak to the absorbance at 450 nm.2.4. Experimental procedure
In 50 mL of an aqueous sample solution (likely to contain both AuNPs and gold ions), 0.5 mL of the 1 mol L−1 KOH solution and 0.5 mL of the Mg2+/Al3+ solution (75/25 mmol L−1) were added sequentially. The solution was mixed by vortex agitation for 1 min and centrifuged at 6000 rpm (4200 g) for 25 min. The precipitate was collected at the bottom of the vial and redissolved in 0.5 mL of 2.5 mol L−1 HCl.To separate gold ions from AuNPs and remove the excess amount of K+, Mg2+ and Al3+, the extract obtained after dissolution of the LDH with HCl was transferred to a graduated Eppendorf vial with 1.0 mL distilled water (a total volume of 1.5 mL). The solution was centrifuged at 18![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm (15120 g) for 20 min to precipitate the AuNPs and leave gold ions and other co-existing species in the suspension and in the supernatant solution. Then 1.4 mL of the aqueous supernatant was carefully removed with a glass Pasteur pipette and slowly replaced with 1.4 mL of 6 M HNO3 in order to minimize the redispersion of AuNPs. The separation procedure was repeated two more times to completely remove dissolved ions (in total three times). An aliquot of 20 μL of the final solution was used for the analysis by ETAAS according to the temperature program given in Table S1.†
000 rpm (15120 g) for 20 min to precipitate the AuNPs and leave gold ions and other co-existing species in the suspension and in the supernatant solution. Then 1.4 mL of the aqueous supernatant was carefully removed with a glass Pasteur pipette and slowly replaced with 1.4 mL of 6 M HNO3 in order to minimize the redispersion of AuNPs. The separation procedure was repeated two more times to completely remove dissolved ions (in total three times). An aliquot of 20 μL of the final solution was used for the analysis by ETAAS according to the temperature program given in Table S1.†
2.5. Real samples
Genuine water samples (river and lake water and effluent wastewater) were filtered through 0.45 μm glass fiber filters and spiked with known amounts of AuNPs. The samples were used without further treatment.3. Results and discussion
3.1. Characterization of LDHs and the mechanism of AuNP extraction
The LDHs that were formed under the experimental working conditions conformed to the characteristics of LDHs, as published previously, exhibiting the typical IR bands observed in Mg2+–Al3+ LDH (Fig. 1a) with characteristic peaks at 3450 cm−1 of the O–H vibrations, the O–H bending vibrations at approximately 1630 cm−1, the NO3 vibrations at 1380 cm−1 and the Mg–O–Al vibrations at 450 cm−1.24 The SEM images (Fig. 1b) show that LDHs exhibit a plate-like shape and generally tend to have a hexagonal-like morphology which is typical for many LDHs.33,34 | ||
| Fig. 1 (a) ATR-IR spectra of the Mg2+/Al3+ LDH; (b) SEM images of LDH at different magnification (from left to right: 65×, 100×, 300×, 2000×). | ||
3.2. Optimization of the experimental procedure for the extraction of AuNPs
The efficiency of LDHs in extracting AuNPs from water samples was optimized by investigating the concentration and ratio of the precursor metal ions (Mg2+ and Al3+), the concentration of KOH, the extraction time and temperature as well as parameters related to the collection and the elution of AuNPs (i.e. the centrifugation and elution steps). Optimization experiments were performed in 10 mL aqueous solutions fortified with 15.8 nmol L−1 of citrate-capped AuNPs (4 nm). Due to the fact that AuNPs exhibit a unique peak wavelength related to their plasmon resonance, the optimum experimental conditions producing the highest signal were determined spectrophotometrically by recording the absorbance of the HCl extract solutions at the peak wavelength, against a reagent blank. Once the optimum conditions for the extraction of AuNPs have been established, the method was transferred to ETAAS by isolating the AuNPs (20 nm) by means of ultracentrifugation in order to relieve the extract from co-extracted species that may interfere with the analysis and accomplish high sensitivity.3.3. Effect of the Mg2+ and Al3+ concentration and ratio
The concentration and ratio of bivalent and trivalent metal ions affect both the structure and the surface charge of the LDHs. A ratio of M2+/M3+ between 2 and 4 is necessary for the formation of a LDH,23 therefore the influence of Mg2+ and Al3+ concentrations was initially investigated at a Mg2+/Al3+ ratio of 3 by varying the total cation concentration from 1.0 to 10.0 mmol L−1 (Fig. 2a). The absorbance signal was found to increase with the cation concentration up to 5 mmol L−1 but decreases at higher concentrations. On the basis of these observations, the optimum concentration of Mg2+ and Al3+ was set at 5 mmol L−1 (3.75 mmol L−1 of Mg2+ and 1.25 mmol L−1 of Al3+). | ||
| Fig. 2 Effect of the Mg2+/Al3+ concentration (a) and ratio (b) on extraction. Conditions: 15.8 nmol L−1 of citrate-capped AuNPs (4 nm), 100 mmol L−1 KOH, room temperature, 1 min mixing time, centrifugation for 30 min at 6000 rpm (4200 g), dissolution in 0.5 mL of 0.5 mol L−1 HCl. | ||
With regard to the Mg2+/Al3+ ratio, it was observed that the higher extraction efficiency was attained at a Mg2+/Al3+ ratio of 2:1 and above (Fig. 2b). Since a 3:1 molar ratio is commonly used in LDH-mediated extractions we adopted a molar ratio of 3 as optimum.
3.4. Effect of the KOH concentration
The addition of alkali is not only an essential prerequisite for the formation of LDHs by coprecipitation but it also determines the surface charge of AuNPs and therefore their ability to interact with the positively charged LDH surface. The influence of the KOH concentration was investigated from 10–100 mmol L−1 by rapidly adding the KOH solution into the sample containing the AuNPs and the soluble salts of Mg2+ and Al3+. From the results depicted in Fig. S1† it is made clear that the highest absorbance signal is obtained between 30 and 50 mmol L−1 of hydroxide ions while at higher concentrations the signal decreases slightly possibly due to the strong competition of OH− anions with the negatively charged citrate-capped AuNPs for the positive surface charge of the LDH layers. A KOH concentration of 50 mmol L−1 was therefore used throughout the remaining work for the formation of LDHs.3.5. Effect of the extraction time and temperature
The extraction time and temperature were investigated in the range from 2 to 45 min and 10–50 °C, respectively (Fig. S2†). The data show that extraction rapidly reaches equilibrium at room temperature possibly because the in situ formation of LDHs facilitates the spontaneous interaction of AuNPs with the LDHs. Longer extraction times or an increase in the temperature of the solution offer no improvement in the extraction efficiency as evidenced by the fact that the absorbance signal reaches a plateau or decreases, respectively. These conditions are particularity favorable for sample throughput because a large number of samples can be prepared simultaneously without the need for strict control of the reaction time and temperature.3.6. Effect of ionic strength
The addition of inorganic electrolytes to regulate the ionic strength of the sample solution may affect the extraction efficiency in two different ways. The first is by counterbalancing the negative surface charge of AuNPs, which may facilitate their aggregation and reduce their electrostatic interaction with the positively charged LDH surface. The second is by affecting the structure and morphology of LDHs as well as the selectivity of anion exchange. In our work we prepared LDHs using nitrate as counterions by using nitrate salts of Mg2+ and Al3+. Therefore, we studied the influence of ionic strength by using NaNO3 in order to avoid anion exchange reactions with other anions. The addition of NaNO3 was examined in the concentration range from 0 to 500 mmol L−1 in two experiments. In the first experiment NaNO3 was added after the formation of LDHs while in the second experiment NaNO3 was added before the formation of LDHs. The data in Fig. 3 show that the addition of the NaNO3 concentration induces a decrease in the absorbance signals. This is probably because the excess amount of anions adsorbs on the oppositely charged surface of the LDH screening the surface charge of the LDH.34,35 This in turn reduces the electrostatic interactions between the negatively charged citrate-capped AuNPs and the positively charged LDH, thus reducing the extraction efficiency. In parallel, as the concentration of Na+ counter-ions increases the negative surface charge of citrate-capped AuNPs is counterbalanced. Therefore the electrostatic interactions between citrate-capped AuNPs and LDHs are significantly suppressed. | ||
| Fig. 3 Effect of ionic strength on extraction. Conditions: 15.8 nmol L−1 of citrate-capped AuNPs (4 nm), room temperature, Mg2+ and Al3+ concentration 6.7 mmol L−1 (1/3), 50 mmol L−1 KOH, 1 min mixing time, centrifugation for 30 min at 6000 rpm (4200 g), dissolution in 0.5 mL of 0.5 mol L−1 HCl. | ||
These mechanisms are verified by the fact that the reduction in the extraction efficiency is more significant when NaNO3 is added before the formation of LDHs because Na+ ions can counterbalance the surface charge of citrate-capped AuNPs before they can interact with the positively charged surface of the LDHs. On the other hand, when NaNO3 is added after the formation of LDHs, the influence of NaNO3 is less significant because some of the citrate-capped AuNPs have already interacted with LDHs. Hence, the reduction in the extraction efficiency may be due to the competition for the remaining citrate-capped AuNPs. On the basis of these observations we concluded that the method could be applied in water samples of low ionic strength (e.g. river, lake water etc.) but not in samples with a high salt content such as seawater, estuarine water etc.
3.7. Extraction and elution of AuNPs
The final step in the extraction of AuNPs involves the separation of the LDH from the bulk sample phase and its dissolution in order to release the entrapped AuNPs. From Fig. S3† it can be concluded that centrifugation for 25 min at 6000 rpm (4200 g) is necessary to quantitatively collect the LDH. Lower centrifugation times do not effectively separate the LDH which remains in the solution as evidenced by the fact that the aqueous (sample) solutions remain turbid. As a result the extraction efficiency of AuNPs decreases and the recorded absorbance value of AuNPs in the extract solution (Fig. S3†) also decreases.The dissolution of the LDH collected after centrifugation is a critical step in the overall process. The LDH must be quantitatively dissolved in order to release the extracted AuNPs and not interfere with the measurements. The dissolution of LDH is easily accomplished by the addition of HCl but the concentration of HCl necessary to dissolve the LDH was found to depend on the amount of LDH formed during extraction, which in turn is determined by the sample volume that is extracted. For example, to maintain a constant concentration of 5 mmol L−1 for Mg2+/Al3+ (at a molar ratio of 3) 50 μmol of Mg2+/Al3+ ions are required for 10 mL of aqueous sample solution and 250 μmol for 50 mL of the aqueous sample solution. Through trial-and-error tests we found that the concentration of HCl increases almost analogous to the amount of LDH. Specifically, the LDH formed from 50 μmol of Mg2+/Al3+ (1/3) cations could be completely dissolved in 0.5 mL of 0.5 mol L−1 HCl while the LDH formed from 250 μmol of Mg2+/Al3+ (1/3) cations required 0.5 mL of 2.5 mol L−1 HCl to be dissolved completely. On the basis of these observations and in order to maintain a uniform experimental procedure, irrespectively of the sample volume, we dissolved the LDH in 0.5 mL of 2.5 mol L−1 HCl.
3.8. Extraction of AuNPs with different sizes and coatings
AuNPs are used in a plethora of different applications, therefore, it is expected that AuNPs will occur in the environment in a large variety of different sizes, morphologies and surface coatings. In addition, AuNPs undergo a series of physicochemical transformations in the environment that change their initial structural and surface properties.22 Therefore, to enable the determination of AuNPs in environmental samples, analytical methods should be able to extract and determine the total concentration of AuNPs irrespectively of their size and surface coating.The generic utility of the method in the extraction of AuNPs of different sizes and surface coatings was evaluated by extracting 10 mL of aqueous solutions containing citrate-capped AuNPs of 4–60 nm and AuNPs (4 nm) coated with various coatings (in addition to citrate) such as polymers (PVP and PVA) and biomolecules (cysteine). These surface coatings cover a wide range of AuNP capping agents that are used to stabilize AuNPs through electrostatic, steric and electro-steric interactions.
Table 1 presents the calibration functions of the method with citrate-capped AuNPs of variable size. To enable a direct comparison the functions have been calculated using the molar concentration of gold ions (that was used to prepare the AuNPs) and employing logarithmic curves since for larger AuNPs the response deviated significantly from linearity. The slopes of the calibration functions decreased with increasing AuNP size which is reasonable because (a) AuNPs of different sizes contain different amounts of gold atoms (b) AuNP suspensions are typically polydispersed and (c) AuNPs exhibit different absorbance responses that red-shift with increasing AuNP size. Similar observations have been reported using other methods.11,31 From these observations we concluded that this method is suitable for obtaining an estimate of the total concentration of AuNPs. This is made by extrapolating the measured signal to that obtained from AuNPs of a specific size (i.e. the concentration is expressed as equivalents of AuNPs of a specific size that was used for calibration). This approach is common in all generic analytical methods that measure the total concentration of a specific class of compounds (e.g. assays for total antioxidant capacity as gallic acid equivalents, methods of the total phenolic content as trolox equivalents etc.) and has been widely adopted for the analysis of metal nanoparticles by various methods and techniques which calculate the nanoparticle concentration on the basis of an average nanoparticle diameter (usually between 15–20 nm).11,16,18,31
lnx + b) obtained from the extraction of citrated-capped AuNps of variable average size distribution
| Size (nm) | Slope | Intercept | Concentration range (as Au3+ mol L−1 × 10−6) | R 2 |
|---|---|---|---|---|
| 4 | 0.19 | 2.47 | 3–20 | 0.991 |
| 7 | 0.16 | 2.20 | 1.5–10 | 0.98 |
| 10 | 0.17 | 2.20 | 3–20 | 0.98 |
| 14 | 0.11 | 1.44 | 3–30 | 0.99 |
| 20 | 0.1 | 1.32 | 2–30 | 0.994 |
| 30 | 0.09 | 1.30 | 5–30 | 0.997 |
| 50 | 0.08 | 1.10 | 2–30 | 0.98 |
| 60 | 0.05 | 0.76 | 3–30 | 0.996 |
To study the extraction efficiency of AuNPs stabilized with different surface coatings we prepared AuNPs coated with PVP, PVA, cysteine and citrate ions and extracted them under the optimum experimental conditions. The dose–response curves showed that citrate-capped AuNPs were more effectively extracted than AuNPs coated with other molecules. This was attributed to the surface charge of citrate-capped AuNPs as compared to other coatings such as PVP, PVA and cysteine. Although all AuNPs that were examined in this work have a negative charge36–38 citrate-capped AuNPs have lower zeta potential. In addition, citrate is a small molecule as compared to large molecules such as PVP and PVA where steric repulsion forces are significant. Finally cysteine, although it has a free carboxyl group like citrate, also has an ammonium moiety which is positively charged under the working conditions (pKa = 8.2) and may repel the positively charged LDH. When citrate-capped AuNPs were added to tap water the dose–response curves were similar to those observed for other AuNP coatings. This was attributed to the complexation of alkaline earth metals with the carboxylate groups of citrate ions that neutralize the highly negative surface charge of citrate-capped AuNPs.39 In contrast, there was no significant difference in the dose–response curves of PVP, PVA and cysteine-capped AuNPs in tap and distilled water. Therefore, in real samples it should be expected that a uniform response should be produced for AuNPs with variable coatings (Fig. 4).
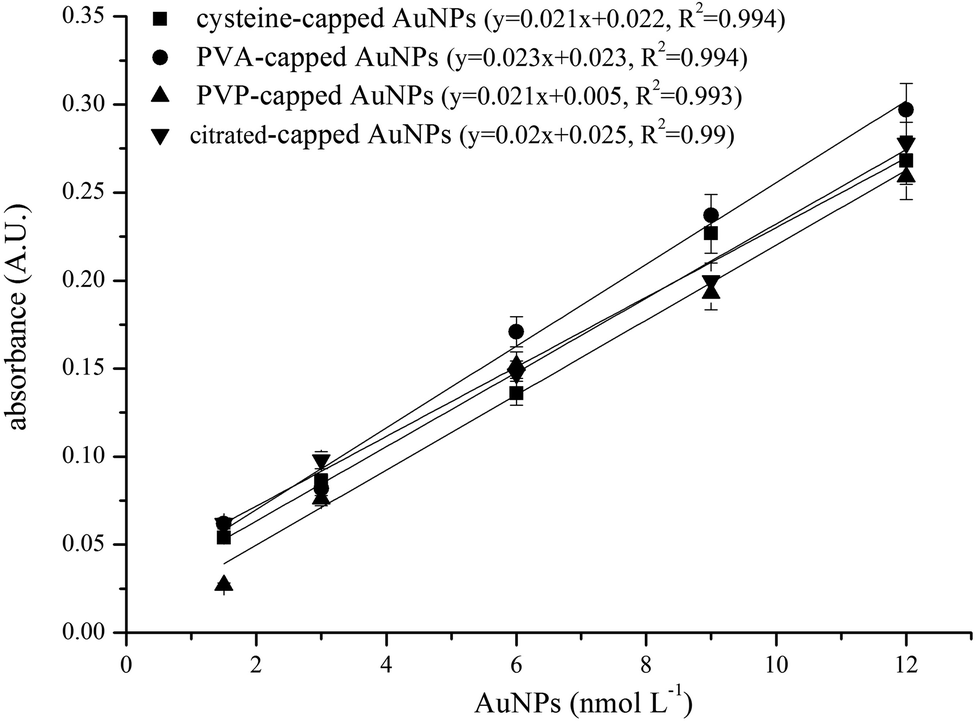 | ||
| Fig. 4 Dose–response curves obtained by extracting 10 mL of tap water spiked with increasing concentrations of AuNPs (4 nm) with different surface coatings. | ||
3.9. Combination of the method with atomic spectroscopy and species selectivity
Spectrophotometry may be an easy and convenient method for the determination of AuNPs due to their characteristic absorbance in the visible region of the electromagnetic spectrum. However, the sensitivity of spectrophotometry is limited to the low nmol L−1 concentration levels. Specifically, the calibration function obtained from spectrophotometric detection by extracting 50 mL of aqueous solutions containing increasing concentrations of citrate-capped AuNPs (4 nm, 8 calibration points) was linear in the range of 0.14–2.1 nmol L−1 (or 0.09–1.4 mg L−1 gold ions) according to the calibration function y = 0.28x − 0.02, R2 = 0.998. Such concentrations may eventually become relevant in the wastewater of laboratories or industries producing large amounts of AuNPs but the concentrations of AuNPs released into the environment are not expected to exceed the ng L−1 or pg L−1 levels. Therefore, more sensitive analytical techniques based on atomic spectroscopy are required for the environmental surveillance of AuNPs.The application of ETAAS was examined as a means to improve the sensitivity of the method and enable the determination of AuNPs at the ng L−1 and pg L−1 levels. ETAAS, however, cannot discriminate between AuNPs and dissolved Au ions that may be present in natural waters. The presence of dissolved Au ions in natural waters may introduce a “Trojan-horse-like” co-extraction effect where Au ions are misinterpreted leading to false-positive estimation of the AuNP concentration in real samples. To assess the selectivity of the method against Au species we extracted aqueous solutions containing citrate-capped AuNPs (20 nm), AuCl4− and mixtures of 1:1 AuCl4−/citrate-capped AuNPs (20 nm) at equimolar gold concentration levels under the optimum experimental conditions. The results showed that both AuNPs and AuCl4− were efficiently extracted using the LDH. In fact, AuCl4− ions could be quantitatively extracted at concentrations as high as 6 mg L−1 due to anion exchange with the interlayer nitrate ions which means that gold ions in environmental samples will also be quantitatively extracted and misinterpreted as AuNPs.
To circumvent this problem we introduced a cleanup step before ETAAS analysis. In this step, the acidic extract obtained after dissolution of the LDH with HCl was ultracentrifuged three times at 18000 rpm (15120 g) for 20 min in order to precipitate the AuNPs.40 The AuNPs that were attached at the bottom of the Eppendorf vial were redispersed in 6 M HNO3 and an aliquot of this sample was directly analyzed by ETAAS. Additionally, due to the clean-up step, no interferences from other ions (such as Ca2+, Fe3+, Cu2+, and Zn2+) that may also precipitate at the working pH value was observed since they were also dissolved and separated in the supernatant solution.
3.10. Analytical figures of merit
To evaluate the analytical merits of the method we extracted 50 mL of aqueous solutions containing increasing concentrations of citrated-capped AuNPs (20 nm) and analyzed them with ETAAS. Parameters such as the linearity of the calibration curve, reproducibility, and recoveries from various water samples were evaluated.The experimental results show that the calibration curves obtained from 8 calibration points are described by a first order polynomial in the range of 50–500 fmol L−1 (12–120 ng L−1 gold ions) according to the equation y = 5 × 10−4x − 0.01, R2 = 0.998. The linear range obtained with ETAAS covers the concentration ranges that are predicted for nanomaterials in environmental samples based on probabilistic or material flow algorithms.4,41 The detection limit, defined as three times the signal to noise ratio, was 19 fmol L−1, corresponding to 4.6 ng L−1 gold ions (calculated on the basis of spherical AuNPs of an average diameter of 20 nm), which is better than or comparable to the LODs reported in previous studies.11–14,17–20,31,42 The LOD can conceivably be improved by increasing the ratio of the sample to extractant volume (i.e. increasing the sample volume or decreasing the volume of HNO3 at the final step of the procedure) or by resorting to more sensitive detectors such as ICP-MS. We assume that higher ratios combined with ICP-MS could bring the LOD to the attomolar levels providing a significant advancement to the early monitoring of AuNPs in the environment. The RSD of the measurements for 5-fold determination of AuNPs at a concentration level of 200 fmol L−1 was evaluated in spiked distilled and river water samples. The RSD ranged from 7.8 to 8.9% in distilled water and river water, respectively, which was considered to be acceptable for the analysis of real samples.
3.11. Method application and recoveries
Although nanomaterials are already considered as emerging environmental pollutants their release into the environment is still limited and real positive samples are not available. Therefore, spiking experiments were used to assess the applicability of the method in the analysis of real samples. Genuine water samples of variable matrix complexity (river water, lake water and effluent wastewater) were spiked with AuNPs and extracted under the optimum experimental conditions. Taking into consideration our previous findings regarding the influence of surface charge of AuNPs on the extraction efficiency and in order to compensate for unrecognized variations in the enrichment procedure, both instrumental and method calibration were performed simultaneously using the same standard solutions composed of 50 mL tap water fortified with increasing concentrations of citrate-capped AuNPs (20 nm).Despite the very low spiking levels of AuNPs (i.e. femtomolar levels), the recoveries ranged from 71–91.5% (Table 2), which is within the range of recovery rates reported in other studies.11–14,17–20 Therefore, the method is suitable for the determination of trace levels of AuNPs in environmental samples.
| Sample | Spiked AuNPs (fM) | Found AuNPs (fM) | Recovery (%) | RSD (%, n = 3) |
|---|---|---|---|---|
| River water | 100.0 | 91 | 91 | 6.7 |
| 200.0 | 183 | 91.5 | 6.3 | |
| Lake water | 100.0 | 71 | 71 | 7.4 |
| 200.0 | 160 | 80 | 7.1 | |
| Effluent wastewater | 200.0 | 157 | 78.5 | 8.2 |
| 400.0 | 340 | 85 | 7.9 |
4. Conclusion
In this work we demonstrated for the first time the ability of layered double hydroxides to extract AuNPs from environmental water samples. The extraction is accomplished in a single step through electrostatic interactions between the positively charged AuNPs and LDHs by in situ formation and dissolution of LDHs in the sample by regulating the pH of the solution. Additionally, by using this method both AuNPs and Au ions can be extracted while species separation is accomplished by ultracentrifugation. In combination with ETAAS the method enables the determination of AuNPs at the femtomolar concentration levels with satisfactory analytical features and sensitivity that are comparable with those of more complex methods or advanced analytical techniques. A disadvantage of the method is the need for ultracentrifugation prior to detection. However, the simplicity of the experimental procedure, the high sample throughput (36 or more samples can be extracted simultaneously depending on the capacity of the centrifuge) and the very low detection limits (femtomolar and potentially attomolar levels with more sensitive detectors) make the method an attractive alternative for routine applications.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research has been financially supported by the General Secretariat for Research and Technology (GSRT) and the Hellenic Foundation for Research and Innovation (HFRI) (Scholarship Code: 763).References
- C. O. Hendren, X. Mesnard, J. Droge and M. R. Wiesner, Environ. Sci. Technol., 2011, 45, 2562–2569 CrossRef CAS PubMed.
- F. Piccinno, F. Gottschalk, S. Seeger and B. Nowack, J. Nanopart. Res., 2012, 14, 1109 CrossRef.
- T. Sun, D. M. Mitrano, N. A. Bornhoft, M. Scheringer, K. Hungerbuehler and B. Nowack, Environ. Sci. Technol., 2017, 51, 2854–2863 CrossRef CAS PubMed.
- B. Giese, F. Klaessig, B. Park, R. Kaegi, M. Steinfeldt, H. Wigger, A. von Gleich and F. Gottschalk, Sci. Rep., 2018, 8, 1565 CrossRef PubMed.
- European Commission, Nanomaterials, Available from: https://ec.europa.eu/environment/chemicals/nanotech/index_en.htm, 2019, accessed 21 August 2019 Search PubMed.
- OECD, Safety of manufactured nanomaterials, Available from: http://www.oecd.org/env/ehs/nanosafety/, 2019, accessed 21 August 2019 Search PubMed.
- E. M. Heithmar, Screening methods for metal-containing nanoparticles in water, United States Environmental Protection Agency (EPA), Washington, 2011 Search PubMed.
- H. Zanker and A. Schierz, Annu. Rev. Anal. Chem., 2012, 5, 107–132 CrossRef CAS PubMed.
- B. F. Da Silva, S. Perez, P. Gardinalli, R. K. Singhal, A. A. Mozeto and D. Barcelo, TrAC, Trends Anal. Chem., 2011, 30, 528–540 CrossRef.
- J. F. Liu, R. Liu, Y. G. Yin and G. B. Jiang, Chem. Commun., 2009, 28, 1514–1516 RSC.
- G. Hartmann and M. Schuster, Anal. Chim. Acta, 2013, 761, 27–33 CrossRef CAS PubMed.
- G. Z. Tsogas, D. L. Giokas and A. G. Vlessidis, Anal. Chem., 2014, 86, 3484–3492 CrossRef CAS PubMed.
- T. G. Choleva, F. A. Kappi, G. Z. Tsogas, A. G. Vlessidis and D. L. Giokas, Talanta, 2016, 151, 91–99 CrossRef CAS PubMed.
- Y. Liu, M. He, B. Chen and B. Hu, Spectrochim. Acta, Part B, 2016, 122, 94–102 CrossRef CAS.
- S. M. Majedia, B. C. Kelly and H. K. Lee, Anal. Chim. Acta, 2013, 789, 47–57 CrossRef PubMed.
- T. G. Choleva, G. Z. Tsogas and D. L. Giokas, Talanta, 2019, 196, 255–261 CrossRef CAS PubMed.
- L. Li, K. Leopold and M. Schuster, Chem. Commun., 2012, 48, 9165–9167 RSC.
- S. Su, B. Chen, M. He, Z. Xiao and B. Hu, J. Anal. At. Spectrom., 2014, 29, 444–453 RSC.
- L. Li and K. Leopold, Anal. Chem., 2012, 84, 4340–4349 CrossRef CAS PubMed.
- L. Zhang, B. Chen, M. He, X. Liu and B. Hu, Anal. Chem., 2015, 87, 1789–1796 CrossRef CAS PubMed.
- G. V. Lowry, K. B. Gregory, S. C. Apte and J. R. Lead, Environ. Sci. Technol., 2012, 46, 6893–6899 CrossRef CAS PubMed.
- Q. Wang and D. O'Hare, Chem. Rev., 2012, 112, 4124–4155 CrossRef CAS PubMed.
- G. Mishra, B. Dasha and S. Pandey, Appl. Clay Sci., 2018, 153, 172–186 CrossRef CAS.
- S. Tang and H. K. Lee, Anal. Chem., 2013, 85, 7426–7433 CrossRef CAS PubMed.
- H. Abdolmohammad-Zadeh, Z. Rezvani, G. H. Sadeghi and E. Zorufi, Anal. Chim. Acta, 2011, 685, 212–219 CrossRef CAS PubMed.
- N. Gissawong, S. Sansuk and S. Srijaranai, Spectrochim. Acta, Part A, 2017, 173, 994–1000 CrossRef CAS PubMed.
- S. Seidi and S. E. Sanati, Microchim. Acta, 2019, 186, 297 CrossRef PubMed.
- X. Wang, W. Zhou, C. Wang and Z. Chen, Talanta, 2018, 186, 545–553 CrossRef CAS PubMed.
- X. Zhao, S. Liu, P. Wang, Z. Tang, H. Niu, Y. Cai, F. Wu, H. Wang, W. Meng and J. P. Giesy, J. Chromatogr. A, 2015, 1414, 22–30 CrossRef CAS PubMed.
- X. Li, Y. Sun, L. Yuan, L. Liang, Y. Jiang, H. Piao, D. Song, A. Yu and X. Wang, Microchim. Acta, 2018, 185, 336 CrossRef PubMed.
- S. P. Mandyla, G. Z. Tsogas, A. G. Vlessidis and D. L. Giokas, J. Hazard. Mater., 2017, 323, 67–74 CrossRef CAS PubMed.
- W. Haiss, N. T. K. Thanh, J. Aveyard and D. G. Fernig, Anal. Chem., 2007, 79, 4215–4221 CrossRef CAS PubMed.
- S. Murath, Z. Somosi, I. Y. Toth, E. Tombacz, P. Sipos and I. Palinko, J. Mol. Struct., 2017, 1140, 77–82 CrossRef CAS.
- O. Saber and H. Tagaya, Mater. Chem. Phys., 2008, 108, 449–455 CrossRef CAS.
- M. Pavlovic, R. Huber, M. Adok-Sipiczki, C. Nardin and I. Szilagyi, Soft Matter, 2016, 12, 4024–4033 RSC.
- H. M. Zakaria, A. Shah, M. Konieczny, J. A. Hoffmann, A. J. Nijdam and M. E. Reeves, Langmuir, 2013, 29, 7661–7673 CrossRef CAS PubMed.
- K. Rahme, G. Minassian, M. Sarkis, M. Nakhl, R. El Hage, E. Souaid, J. D. Holmes and E. Ghanem, J. Nanomater., 2018, 9301912 CAS.
- T. Das, V. Kolli, S. Karmakar and N. Sarkar, Biomedicines, 2017, 5, 19 CrossRef PubMed.
- N. P. Koutsoulis, D. L. Giokas, A. G. Vlessidis and G. Z. Tsogas, Anal. Chim. Acta, 2010, 669, 45–52 CrossRef CAS PubMed.
- Nanopartz Inc, https://www.nanopartz.com/gold-nanoparticles-properties-centrifuge-speeds.asp, accessed, 13 November 2019.
- I. Mahapatra, T. Y. Sun, J. R. A. Clark, P. J. Dobson, K. Hungerbuehler, R. Owen, B. Nowack and J. Lead, J. Nanobiotechnol., 2015, 13, 93 CrossRef PubMed.
- J. F. Liu, J. B. Chao, R. Liu, Z. Q. Tan, Y. G. Yin, Y. Wu and G. B. Jiang, Anal. Chem., 2009, 81, 6496–6502 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ay02321f |
| This journal is © The Royal Society of Chemistry 2020 |