
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLoop-mediated isothermal amplification (LAMP) – review and classification of methods for sequence-specific detection
Lisa
Becherer
 ab,
Nadine
Borst
ab,
Mohammed
Bakheit
c,
Sieghard
Frischmann
c,
Roland
Zengerle
ab and
Felix
von Stetten
*ab
ab,
Nadine
Borst
ab,
Mohammed
Bakheit
c,
Sieghard
Frischmann
c,
Roland
Zengerle
ab and
Felix
von Stetten
*ab
aLaboratory for MEMS Applications, IMTEK - Department of Microsystems Engineering, University of Freiburg, Georges-Koehler-Allee 103, 79110 Freiburg, Germany. E-mail: Felix.von.Stetten@Hahn-Schickard.de
bHahn-Schickard, Georges-Koehler-Allee 103, 79110 Freiburg, Germany
cMast Diagnostica GmbH, Feldstraße 20, 23858 Reinfeld, Germany
First published on 9th January 2020
Abstract
In the course of the last 20 years, isothermal nucleic acid amplification tests have emerged as an important diagnostic tool, not only for clinical applications, but also for food quality control and environmental monitoring. Loop-mediated isothermal amplification (LAMP) is well known for its robust and highly sensitive and specific amplification of target DNA, which is achieved by utilizing up to six primers. Moreover, LAMP excels through its isothermal and energy efficient amplification requirements, rendering it a prime candidate for low-cost diagnostics and analysis at the point of need. Recently, methods for sequence-specific detection have gained more importance because, unlike sequence-independent detection methods, they are highly specific towards the target DNA. In the last 13 years, a variety of sequence-specific methods have emerged, based on a very diverse range of sensing techniques, including optical, magnetic, piezoelectric, electrochemical and magnetoresistive sensing. To give structure to the diverse multitude of sequence-specific methods, we created a systematic classification and provide a critical comparative evaluation according to a catalogue of criteria (analytical performance, multiplexing, quantification and instrumental requirements). Fluorescence-based detection, making up half of the methods, can be processed on open platforms and satisfies all the criteria listed before. Instrumental requirements are discussed in terms of complexity, portability and fluidic cartridges. In addition, the technological readiness level and the kind of platform (open versus method-tailored) are evaluated, the latter playing an important role in the miniaturization and automation of operational process steps. We also observe an increase in the use of smartphone-integrated sensors to improve LAMP-based point-of-need testing. In summary, recent developments in methods for the sequence-specific detection of LAMP demonstrate high potential for many future applications.
 Lisa Becherer | Lisa Becherer studied chemistry at the University of Freiburg. In 2015 she graduated as a Master of Science with a master's thesis on spin–spin interactions on DNA aptamer complexes. She currently works as a PhD candidate at Hahn-Schickard and at the Laboratory for MEMS Applications at IMTEK, University of Freiburg. Her research involves nucleic acid analysis with focus on isothermal amplification and digital amplification on centrifugal microfluidic cartridges. |
 Nadine Borst | Dr Nadine Borst studied biochemistry at the University of Regensburg and completed her PhD at the Technical University of Munich in cooperation with the Fraunhofer Institute for Interfacial Engineering and Biotechnology. There, she worked in the field of enzyme engineering and assay development. In 2016, Nadine Borst joined Hahn-Schickard and since February 2017 she is heading the Nucleic Acid Analysis group. Her current research is focused on real-time and digital assays, isothermal amplification and single cell analysis on lab-on-a-chip platforms. |
 Mohammed Bakheit | Dr Mohammed Bakheit obtained his Bachelor and Master's degrees in the Faculty of Veterinary Medicine, University of Khartoum and doctoral degree in the Free University of Berlin. As a postdoc, he worked in diagnostics (both serological and molecular), first in the National Reference Center for Protozoan Diseases (NRCPD), Obihiro University focusing on methods for the diagnosis of Cryptosporidium species infection using LAMP. In his second postdoc term at Research Center Borstel he participated further in development of isothermal diagnostics for tick-borne diseases. Since 2014, he has been working for the product development team of Mast Diagnostica GmbH using LAMP. |
 Sieghard Frischmann | Dr Sieghard Frischmann studied biology at the Friedrich-Alexander-Universität Erlangen and completed his PhD at the University of Hamburg, Universitätsklinik Eppendorf. Sieghard Frischmann joined Mast in 1992 and now he is head of product development and production at Mast. He is leading a team for the development of molecular diagnostic (based on the LAMP) and protein technologies. He works as a Medical Product Safety Manager in the terms of the EN ISO 13485 standard and the German Medical Product Law. He leads the Regulatory Affairs Team at Mast and is responsible for product registration worldwide. |
 Roland Zengerle | Prof. Dr-Ing. Roland Zengerle is full professor at the Department of Microsystems Engineering at the University of Freiburg, director at the “Hahn-Schickard-Institut für Mikroanalysesysteme”. The research of Dr Zengerle is focused on Microfluidics, lab-on-a-chip as well as Electrochemical Energy Systems. Dr Zengerle co-authored more than 380 papers. From 2004 until 2014, he was member of the editorial board of the Springer Journal of Microfluidics and Nanofluidics and since 2017, he is a Member of the Advisory Board of the scientific journal “Lab on a Chip”. Since 2011, Dr Zengerle is a member of the German national academy of sciences, Leopoldina. |
 Felix von Stetten | Apl. Prof. Dr Felix von Stetten completed his PhD in Microbiology in 1999 from the Technical University of Munich. Thereafter, he joined in the diagnostic industry, where he was involved in the development of methods for sample preparation, real-time PCR and DNA-arrays. Subsequently, he joined the Laboratory for MEMS Applications at IMTEK, University of Freiburg, where he was involved in lab-on-a-chip-research and the development of molecular diagnostic techniques. In 2008, he became head of the Hahn-Schickard lab-on-a-chip division and in 2016 associate director of the Hahn-Schickard-Institut für Mikroanalysesysteme. |
1. Introduction
Since the revolutionary development of the polymerase chain reaction (PCR) in the 1980s,1,2 nucleic acid amplification tests (NAATs) have become an indispensable tool throughout the entire life sciences field, and have even grown to be the gold standard of nucleic acid analysis, especially in clinical applications,3–6 but also for food quality control7 and environmental monitoring.8 A remarkable trend, emerging between 1995 and 2005, can also be observed in the development of isothermal NAATs, which was provoked by the limitations of PCR. The complex and expensive devices required for thermal cycling and real-time detection during PCR restrict the use of this amplification method. Isothermal NAATs enable amplification reactions at constant and moderate temperatures. Simple and low-cost devices, as well as fast processing times compared to PCR, make isothermal NAATs increasingly attractive and open up new application opportunities in the field of point-of-care (POC)/point-of-need (PON) testing.9–11The development of rolling circle amplification (RCA),12 the pioneering isothermal NAAT method, laid the cornerstone for ongoing research into numerous alternative methods enabling isothermal amplification of DNA. These include loop-mediated isothermal amplification (LAMP),13 recombinase polymerase amplification (RPA),14 nucleic acid sequence based amplification (NASBA),15 strand displacement amplification (SDA),16 helicase dependent amplification (HDA)17 and transcription-mediated amplification (TMA)18 to just name the most important ones. LAMP and RCA rank among the most frequently used methods described in literature. The percentage of publications listed in Web of Science that have LAMP in their title, illustrated in Fig. 1, demonstrates the prevalence of LAMP among isothermal NAATs (status as of 2019).
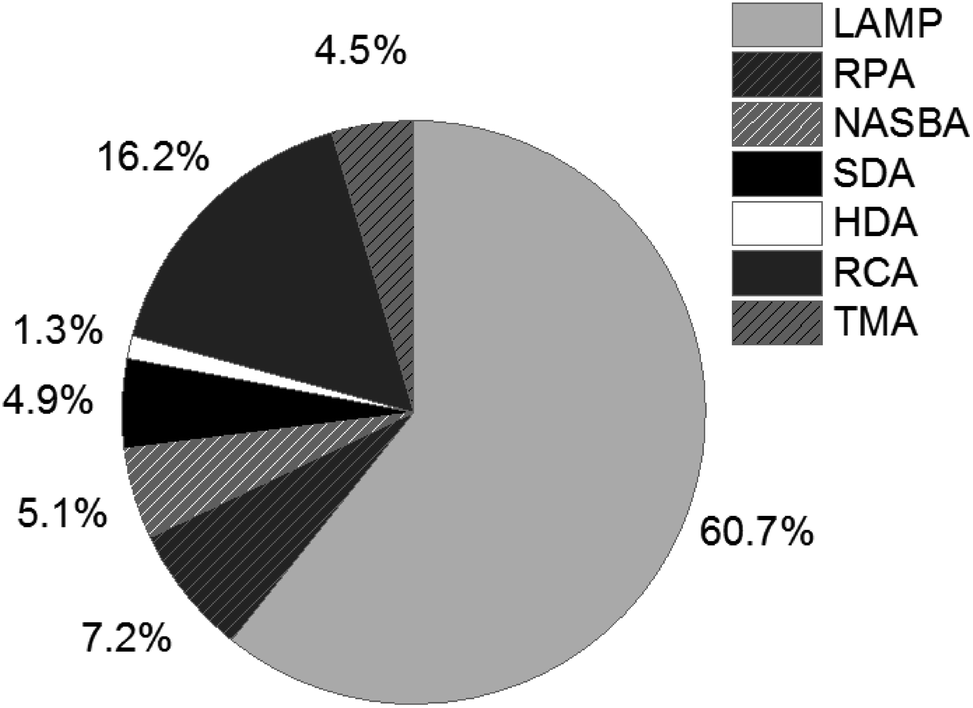 | ||
| Fig. 1 Percentage of publications listed in Web of Science (status as of 2019), refined search for appearance in title. | ||
LAMP was first described by Notomi et al. in 200019 and was optimized with additional primers for accelerated amplification by Nagamine et al.20 It has proven itself through its high sensitivity and specificity and its tolerance to PCR-inhibiting substances.21,22 The outstanding specificity is accomplished by using four to six specific primers, which recognize six to eight different regions in the target DNA sequence. A standard method for the detection of LAMP is the measurement of the turbidity caused by precipitated magnesium pyrophosphate,23,24 as well as endpoint detection with the naked eye.25–27 Other target sequence-independent detection methods rely on gel electrophoresis,28 metal indicators for calcium,29 colorimetric LAMP,30 coffee-ring effect on colloid-crystal substrates,31 paper-based rapid detection of LAMP–magnetic bead aggregates,32 melting and annealing curve analysis,33 intercalating fluorescent dyes such as SYBR green,34–36 bioluminescence through pyrophosphate conversion37,38 or electrochemiluminescence.39,40
LAMP stands out in terms of its high analytical specificity towards the target analyte due to the utilization of six primers. However, the large number of long primers means there is a risk of primer dimers forming.41,42 Sequence-independent detection methods, listed above, indicate extended primer dimers as false positive results. In contrast, methods for sequence-specific detection excel through the use of target-specific probes or modified primers as biorecognition elements. Such sequence-specific detection enables the accurate identification of amplicons without being affected by unspecific products. Moreover, sequence-specific detection allows for differentiation between and identification of multiple targets simultaneously in one assay (one-pot multiplexing43). Appropriate selection and adaption of biorecognition elements plays an essential role in biosensing performance, especially with respect to sensitivity and specificity.44 Recent advancements have provided automated systems with integrated sample handling, sample preparation and biosensing.45 This integration of several laboratory processes into miniaturized total chemical analysis systems (μTAS) offers tremendous advantages, such as lower costs due to reduced volumes and consumption of reagents, compact system size, and high throughput.46,47 Furthermore, miniaturization reduces the complexity of the instrumentation and its operation and, combined with the isothermal reaction conditions enabled by LAMP, paves the way for on-site testing in the environmental field48,49 or close to the patient at the PON.50,51
Methods for real-time monitoring and endpoint detection of LAMP were summarized in a brief review by Zhang et al.52 These methods comprise diverse sequence-independent and sequence-specific methods. Furthermore, Mayboroda et al.43 reviewed multiplex methods for isothermal amplification in general. As a completion of the previous work and in consideration of the novel methods published after 2014, we provide the first comprehensive, systematic classification and critical analysis of such methods. In this review, we place particular focus on methods for sequence-specific detection. First, we give a short introduction to the principle of a LAMP reaction. The terms used throughout this review are defined in Table 1. In chapter 3, we classify the sequence-specific detection methods for LAMP according to the transducing elements and sensing techniques they use. We define the assessment criteria which we use to evaluate the different methods at the beginning of chapter 4. The main section of chapter 4 gives a detailed description and illustration of the individual methods, and related instrumentation is discussed in chapter 5. We compare the different methods according to our assessment criteria and their related instrumentation in a general discussion in chapter 6, and provide a summarizing overview in Table 2.
| Term | Definition |
|---|---|
| Analytical sensitivity | The analytical sensitivity of an NAAT describes the minimum number of target copies in a sample that can be measured accurately.53 It is expressed as the limit of detection (LOD), describing the number of copies which can be detected with a probability of 95% |
| Analytical specificity | The analytical specificity of an NAAT describes the probability of detecting the correct target sequence rather than non-specific targets53 |
| Biorecognition element | Element in a biosensor which specifically interacts with the target analyte |
| Biorecognition event | Interaction between biorecognition element and target analyte |
| Biosensor | “A device that uses specific biochemical reactions mediated by isolated enzymes, immunosystems, tissues, organelles or whole cells to detect chemical compounds usually by electrical, thermal or optical signals”. (B. Nagel, H. Dellweg and L. M. Gierasch 1992)54,55 |
| Diagnostic sensitivity | This is also referred to as clinical sensitivity, and describes the percentage of positive test results which are correctly identified as positive53 |
| Diagnostic specificity | This is also referred to as clinical specificity and describes the percentage of negative test results which are correctly identified as negative53 |
| FRET | Förster resonance energy transfer. The energy of an excited fluorophore (donor) is transferred radiation-free to a second light-sensitive molecule (acceptor), also called a quencher |
| Heterogeneous detection | Planar sensor surfaces, connected to the transducing element, are used for heterogeneous detection. During signal detection, biorecognition elements capturing the target or a mediator, are in close proximity or linked to the sensing surface |
| Homogeneous detection | Signals are generated within and detected from the entire volume of the reaction mix. During signal transduction, biorecognition elements are homogeneously distributed in the reaction volume, either dissolved in solution or immobilized on suspended particles |
| Imprecision | Describes the repeatability (intraassay variance) and reproducibility (interassay variance) of an NAAT53 |
| Method-tailored demonstrators | Custom-made instrumentation, comprising devices and/or disposables, which was developed for a special method and application |
| Miniaturized total chemical analysis systems (μTAS) | μTAS are devices which provide automated sample handling. All steps for the analysis of a sample are included (sample preparation, (bio)chemical reaction and detection) |
| One-pot multiplexing | Multiple target analytes are parallelly amplified in parallel in a single reaction, comprising multiple primer sets, and detected simultaneously43 |
| Open platforms | Commercially available instrumentation, which can beis adaptative to different protocols and reagents. The instrumentation comprises devices and related disposable cartridges, such as chips or disks |
| Signal transduction | The process of converting a biorecognition event into an electrical signal with a transducing element |
| Transducing element | Element in a biosensor, which converts the biorecognition event into an electrical measurable electrical signal |
| TRL | Technological readiness level. This scale indicates the development status of a technology |
2. LAMP mechanism
The principle of LAMP is depicted in Fig. 2. The optimum temperature range of LAMP is between 60–65 °C. LAMP utilizes two outer primers (forward outer primer, F3, and backward outer primer, B3), two inner primers (forward inner primer, FIP, and backward inner primer, BIP) and a DNA polymerase with strand-displacement activity.19 LAMP can be accelerated by a factor of two by using an additional pair of primers, the loop primers (forward loop primer, LF, and backward loop primer, LB).20 Initially, the inner primer FIP, which contains two target sequences specific to two different regions in the template DNA, hybridizes to the target DNA and starts complementary strand synthesis (A). The outer primer F3 starts strand displacement of the elongated FIP primer, releasing single stranded (ss) DNA that serves as a template for the backward primers (B). The BIP primer starts strand synthesis at the ssDNA and is subsequently displaced by the B3 primer (C). Both the 3′ and the 5′ end are complementary to sequences further inwards, enabling the formation of a stem-loop DNA structure (C and D). The stem-loop structure is the starting point for exponential amplification. Self-priming and the elongation of its 3′ end (F1) induces displacement of the 5′ end (B1c), subsequent unfolding of the hairpin structure, and backfolding of the newly synthesized strand. Repetition of the self-priming pathway generates long amplicons with cauliflower-like structures. Additionally, inner and outer primers hybridize to the loop structures and initialize strand synthesis and subsequent displacement. Loop primers, annealing to the loops in the stem-loop structure (D), further accelerate LAMP.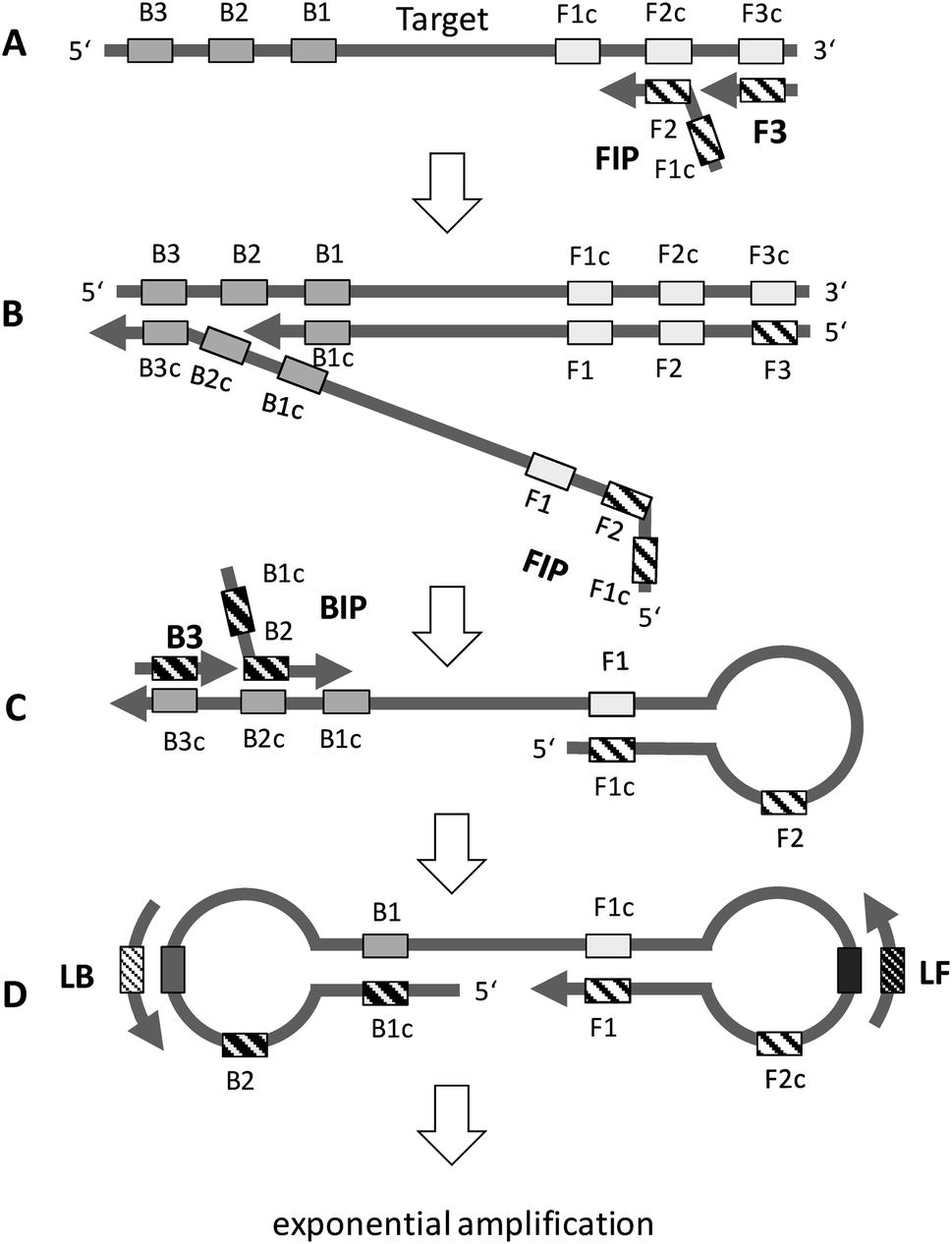 | ||
| Fig. 2 General principle of LAMP. See text for explanation (redrawn according to ref. 19 and 20). | ||
3. Classification of detection methods
We have divided the methods used for sequence-specific detection of LAMP into groups and sub-groups according to the techniques used for signal detection. For a visual illustration of this classification, we provide a graphical overview of all the methods discussed within this review (Fig. 3). The methods are classified into homogeneous and heterogeneous detection (Fig. 3, black boxes). We further differentiate between particle-free and particle-based methods (Fig. 3, dark grey boxes) and classify the methods according to the transducing elements (Fig. 3, grey boxes) and sensing techniques (Fig. 3, light grey boxes) used. This systematic scheme is applicable to real-time as well as to endpoint detection of LAMP.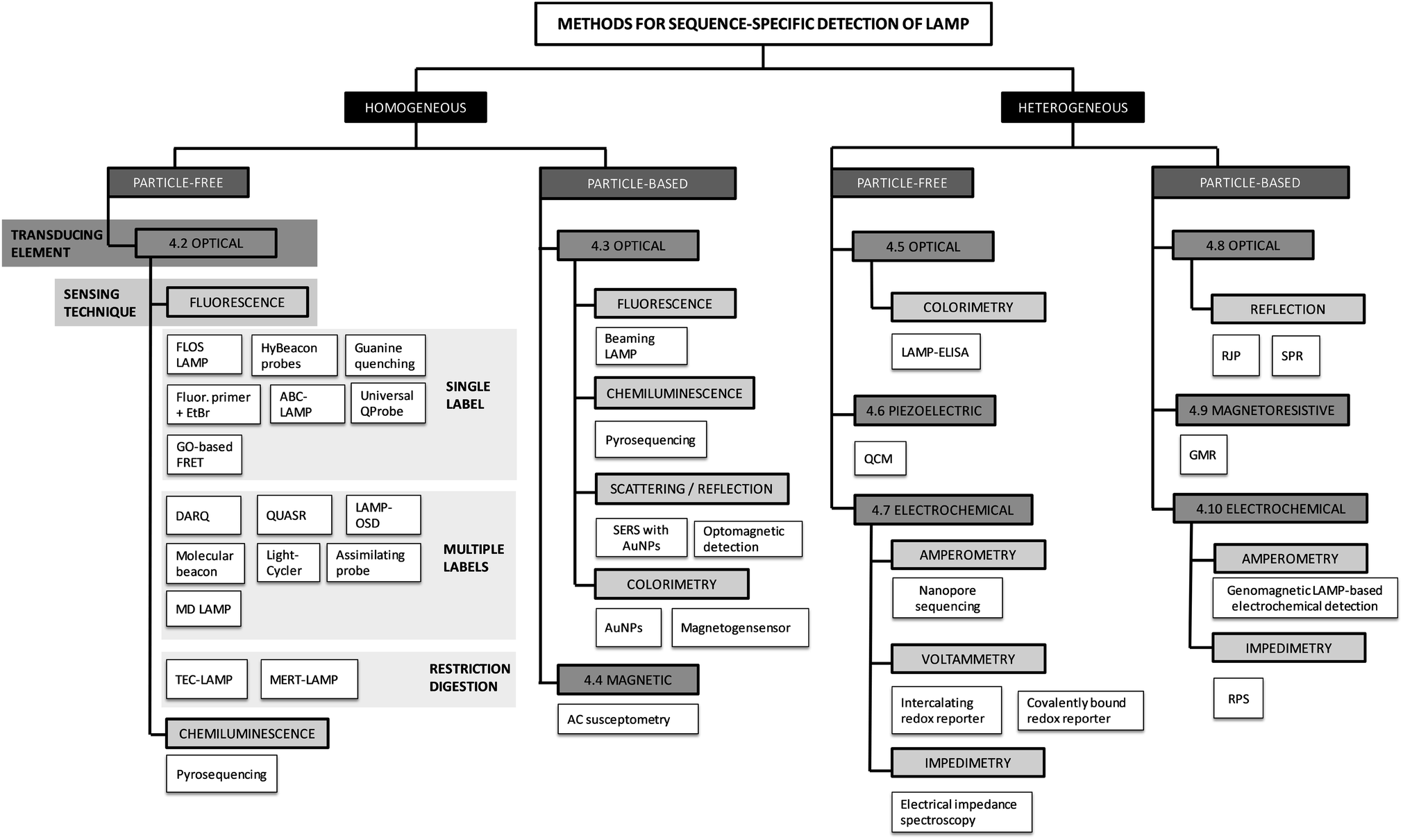 | ||
| Fig. 3 Overview and grouping of the methods for sequence-specific detection of LAMP. The abbreviations used herein are explained in the following: fluorescence of loop primer upon self-dequenching (FLOS), alternately binding quenching probe competitive LAMP (ABC-LAMP), quenching probe (QProbe), graphene oxide (GO), detection of amplification by release of quenching (DARQ), quenching of unincorporated amplification signal reporters (QUASR), one-step strand displacement (OSD), mediator displacement (MD), multiple endonuclease restriction real-time (MERT), Tth endonuclease cleavage (TEC), surface-enhanced Raman spectroscopy (SERS), gold nanoparticles (AuNPs), enzyme-linked immunosorbent assay (ELISA), quartz crystal microbalance (QCM), retroreflective Janus particle (RJP), surface plasmon resonance (SPR), giant magnetoresistive (GMR) measurement, resistive pulse sensing (RPS). | ||
The major divide is into homogeneous and heterogeneous methods. Homogeneous detection methods rely on signals generated within and detected from the entire volume of the reaction mix. During signal transduction (see definitions), biorecognition elements (see definitions) are homogeneously distributed in the reaction volume, either dissolved in solution (particle-free detection) or immobilized on suspended particles (particle-based detection). In contrast, heterogeneous detection methods make use of planar sensor surfaces connected to the transducing element. For signal detection, biorecognition elements which capture the target analyte are in close proximity or linked to the sensing surface. For the next two levels of the systematic classification of the methods, we rely on the differentiation of the signal transduction and sensing techniques. Signal transduction, the conversion of the biorecognition event into a measurable signal, can be classified according to the transducing element (Fig. 3, grey boxes): optical, piezoelectric, magnetic, magnetoresistive or electrochemical. We further divided the methods using each type of transducer into sub-groups depending on the sensing technique used (Fig. 3, light grey boxes).
4. Methods for sequence-specific detection of LAMP
4.1. Assessment criteria
In this review we assess 34 different methods for sequence-specific detection of LAMP. In order to compare these methods, we define assessment criteria, listed in Table 2. Data that is available and published for each method is indicated with an “X”. The first criterion is the analytical performance. This was assessed according to the MIQE guidelines,53 which define the following criteria: analytical sensitivity, analytical specificity and imprecision (accuracy, repeatability and reproducibility). The second criterion is the ability to cope with complex sample material. To assess the second criterion, we evaluated published clinical studies reporting on the diagnostic sensitivity and diagnostic specificity, or demonstrating proof of concept with real sample material. Organisms and target genes utilized to assess the analytical performance or the diagnostic sensitivity and specificity are listed in Table 3. The third, fourth and fifth assessment criteria were the capability regarding multiplexing, quantification and detection by naked eye. The final criterion was the instrumental requirements. Method-tailored and open platforms that are used to apply a certain method are considered separately (platform section in Table 2) and are evaluated in chapters 5 and 6.4.| Methods for sequence-specific detection of LAMP | Analytical performance | Complex sample material | References validation | Multiplexing | Quantification | Detection by naked eye | Instrumental platform | Compatible method(s) | TRL | Open (O)/method-tailored (M) | Portable devices | Cartridge | Reusable sensor/cartridges | Complexity | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Sensitivity | Specificity | Imprecision | Proof of concept | Diag. sensitivity | Diag. specificity | Reagents preparation | Operational steps | Training, expertise | ||||||||||||
| a A “X” indicates available data, published in literature. Brackets illustrate, that quantification and/or multiplexing is possible and has been shown for similar sensing techniques. b The complexity of the practical operation on the platform is rated by score factors. Score factor 1 indicates a low level of complexity and score factor 3 a high level of complexity. | ||||||||||||||||||||
| 4.2.1. FLOS LAMP & 4.2.2. HyBeacon probes56–62 | X | X | X | X | 59 and 61 | X | X | X | Standard isothermal amplification device with integrated fluorescence reader | 4.2.1.–4.2.16. | 9 | O | X | 1 | 1 | 1 | ||||
| Visual detection by adding polyethylenimine (PEI)60,62 | ||||||||||||||||||||
| 4.2.3. Guanine quenching63 | X | X | X | X | 63 | (X) | X | Standard isothermal amplification device with integrated fluorescence reader (see Table 2, row 4.2.1., instrumental platform) | ||||||||||||
| 4.2.4. ABC-LAMP64 | (X) | X | ||||||||||||||||||
| 4.2.5. Fluoro-primer with EtBr65 | X | 65 | X | X | ||||||||||||||||
| 4.2.6. Universal QProbe66 | X | 66 | (X) | (X) | ||||||||||||||||
| 4.2.7. GO-based FRET67 | X | X | 67 | (X) | X | |||||||||||||||
| 4.2.8. DARQ27,68–70,72 | X | X | X | X | X | 69 | X | X | ||||||||||||
| 4.2.9. QUASR73–82 | X | X | 74–76 | X | X | Custom-made portable “LAMP box” with smartphone readout74,75 | 4.2.1.–4.2.16. | 4 | M | X | 1 | 1 | 1 | |||||||
| Custom-made centrifugal microfluidic platform for the orthogonal detection of proteins and nucleic acids82 | 4.2.1.–4.2.16. | 4 | M | X | X | 1 | 1 | 1 | ||||||||||||
| 4.2.10. LAMP-OSD83–86 | X | X | X | X | X | 86 | X | X | Custom-made filter-based sample-to-answer platform by visual endpoint detection with smartphones85,86 | 4.2.1.–4.2.16. | 4 | M | X | 1 | 2 | 2 | ||||
| 4.2.11. Molecular beacon87–91 | X | X | X | X | X | 87 | X | X | Custom-made digital microfluidic (DMF) system with readout under a fluorescence microscope89 | 4.2.1.–4.2.16. | 4 | M | X | 1 | 2 | 2 | ||||
| 4.2.12. LightCycler94,95 | X | X | X | 94 and 95 | (X) | X | Standard isothermal amplification device with integrated fluorescence reader (see Table 2, row 4.2.1., instrumental platform) | |||||||||||||
| 4.2.13. Assimilating probe96–100 | X | 97 and 98 | X | X | Custom-made power-free device for incubation at LAMP conditions combined with fluorometer97,98 | 4.2.1.–4.2.16. | 4 | M | X | 1 | 1 | 1 | ||||||||
| Custom-made small handheld device for incubation and detection99 | 4.2.1.–4.2.16. | 4 | M | X | 1 | 1 | 1 | |||||||||||||
| 4.2.14. MD LAMP101,102 | X | X | X | X | X | X | 101 and 102 | X | X | Standard isothermal amplification device with integrated fluorescence reader (see Table 2, row 4.2.1., instrumental platform) | ||||||||||
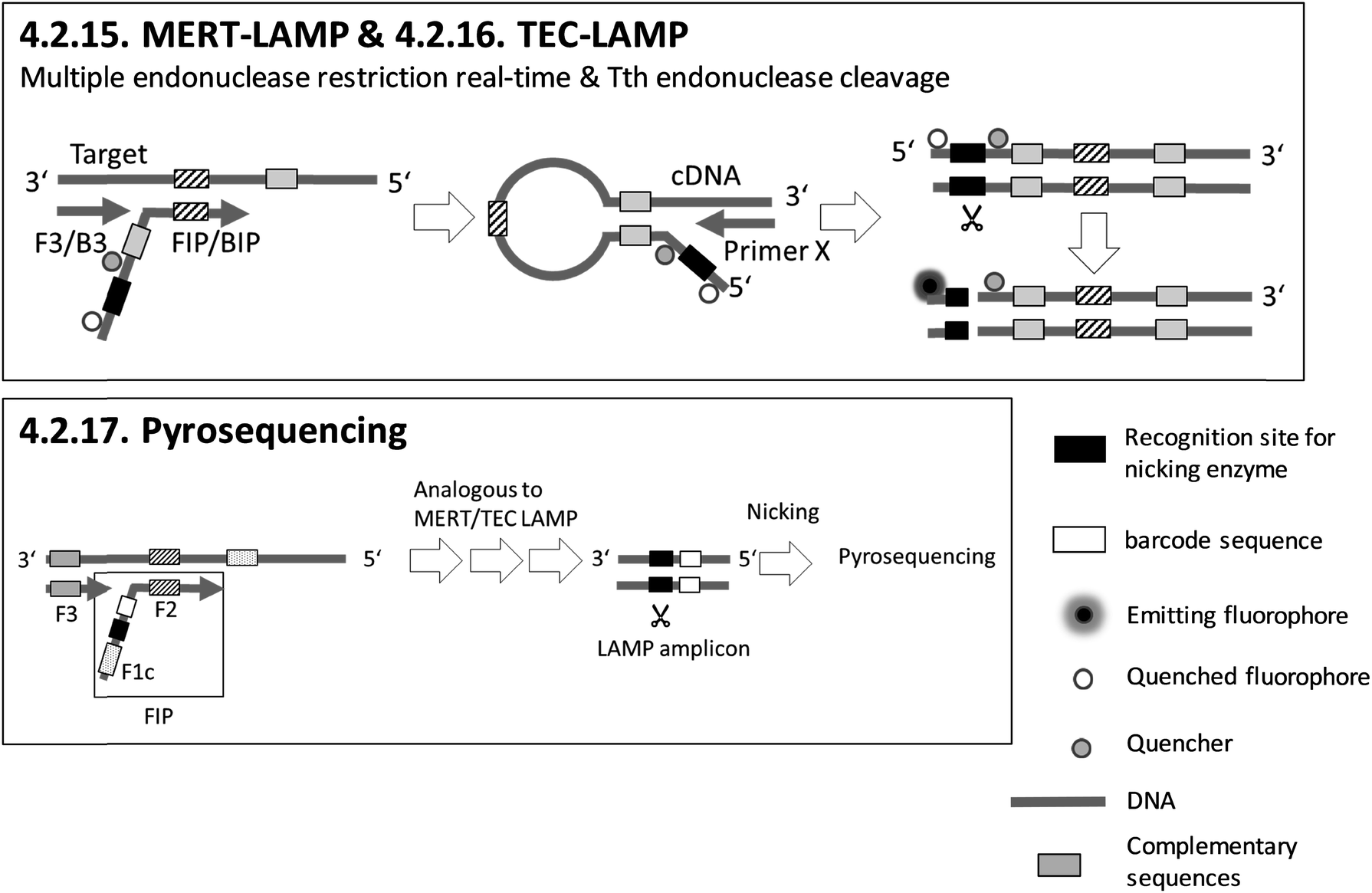
| 4.2.15. MERT-LAMP103,104 | X | X | X | 103 and 104 | X | X | X | |||||||||||||
| 4.2.16. TEC-LAMP105 | X | X | X | X | X | 105 | X | X | ||||||||||||
| 4.2.17. Pyrosequencing106 | X | Pyrosequencer (Hitachi, Ltd., Central Research Laboratory, Japan)106 | 4.2.17. | 9 | O | X | X | 1 | 3 | 3 | ||||||||||
| 4.3.1. BEAMing LAMP107 | X | X | X | 107 | (X) | X | Emulsification in a 48-well TissueLyser, Flow cytometry107 | 4.3.1. | 9 | O | 2 | 3 | 3 | |||||||
| 4.3.2. Surface enhanced Raman spectroscopy (SERS) with AuNPs111 | X | X | X | 111 | X | Confocal micro-Raman spectroscopic system equipped with a Leica microscope111 | 4.3.2. | 9 | O | 2 | 3 | 3 | ||||||||
| 4.3.3. Optomagnetic detection112,113 | X | X | 113 | X | Custom made chip and measuring station for optical detection112 | 4.3.3. | 4 | M | X | 2 | 1 | 2 | ||||||||
| 4.3.4. AuNPs114–124 | X | X | X | X | 114 | X | Visual detection, in tube assay | |||||||||||||
| 4.3.5. Magnetogenosensor148 | X | X | 148 | X | Visual detection, lateral flow dipsticks 4.3.4.1. | |||||||||||||||
| 4.4.1. AC susceptometry150 | X | X | 150 | X | AC susceptometer150 | 4.4.1. | 9 | O | X | 2 | 2 | 2 | ||||||||
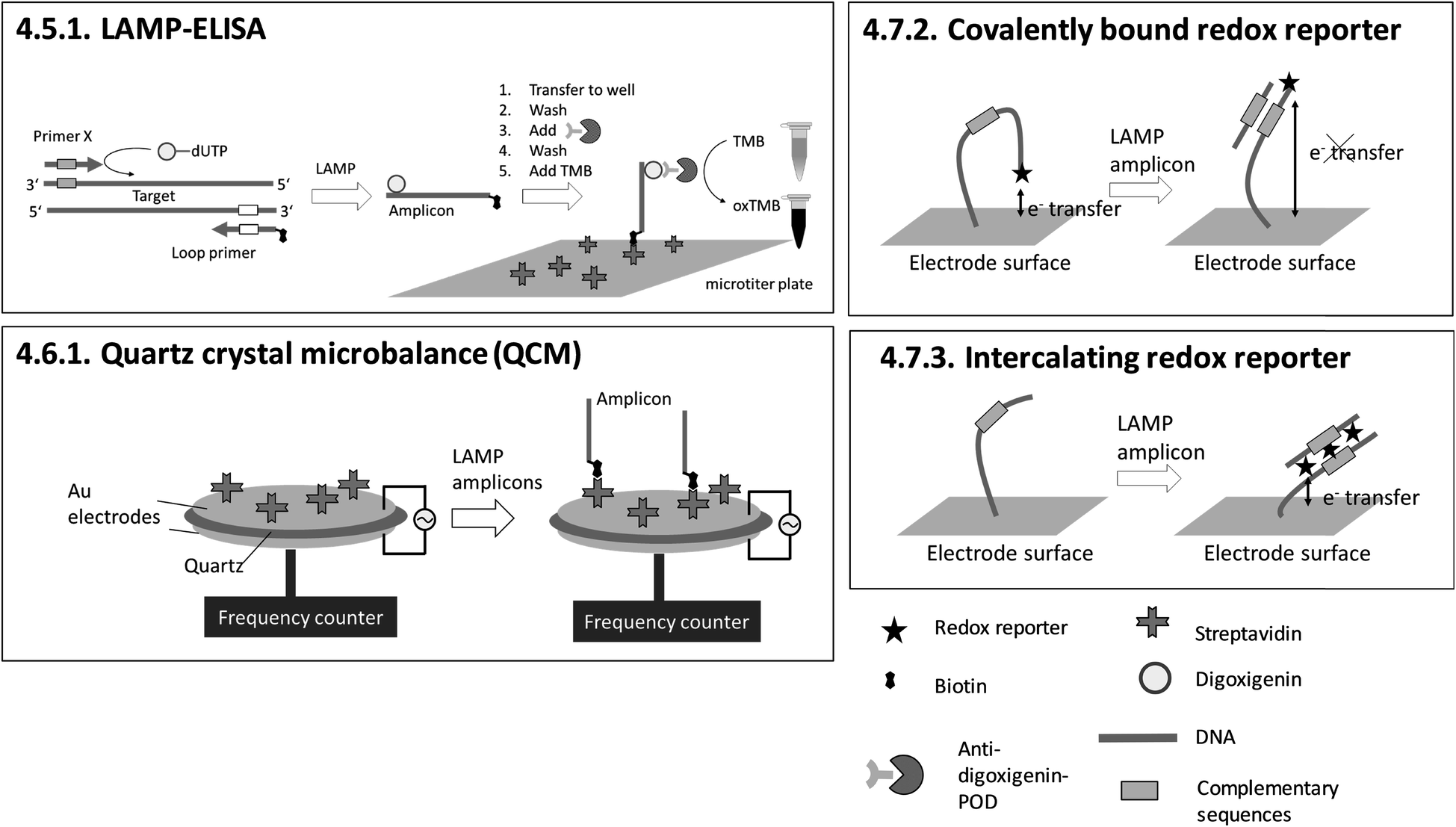
| 4.5.1. LAMP-ELISA151,152 | X | X | X | 151 and 152 | X | Microtiter plate reader152 | 4.5.1. | 9 | O | X | 1 | 3 | 3 | |||||||
| 4.6.1. Quartz crystal microbalance (QCM)153 | X | X | X | X | 153 | X | Commercially available quartz crystals enclosed by electrodes and custom-made QCM system for LAMP and readout153 | 4.6.1. | 4 | M/O | X | X | 1 | 2 | 2 | |||||
| 4.7.1. Nanopore sequencing154–157 | X | X | X | 154–157 | X | (Minion) nanopore sequencer154–157 | 4.7.1. | 9 | O | X | X | 1 | 3 | 3 | ||||||
| 4.7.2. Covalently bound redox reporter158 | X | X | 158 | X | (X) | Custom-made microfluidic chip and a potentiostat for square-wave voltammetry158 | 4.7.2. and 4.7.3. | 4 | M/O | X | X | X | 2 | 1 | 2 | |||||
| 4.7.3. Intercalating redox reporter159,160,162 | X | X | 159, 160 and 162 | X | (X) | Custom-made microfluidic chip and Genelyzer™ (Toshiba) for linear sweep voltammetry159,160 | 4.7.2. and 4.7.3. | 4 | M/O | X | X | 2 | 2 | 2 | ||||||
| 4.7.4. Electrical impedance spectroscopy163 | X | X | X | 163 | (X) | X | Custom-made measuring station for electrical impedance spectroscopy163 | 4.7.4. | 4 | M | X | X | 2 | 2 | 2 | |||||
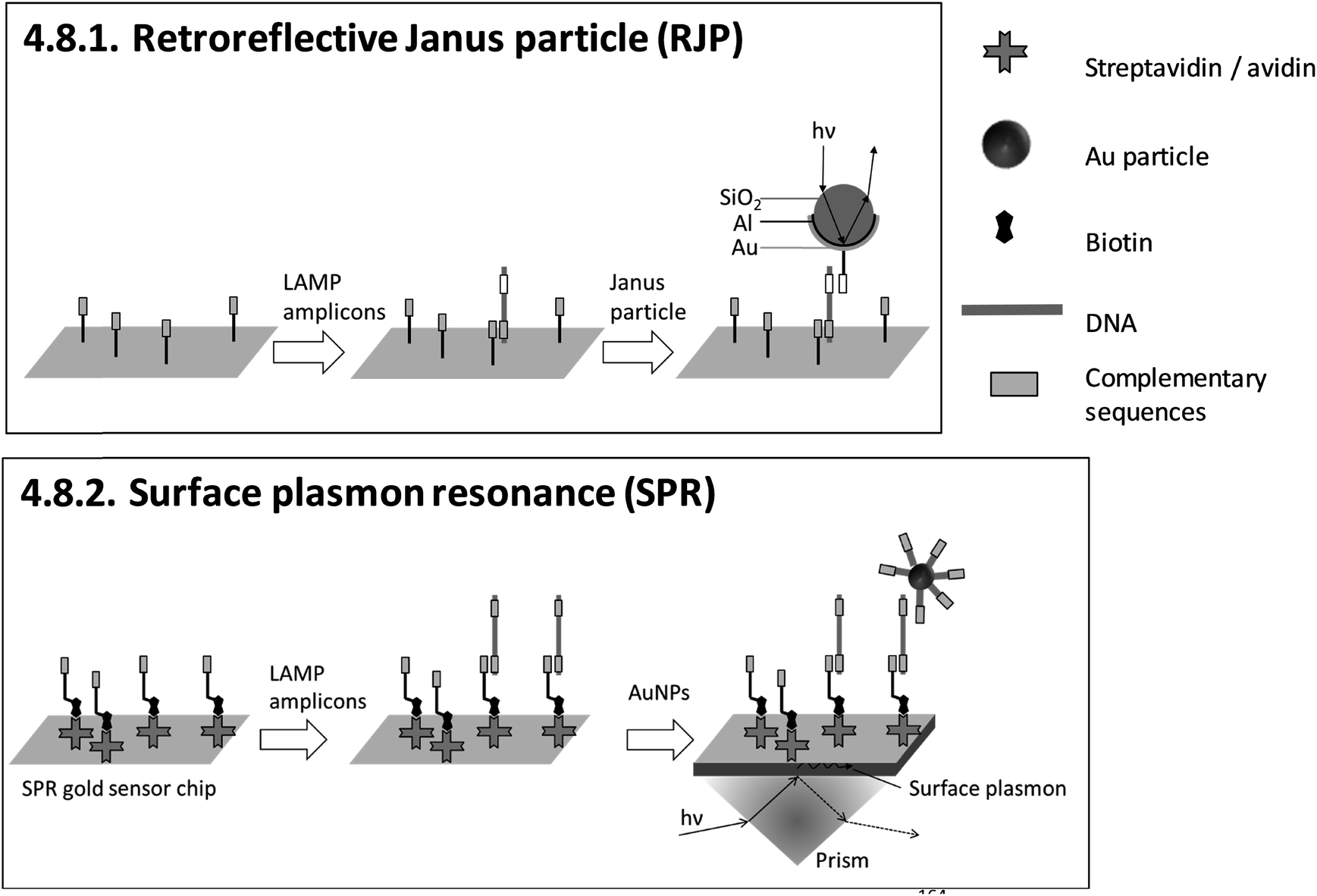
| 4.8.1. Retroreflective Janus particle (RJP)164,165 | X | 164 | X | Commercially available RJP-quantifying chips (from AMED, Seoul, Korea) and custom-made optical detection system164,165 | 4.8.1. | 4 | M/O | X | X | 3 | 2 | 2 | ||||||||
| 4.8.2. Surface plasmon resonance (SPR)166–169 | X | X | X | X | 166 | X | X | Commercially available Au sensor chips (from Ssens, Netherland) and custom-made optical detection system166 | 4.8.2. | 4 | M/O | X | X | X | 3 | 2 | 2 | |||
| 4.9.1. Giant magnetoresistive (GMR) measurement170 | X | 170 | X | Custom made GMR detecting system, custom made microfluidic chips170 | 4.9.1. | 4 | M | X | X | X | 3 | 2 | 2 | |||||||
| 4.10.1. Genomagnetic LAMP-based electrochemical detection171,172 | X | X | X | 171 and 172 | X | Commercially available electrochemical chips, magnetic support beneath the chip and a multi potentiostat/galvanostat (all from DropSens, Spain)171,172 | 4.10.1. | 9 | O | X | X | 2 | 3 | 3 | ||||||
| 4.10.2. Resistive pulse sensing (RPS)173 | X | 173 | X | Nanopore sensor (qNano, IZON Science, Christchurch, New Zealand)173 | 4.10.2. | 9 | O | X | 2 | 2 | 2 | |||||||||
| Aedes aegypti (Wolbachia infection)85,86 |
| Ammonia-oxidizing enzyme in environmental bacteria64 |
| Avian reovirus70 |
Bacteriophage MS2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 73 73 |
| Bemisia tabaci 60 |
| BRAF allele (V600E)83 |
| Brucella 72 |
| Caenorhabditis elegans 68 |
| Chikungunya virus73 |
| Chlamydia trachomatis 61 |
| Dengue virus87,112,156 |
| Diarrheal disease82 |
| Escherichia coli 68 |
| Fomitiporia torreyae 59 |
| Fulviformes umbrinellus 59 |
| Fusarium oxysporum f. sp. lycopersici (point mutations in xylem 3 (SIX3) gene)66 |
| Genetically modified maize77–81 |
| Haemophilus ducreyi 101 |
| Haemophilus influenzae 105 |
| hBRCA168 |
| HeLa68 |
| Hepatitis B and C virus62,87,107 |
| Herpes simplex virus 1 (HSV1) US483 |
| Human immunodeficiency virus87,101 |
| HLA-B*15:02 allele163 |
| Human papillomavirus153,171 |
| Human T-lymphotropic virus 1101 |
| Influenza virus63,114 |
| Lambda DNA68 |
| Leptospira 148 |
| Leishmania 155 |
| Listeria ivanovii 103 |
| Listeria monocytogenes 103 |
| Methicillin-resistant Staphylococcus aureus (MRSA)71,166 |
| Middle east respiratory syndrome coronaviruses84 |
| Neisseria meningitide 105 |
| Newcastle disease virus113 |
| Panton–Valentine leukocidin toxin of MRSA173 |
| Plasmodium falciparum 83,154,157 and, P. vivax, P. ovale wallikeri, P. ovale curtisi, P. knowlesi and P. malariae154 |
| Pseudomonas aeruginosa 125 |
| Ralstonia solanacearum 96 |
| Respiratory syncytial virus63 |
| Rheumatoid arthritis (SNPs associated with)159,160 |
| Salmonella 69,111,151,152,158,164 |
| Shiga toxigenic Escherichia coli65 |
| Streptococcus pneumoniae 105 |
| Treponema pallidum 101,106 |
| Trialeurodes vaporariorum 60 |
| Trypanosoma brucei 89 |
| Varicella-zoster virus58 |
| Vibrio cholera 88 |
| Vibrio parahaemolyticus 104 |
| Vibrio vulnificus 104 |
| Vitamin K epoxide reductase 1 (SNP)61 |
| West Nile virus73,87 |
| White spot syndrome virus67,94 |
| Yersinia enterocolitica 162 |
| Zika virus74,150 |
| β2-Tubulin gene region of causal pathogens of Fusarium head blight of wheat (mutant genotypes F167Y, E198Q, and F200Y)95 |
4.2. Homogeneous, particle-free detection – optical
Sections 4.2.1.–4.2.7. relate to single-modified fluorogenic primers and probes.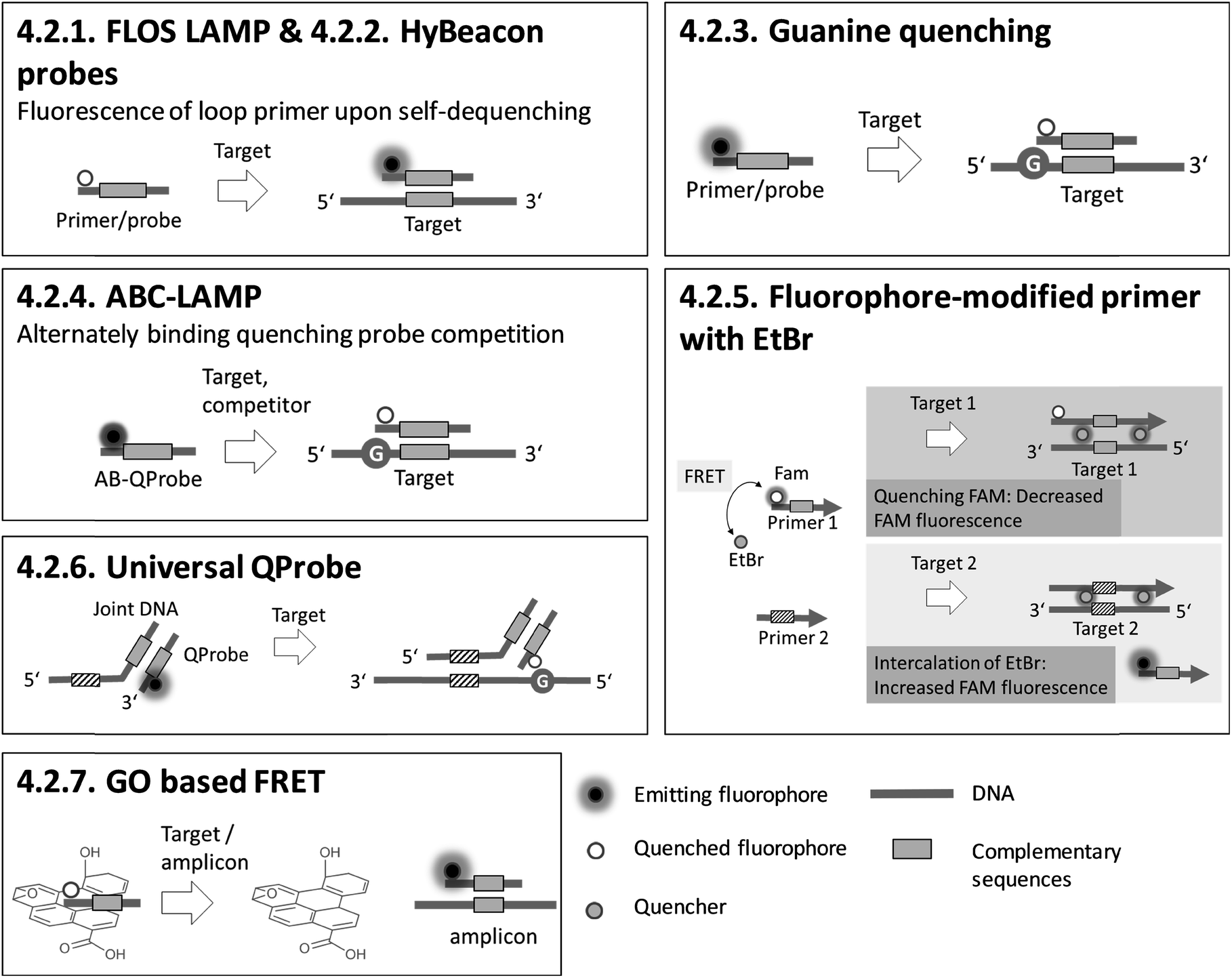
Sections 4.2.8.–4.2.14. relate to multiple-modified fluorogenic primers and probes.

Sections 4.2.15.–4.2.17. relate to detection based on restriction enzymes.
4.3. Homogeneous, particle-based detection – optical
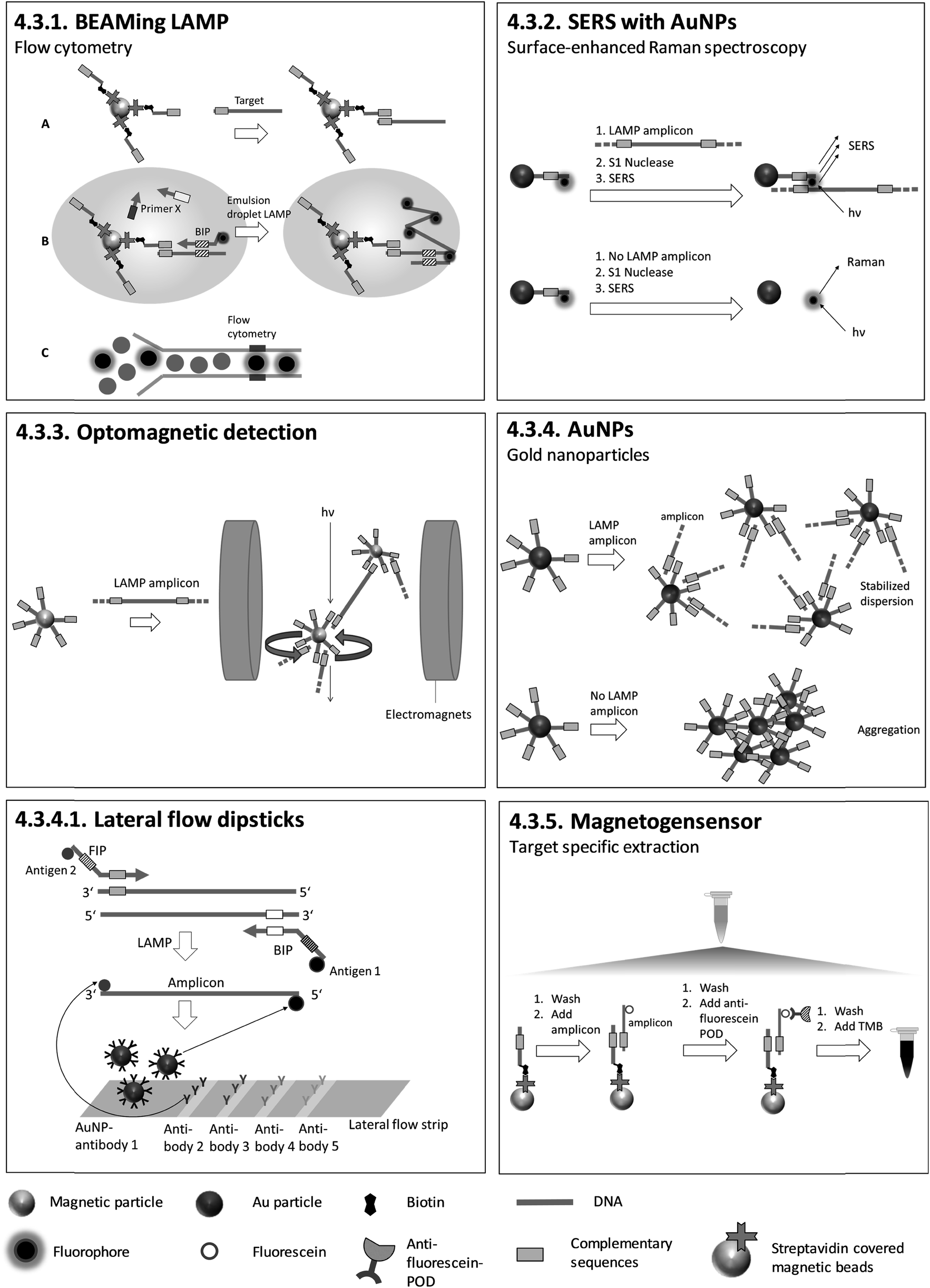
4.3.4.1. Lateral flow dipsticks. During LAMP, primers that are two-fold modified with two different antigens are incorporated into the amplicon. Antigen 1 (identical for each target) is for colour reaction and antigen 2 (individual for each target) is for capturing at the test line. Biotin in combination with streptavidin-modified AuNPs can be used instead of antigen 1. Antibodies for antigen 2 are pre-immobilized on the test lines and Au-NPs labelled with antibodies for antigen 1, for colour reaction, are pre-immobilized on the conjugate pad. After dipping the test strip into a running buffer which includes LAMP products, antibody 1-labeled AuNPs are rehydrated and transported in the running buffer. Antigen 2-tagged amplicons are trapped at specific test lines by antibodies for antigen 2, and AuNPs accumulate on the test lines due to their binding to antigen 1, which is attached to the amplicons. Positive amplification is indicated by a red band which can be observed visually (Fig. 7). The described lateral flow method was utilized by Chen et al.125 after triplex LAMP to identify three different toxin genes of Pseudomonas aeruginosa with an analytical sensitivity of 20 CFU per reaction. Similar lateral flow approaches were utilized to detect various other pathogens.126–146 Lateral flow tests have also been integrated into sample-to-answer microfluidic systems.147 Lateral flow tests are easy to handle and do not require additional equipment other than an incubator for LAMP. The strips are universally applicable as no target-specific probes are immobilized on the surface. These tests can therefore be produced on a large scale, minimizing fabrication costs. However, they are not quantitative, and the opening of LAMP reaction vials risks contamination with amplicons.
4.4. Homogeneous, particle-based detection – magnetic
 | ||
| Fig. 8 Schematic illustration of 4.4.1. AC susceptometry150 (redrawn according to the references). | ||
4.5. Heterogeneous, particle-free detection – optical
4.6. Heterogeneous, particle-free detection – piezoelectric
4.7. Heterogeneous, particle-free detection – electrochemical
Electrical impedance spectroscopy was utilized for the detection of the HLA-B*15:02 allele responsible for adverse drug reactions in humans. Diagnostic sensitivity and specificity were determined with 27 crude blood samples to be 92.9% and 84.6% respectively.
The authors claim that the fabrication of the sensors is simple and low-cost, and that future miniaturization and integration into a PON device is possible.
4.8. Heterogeneous, particle-based detection – optical
4.9. Heterogeneous, particle-based detection – magnetoresistive
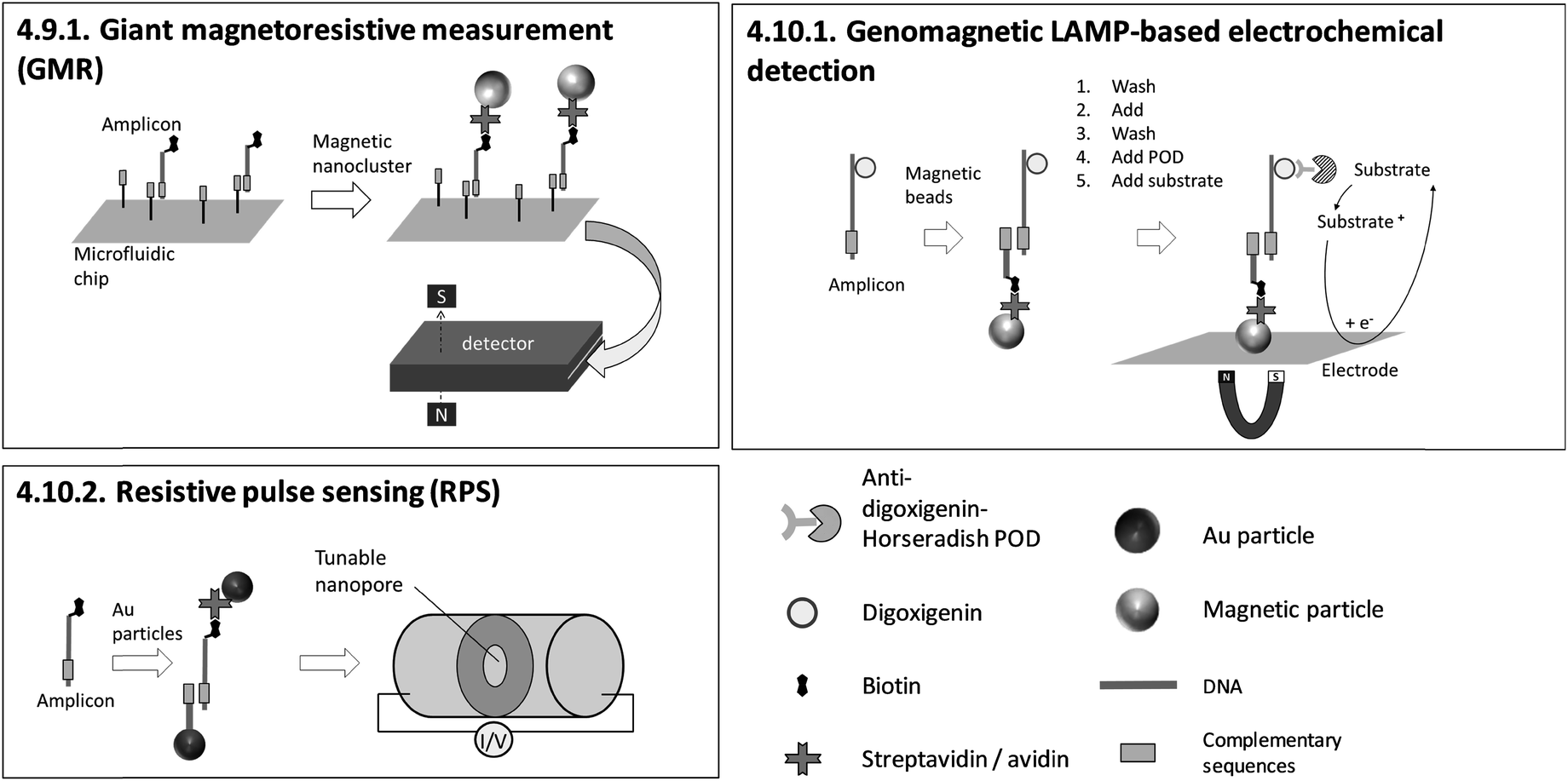
4.10. Heterogeneous, particle-based detection – electrochemical
5. Instrumentation: open versus method-tailored platforms
In this chapter, we provide criteria for the assessment of the operational implementation of LAMP with sequence-specific detection. Table 2 shows platforms on which the individual methods can be implemented. Each platform comprises a device, as well as disposable tubes or cartridges.The technological readiness level (TRL)174 of each platform is listed in a separate column and ranges from level three (demonstration of proof of concept) to nine (works in an operational environment and is ready for commercial deployment). Next, we differentiate between open platforms (O), which can be adapted to various protocols and reagents, and method-tailored platforms (M). All open platforms are already commercially available, and the methods have been adjusted to their requirements. In contrast, method-tailored platforms are custom-made and designed specifically for the applied method. As LAMP is predestined for on-site testing, another criterion for the assessment of a platform is its portability. We also indicated the use of disposable cartridges, like disks or chips, as these represent a substantial cost factor compared to the use of standard reaction tubes. Reusable cartridges or sensors have been shown to reduce costs, and therefore these are also marked in Table 2.
We next evaluated the practical operation. The evaluation was inspired by the key criteria given in the standards of the Clinical Laboratory Improvement Amendments (CLIA) program,175 which regulates clinical laboratory testing on patient specimens in the United States. Based on the available literature, we evaluated the complexity of the methods according to their CLIA score (Table 2). Scores range between one and three. A score of one indicates a low level of complexity, and score of three a high level of complexity.176 We provide scores for the following categories: preparation of reagents, characteristics of the operational steps, training, and expertise.175,176
6. Comparative general discussion
A brief description and visualization of the methods for sequence-specific detection of LAMP has been presented in chapter 4, and criteria for the evaluation of the platforms for the operational implementation of these methods have been presented in chapter 5. In the discussion chapter, we assess the methods and platforms according to the criteria specified in chapters 4.1 and 5 respectively. We also extend the assessment with additional criteria specific to homogeneous or heterogeneous detection methods.First, we evaluate the status of the research with regard to the analytical performance of the methods and their ability to analyze complex sample material. We also highlight any possible shortcomings. In the second step, we evaluate the methods in a comparative manner. Homogeneous and heterogeneous detection methods are considered separately. Third, we focus on the operational implementation of various methods on the different instrumental platforms.
6.1. Analytical performance and analysis of complex sample material
The analytical performance of a large number of methods has been extensively examined in literature, as can be seen from Table 2. The majority of the methods (31 out of 34, 91%) have been evaluated according to analytical sensitivity. Moreover, the analysis of complex sample material has been demonstrated for more than half the methods (21 out of 34). Parameters from field studies, such as diagnostic sensitivity and diagnostic specificity, were reported for more than one third of the methods (12 out of 34). For future studies, we strongly suggest including investigations of repeatability (intraassay variance) and reproducibility (interassay variance), as these parameters were published in only 6% of the cases. The assay variance is a key parameter for the assessment of the precision and robustness of an assay and the variation in results between runs or between different laboratories.536.2. Homogeneous methods
Homogeneous sequence-specific methods are those which enable the detection of signals from the entire reaction volume due to the homogeneous distribution of biorecognition elements. Biorecognition elements are either dissolved in solution or immobilized on suspended particles. Homogeneous, particle-free methods make use of optical transducing elements (fluorescence) only, whereas particle-based methods can be differentiated by their use of optical (fluorescence, chemiluminescence, scattering, reflection or colorimetry) or magnetic transducing elements.Since a large proportion of the homogeneous methods (18 out of 23) are based on fluorescence, we first discuss and compare fluorescence-based methods in detail. For this, we extend the general assessment criteria (quantification, multiplexing and detection by naked eye, chapter 4.1) and focus additionally on the modification of primers and probes, the optimization of fluorescence signals and the adaption to different targets.
Most of the fluorescence-based methods for sequence-specific detection of LAMP allow real-time monitoring of LAMP, and generate rapid and quantitative results using single- or multiple-fluorescence-labelled primers and probes. To obtain high amplification efficiencies and to maximize signal-to-noise ratios, the concentrations and ratios of fluorescence-labelled primers and probes must be adjusted carefully for each new target sequence. Semi-universal and universal methods like 4.2.6. universal QProbe, 4.2.13. assimilating probe and 4.2.14. MD LAMP render elaborate work to optimize the signal redundant, as the fluorophore and quencher are located at universal sequences. Consequently, these methods allow simple adaption to various target panels. 4.2.7. GO based FRET stands out for its excellent fluorescence yields due to efficient quenching properties. However, a drawback is an additional handling step required for adding graphene oxide (GO) after the LAMP reaction.
Adding PEI after LAMP enables instrument-free, visual endpoint detection by naked eye. This method was applied after 4.2.1. FLOS LAMP and 4.2.2. HyBeacon probes. Visual detection by naked eye was also demonstrated for 4.2.9. QUASR. All methods based on fluorescence enable multiplex detection and, apart from one exception, quantification of the target analytes, with 4.2.4. ABC-LAMP placing special focus on precise quantification. 4.2.17. pyrosequencing is particularly suitable for multiplexing. However, the simultaneous amplification of numerous targets by LAMP is not easy to implement, and therefore the efficiency of amplification is the limiting factor.
The next section deals with homogeneous methods based on suspended particles. The general assessment criteria (chapter 4.1) are extended to cover the modification of primers, probes or particles, real-time vs. endpoint detection, quantification, multiplexing and detection by naked eye. In contrast to fluorescence-based methods, only half of the homogeneous, particle-based methods require fluorescence-labelling or further modification of the primers or probes participating in the LAMP reaction. Target-specific probes are immobilized covalently or due to streptavidin-biotin interaction on particle surfaces. Except for wash-free methods like 4.3.3. optomagnetic detection and 4.4.1. AC susceptometry, all the particle-based methods focus on endpoint detection. 4.3.1. BEAMing LAMP and 4.3.2. SERS have a complex workflow comprising many individual manual steps. However, BEAMing LAMP stands out for its precise, digital quantification without the need of standards, while SERS is capable of single molecule detection and therefore enables highly sensitive measurements. Furthermore, BEAMing LAMP is the only homogeneous, particle-based method capable of multiplexing. Visual detection by naked eye is possible with 4.3.5. magnetogenosensor and 4.3.4. AuNPs. Moreover, a breakthrough was achieved by detecting LAMP products with AuNPs in 4.3.4.1. lateral flow dipsticks after LAMP. Lateral flow dipsticks are easy to handle and universally applicable due to their probe-free sensing surfaces.
6.3. Heterogeneous methods
Heterogeneous detection methods make use of planar sensing surfaces, in some instances combined with suspended particles. In contrast to homogeneous methods, the biorecognition elements are in close proximity or linked to the sensing surface during signal transduction. Heterogeneous methods make use of various transducing elements, namely optical (colorimetry, reflection), piezoelectric, electrochemical (amperometry, voltammetry, impedimetry) and magnetoresistive elements.In addition to the general assessment criteria defined in chapter 4.1 (quantification, multiplexing and detection by naked eye), heterogeneous detection methods are evaluated according to the modification of primers, probes, particles or sensing surfaces, the hybridization time, and real-time vs. endpoint detection. These criteria apply equally to particle-free and particle-based methods, and these are therefore considered jointly.
Sensing and particle surfaces are coated either with streptavidin or with covalently binding, target-specific probes. The insertion of biotin into amplicons by biotin-labelled primers is unproblematic regarding the efficiency of amplification due to the small size of biotin. However, for 4.5.1. LAMP-ELISA and 4.10.1. genomagnetic LAMP-based electrochemical detection, digoxigenin-labelled dUTPs are inserted into amplicons. The use of digoxigenin-labelled dUTPs causes additional optimization work, since the amplification efficiency is decreased for high dUTP concentrations.177 In contrast, biotin- and fluorescence label-free methods (4.7.1.–4.7.4., 4.8.1., 4.8.2.) are advantageous with regard to assay optimization, as no modification of primers is required at all.
The dependency between signal intensity and target concentration allows rough quantification by endpoint detection. Precise quantification can be achieved by real-time monitoring. In general, slow diffusion rates of target molecules and amplicons migrating towards the sensing surface178 increase the time for hybridization. Long hybridization times pose a general obstacle for real-time monitoring and therefore for precise quantification. 4.10.1. genomagnetic LAMP-based electrochemical detection is limited to endpoint detection, but it circumvents long hybridization times by accumulating amplicons, bound to magnetic beads, on the electrode surfaces with the help of magnetic forces. A pioneer of real-time monitoring is 4.6.1. QCM, albeit with a pre-incubation step in a chamber separated from the sensing surface before polymerase is added. A further important issue concerning precise quantification arises when using biotinylated primers. For 4.5.1. LAMP-ELISA, 4.6.1. QCM, 4.10.1. GMR and 4.10.2. RPS, biotinylated amplicons compete with excess biotinylated primers for binding to streptavidin-coated sensing surfaces or particles, affecting the sensitivity of the assay.
An advantage of heterogeneous detection methods, which are based on planar sensing surfaces, is the simultaneous detection of multiple targets by spatially resolved measurements,158–160,166 or alternatively by wavelength division179,180 or spectral analysis of multiple light beams.181 Multiplex detection and differentiation between targets was demonstrated exclusively for 4.7.2. and 4.7.3. electrochemical detection with redox reporters, and 4.8.2. SPR. Another achievement is 4.7.1. nanopore sequencing, which provides detailed genotyping analysis. However, this is at the expense of requiring elaborate post-LAMP DNA library preparation. 4.5.1. LAMP-ELISA allows nearly instrument-free handling and surpasses almost all methods with regard to high throughput by using microtiter plates. No heterogeneous method allows visual detection by naked eye.
6.4. Instrumental platforms
More than two thirds of all methods are implemented on open instrumental platforms. Fluorescence-based methods make up a large proportion of these. Except for 4.3.1. BEAMing LAMP, all fluorescence-based methods are compatible with standard isothermal amplification devices with an integrated fluorescence reader. The open instrumental platforms listed in Table 2 are commercially available and work in operational environments (TRL 9). For method-tailored platforms, only demonstrations have been presented so far. These have only been validated in the laboratory environment and are still assigned low TRLs (TRL 4). Open instrumental platforms can also be combined with method-tailored cartridges (disks or chips), and method-tailored platforms can be combined with commercially available and universal cartridges (Table 2, O/M). In order to bring these methods to the market, higher TRLs, i.e. validation in operational environments must be achieved.On-site testing at the PON requires portable instrumentation based on simple equipment. Platforms designed specifically to meet the demands for use at the PON182,183 have mainly been presented for fluorescence-based detection. Simple readout via smartphone, replacing bulky and expensive optical components, has been implemented by a portable LAMP box74,75 or combined with filter-based sample preparation.85,86 The integration of small smartphones into diagnostic platforms can reduce costs considerably. In future, exploitation of the variety of sensors hosted in smartphones, including optical sensors, wireless data transfer and a global positioning system, could also be beneficial in the context of telemedicine and e-health. Testing in the field requires devices which do not require electric power or any other external energy supply. The only devices which do not require power supply are so far method-tailored and comprise power-free devices for incubation at LAMP conditions combined with a fluorometer or smartphone readout.97,98 Both devices follow the same clever approach of generating heat for LAMP incubation with an exothermic reaction.
Devices with microfluidic cartridges enable fully automated LAMP with subsequent detection (4.2.9., 4.2.11., 4.7.2., 4.9.1.), facilitating on-site sample processing.82,89,158,170 Compared to methods based on fluorescence, electrochemical readout seems more amenable to miniaturization due to the independence of optical path lengths, but suffers from the additional costs of disposables with integrated electrodes.
Probe-free or streptavidin-coated universal sensor surfaces (4.5.1., 4.6.1., 4.7.1., 4.10.1., 4.10.2.) are attractive as these methods reduce costs by enabling cheap, large-scale fabrication of target-independent disposables. One highlight is 4.10.1. genomagnetic LAMP-based electrochemical detection, which uses unmodified and inexpensive carbon electrodes. Moreover, instrumentation providing reusable sensing surfaces (4.6.1., 4.7.2., 4.8.2., 4.9.1., 4.10.2.) enables further cost reduction. However, we would like to point out that carryover contamination with amplicons due to the reuse of sensors poses a tremendous risk to reliable results. Therefore, LAMP and detection should be conducted in separate chambers, as is demonstrated for all methods except 4.6.1. QCM.
We now evaluate the complexity of the practical operation of the methods. The majority of fluorescence-based detection methods used standard isothermal amplification devices with an integrated fluorescence reader and enable real-time detection of LAMP. These stand out due to a low degree of complexity. Scores for all three categories of complexity, comprising first the preparation of reagents, second the characteristics of the operational steps and third training and expertise, are one. First, biorecognition elements can be pre-stored in the primer mix. Second, real-time monitoring in a thermal cycler doesn't require post-LAMP steps. Consequently, the complexity of the operational steps and the required training and expertise is low.
Time-consuming operational steps comprising the addition of particles, incubation and washing steps, require trained staff and therefore have a higher level of complexity. Particle-based methods at a developmental stage involve elaborate particle modification, raising their complexity score for reagent preparation and expertise to two and two/three respectively. Furthermore, the modification of particles and sensor surfaces increases the complexity of the reagent preparation even more. Large-scale fabrication and the automation of operational steps in the course of a market launch will lower their complexity and increase their user-friendliness.
7. Conclusion and future prospects
LAMP is gaining more and more attention as field-friendly analytical tool, especially for PON applications, due to its fast processing times and simple instrumentation.9 Methods for sequence-specific detection of LAMP make full use of the specificity towards the target analyte for which LAMP is especially renowned, and are thus preferable to sequence-independent methods. For the assessment and comparison of sequence-specific methods, we provided a catalogue of assessment criteria covering the most important aspects (Table 2): analytical performance, handling of complex sample material, multiplex capacity, the ability to quantify the target analyte and detection by naked eye. Additionally, we evaluated the method-related platforms according to their TRL, accessibility (open/method-tailored platforms), portability, disposability of cartridges, reusability and complexity (reagent preparation, operational steps and training/expertise), as summarized in Table 2. The methods differ from each other in many aspects. Focus is placed on precise quantification (4.2.4. ABC-LAMP, 4.3.1. BEAMing LAMP), multiplex detection and genotyping (sequencing 4.2.17., 4.7.1.) or detection by naked eye (adding PEI after LAMP 4.2.1. and 4.2.2., 4.3.4. AuNPs, lateral flow dipsticks 4.3.4.1., 4.3.5. magnetogenosensor, 4.5.1. LAMP-ELISA).Homogeneous detection methods capture the signal from the entire sample volume, predominantly in real time, enabling fast results and quantification without time-consuming washing steps. There is a large variety of homogeneous, fluorescence-based detection methods that have many similarities with regard to their required instrumental platforms and the complexity of their practical operation. Semi-universal (4.2.6., 4.2.13.) and universal (4.2.14.) homogeneous methods feature unique universal properties, and thus can be easily adapted to different targets. Heterogeneous, surface-based detection is mainly restricted to endpoint detection due to slow diffusion processes, but also has advantages, as it is more easily miniaturizable and integrable into μTAS systems. First steps towards real-time monitoring have been made by 4.6.1. QCM, a pioneering method which does not require post-LAMP modifications. Electrochemical detection is another heterogeneous method which is a promising candidate for real-time monitoring. In contrast to PCR, which suffers from thermally unstable bonds between the electrode surface and oligonucleotide probes during cycles,184,185 LAMP allows more moderate thermal conditions and paves the way for simultaneous amplification and heterogeneous detection.
The miniaturization and automation of operational steps in μTAS systems facilitates usage at the PON. So far, innovative solutions have been realized by fluorescence82 and electrochemical detection methods.158–160 In the context of miniaturization, electrochemical detection surpasses optical readout, as no long path lengths, lenses or waveguides are necessary. The latest trends in research show that bulky optical components can be replaced by smartphones.74,75,85,86 Further developments could lead to various heterogeneous detection methods being combined with simple smartphone readout, making expensive and sophisticated devices redundant and thus increasing user-friendliness. An exciting and stimulating demonstration of the smartphone approach is the detection of SPR, with a smartphone as the transducing element.186 In addition, novel technologies in the field of electrochemical detection make use of smartphones for signal processing and data transmission.187–189 Generally speaking, the combination of field-friendly LAMP and smart devices is a forward-looking approach that has the potential to bring diagnostics even closer to the patient.
Some methods, including mainly fluorescence-based methods but also sequencing 4.2.17. and 4.7.1., 4.3.2. SERS, 4.4.1. AC susceptometry, 4.5.1. LAMP-ELISA, 4.10.1. genomagnetic LAMP-based electrochemical detection and 4.10.2. resistive pulse sensing, can be implemented on open platforms which are already commercially available. Method-tailored platforms presented in this review are still in their youth and do not exceed TRL4, but convincing PON concepts have been shown in the laboratory environment. The next steps required to reach higher TRLs and aim for market launch will be the adaption to operational environments and the development of prototypes.
In summary, there are various methods for the sequence-specific detection of LAMP which distinguish themselves by their different intended uses. Our review intends to provide a comprehensive overview and a systematic evaluation of these methods with regard to different user requirements, ranging from the overall performance of the method to the related instrumental platforms. Ultimately, it may serve as a guideline for the selection of the most appropriate methods.
Abbreviations
| ABC-LAMP | Alternately binding quenching probe competitive LAMP |
| AuNPs | Gold nano particles |
| B3 | Backward outer primer |
| BIP | Backward inner primer |
| CLIA | Clinical laboratory improvement amendments |
| DARQ | Detection of amplification by release of quenching |
| DENV | Dengue virus |
| dUTP | 2′-Deoxyuridine, 5′-triphosphate |
| EtBr | Ethidium bromide |
| F3 | Forward outer primer |
| FIP | Forward inner primer |
| FLOS | Fluorescence of loop primer upon self-dequenching |
| FRET | Förster resonance energy transfer |
| GMR | Giant magnetoresistive |
| GO | Graphene oxide |
| HDA | Helicase dependent amplification |
| H(B, C, E)V | Hepatitis B, C, E virus |
| HIV | Human immunodeficiency virus |
| HPV | Human papillomavirus |
| LAMP | Loop-mediated isothermal amplification |
| LAMP-ELISA | LAMP detected by enzyme-linked immunosorbent assay |
| LB | Backward loop primer |
| LF | Forward loop primer |
| MD LAMP | Mediator displacement LAMP |
| MERT-LAMP | Multiple endonuclease restriction real-time LAMP |
| MNP | Magnet nano particle |
| MRSA | Methicillin-resistant Staphylococcus aureus |
| NAAT | Nucleic acid amplification test |
| NASBA | Nucleic acid sequence based amplification |
| NP | Nano particle |
| PEI | Polyethylenimine |
| POC | Point-of-care |
| PON | Point-of-need |
| QCM | Quartz crystal microbalance |
| QUASR | Quenching of unincorporated amplification signal reporters |
| LAMP-OSD | LAMP one-step strand displacement |
| QProbe | Quenching probe |
| RCA | Rolling circle amplification |
| RJP | Retroreflective Janus particle |
| PCR | Polymerase chain reaction |
| RPA | Recombinase polymerase amplification |
| RPS | Resistive pulse sensing |
| SDA | Strand displacement amplification |
| SERS | Surface enhanced Raman spectroscopy |
| SNP | Single nucleotide polymorphism |
| SPR | Surface plasmon resonance |
| TEC-LAMP | Tth endonuclease cleavage LAMP |
| TMA | Transcription-mediated amplification |
| TRL | Technological readiness level |
| UR | Universal reporter |
| WNV | West Nile virus |
| WSSV | White spot syndrome virus |
| μTAS | Miniaturized total chemical analysis systems |
Conflicts of interest
The authors declare no conflict of interest.References
- K. Mullis, F. Faloona, S. Scharf, R. Saiki, G. Horn and H. Erlich, Cold Spring Harbor Symp. Quant. Biol., 1986, 51, 263–273 CrossRef CAS PubMed.
- K. B. Mullis and F. A. Faloona, Methods Enzymol., 1987, 155, 335–350 CAS.
- S. Yang and R. E. Rothman, Lancet Infect. Dis., 2004, 4, 337–348 CrossRef CAS.
- K. Mann and C. M. Ogilvie, Prenatal Diagn., 2012, 32, 309–314 CrossRef CAS PubMed.
- L. S. Kristensen and L. L. Hansen, Clin. Chem., 2009, 55, 1471–1483 CrossRef CAS PubMed.
- S. E. Cavanaugh and A. S. Bathrick, Forensic Sci. Int.: Genet., 2018, 32, 40–49 CrossRef CAS PubMed.
- I. Laube, J. Zagon and H. Broll, Int. J. Food Sci. Technol., 2007, 42, 336–341 CrossRef CAS.
- H. A. Bowers, T. Tengs, H. B. Glasgow, J. M. Burkholder, P. A. Rublee and D. W. Oldach, Appl. Environ. Microbiol., 2000, 66, 4641–4648 CrossRef CAS PubMed.
- Z. K. Njiru, PLoS Neglected Trop. Dis., 2012, 6, e1572 CrossRef PubMed.
- X. Fang, Y. Liu, J. Kong and X. Jiang, Anal. Chem., 2010, 82, 3002–3006 CrossRef CAS PubMed.
- P. Craw and W. Balachandran, Lab Chip, 2012, 12, 2469–2486 RSC.
- M. M. Ali, F. Li, Z. Zhang, K. Zhang, D.-K. Kang, J. A. Ankrum, X. C. Le and W. Zhao, Chem. Soc. Rev., 2014, 43, 3324–3341 RSC.
- Y. Mori, H. Kanda and T. Notomi, J. Infect. Chemother., 2013, 19, 404–411 CrossRef CAS PubMed.
- J. Li, J. Macdonald and F. von Stetten, Analyst, 2018, 144, 31–67 RSC.
- A. Borst, J. Verhoef, E. Boel and A. C. Fluit, Clin. Lab., 2002, 48, 487–492 CAS.
- B. J. Toley, I. Covelli, Y. Belousov, S. Ramachandran, E. Kline, N. Scarr, N. Vermeulen, W. Mahoney, B. R. Lutz and P. Yager, Analyst, 2015, 140, 7540–7549 RSC.
- M. Vincent, Y. Xu and H. Kong, EMBO Rep., 2004, 5, 795–800 CrossRef CAS PubMed.
- L. Comanor, Am. J. Gastroenterol., 2001, 96, 2968–2972 CrossRef CAS PubMed.
- T. Notomi, H. Okayama, H. Masubuchi, T. Yonekawa, K. Watanabe, N. Amino and T. Hase, Nucleic Acids Res., 2000, 28, E63 CrossRef CAS PubMed.
- K. Nagamine, T. Hase and T. Notomi, Mol. Cell. Probes, 2002, 16, 223–229 CrossRef CAS PubMed.
- P. Francois, M. Tangomo, J. Hibbs, E.-J. Bonetti, C. C. Boehme, T. Notomi, M. D. Perkins and J. Schrenzel, FEMS Immunol. Med. Microbiol., 2011, 62, 41–48 CrossRef CAS PubMed.
- H. Kaneko, T. Kawana, E. Fukushima and T. Suzutani, J. Biochem. Biophys. Methods, 2007, 70, 499–501 CrossRef CAS PubMed.
- Y. Mori, K. Nagamine, N. Tomita and T. Notomi, Biochem. Biophys. Res. Commun., 2001, 289, 150–154 CrossRef CAS PubMed.
- Y. Mori, M. Kitao and T. Notomi, J. Biochem. Biophys. Methods, 2004, 59, 145–157 CrossRef CAS PubMed.
- N. Kuboki, N. Inoue, T. Sakurai, F. Di Cello, D. J. Grab, H. Suzuki, C. Sugimoto and I. Igarashi, J. Clin. Microbiol., 2003, 41, 5517–5524 CrossRef CAS PubMed.
- Y. Mori, H. Kanda and T. Notomi, Nat. Protoc., 2008, 3, 877–882 CrossRef PubMed.
- N. A. Tanner and T. C. Evans, Curr. Protoc. Mol. Biol., 2014, 105, 15.14 Search PubMed.
- T. Iwamoto, T. Sonobe and K. Hayashi, J. Clin. Microbiol., 2003, 41, 2616–2622 CrossRef CAS PubMed.
- M. Goto, E. Honda, A. Ogura, A. Nomoto and K.-I. Hanaki, BioTechniques, 2009, 46, 167–172 CrossRef CAS PubMed.
- S. J. Oh, B. H. Park, J. H. Jung, G. Choi, D. C. Lee, D. H. Kim and T. S. Seo, Biosens. Bioelectron., 2016, 75, 293–300 CrossRef CAS PubMed.
- D. Zhang, B. Gao, C. Zhao and H. Liu, Langmuir, 2019, 35, 248–253 CrossRef CAS PubMed.
- S. Roy, I. A. Rahman and M. U. Ahmed, Anal. Methods, 2016, 8, 2391–2399 RSC.
- A.-R. Cho, H.-J. Dong and S. Cho, Korean Journal for Food Science of Animal Resources, 2014, 34, 799–807 CrossRef PubMed.
- T. Cai, G. Lou, J. Yang, D. Xu and Z. Meng, J. Clin. Virol., 2008, 41, 270–276 CrossRef CAS PubMed.
- N. W. Lucchi, A. Demas, J. Narayanan, D. Sumari, A. Kabanywanyi, S. P. Kachur, J. W. Barnwell and V. Udhayakumar, PLoS One, 2010, 5, e13733 CrossRef PubMed.
- H. Maeda, S. Kokeguchi, C. Fujimoto, I. Tanimoto, W. Yoshizumi, F. Nishimura and S. Takashiba, FEMS Immunol. Med. Microbiol., 2005, 43, 233–239 CrossRef CAS PubMed.
- O. A. Gandelman, V. L. Church, C. A. Moore, G. Kiddle, C. A. Carne, S. Parmar, H. Jalal, L. C. Tisi and J. A. H. Murray, PLoS One, 2010, 5, e14155 CrossRef CAS PubMed.
- P. Hardinge, G. Kiddle, L. Tisi and J. A. H. Murray, Sci. Rep., 2018, 8, 17590 CrossRef PubMed.
- S. Roy, S. X. Wei, J. L. Z. Ying, M. Safavieh and M. U. Ahmed, Biosens. Bioelectron., 2016, 86, 346–352 CrossRef CAS PubMed.
- N. F. N. Azam, S. Roy, S. A. Lim and M. Uddin Ahmed, Food Chem., 2018, 248, 29–36 CrossRef CAS PubMed.
- D.-G. Wang, J. D. Brewster, M. Paul and P. M. Tomasula, Molecules, 2015, 20, 6048–6059 CrossRef CAS.
- D. Lee, M. La Mura, T. R. Allnutt and W. Powell, BMC Biotechnol., 2009, 9, 7 CrossRef PubMed.
- O. Mayboroda, I. Katakis and C. K. O'Sullivan, Anal. Biochem., 2018, 545, 20–30 CrossRef CAS PubMed.
- M. A. Morales and J. M. Halpern, Bioconjugate Chem., 2018, 29, 3231–3239 CrossRef CAS PubMed.
- P. Sengupta, K. Khanra, A. R. Chowdhury and P. Datta, Bioelectronics and Medical Devices: From Materials to Devices - Fabrication, Applications and Reliability, 2019, ch. 4, pp. 47–95 Search PubMed.
- P. J. Asiello and A. J. Baeumner, Lab Chip, 2011, 11, 1420–1430 RSC.
- S. Shoji, Electron. Commun. Jpn. Part II Electron., 1999, 82, 21–29 CrossRef.
- H. Suzuki, Mater. Sci. Eng., C, 2000, 12, 55–61 CrossRef.
- L. Marle and G. M. Greenway, TrAC, Trends Anal. Chem., 2005, 24, 795–802 CrossRef CAS.
- A. J. Tudos, G. J. Besselink and R. B. Schasfoort, Lab Chip, 2001, 1, 83–95 RSC.
- P. K. Sorger, Nat. Biotechnol., 2008, 26, 1345–1346 CrossRef CAS PubMed.
- X. Zhang, S. B. Lowe and J. J. Gooding, Biosens. Bioelectron., 2014, 61, 491–499 CrossRef CAS PubMed.
- S. A. Bustin, V. Benes, J. A. Garson, J. Hellemans, J. Huggett, M. Kubista, R. Mueller, T. Nolan, M. W. Pfaffl, G. L. Shipley, J. Vandesompele and C. T. Wittwer, Clin. Chem., 2009, 55, 611–622 CrossRef CAS PubMed.
- IUPAC, Compendium of Chemical Terminology, 2nd edn (the “Gold Book”), Compiled by A. D. McNaught and A. Wilkinson, Blackwell Scientific Publications, Oxford, 2019th edn, 1997 Search PubMed.
- B. Nagel, H. Dellweg and L. M. Gierasch, Pure Appl. Chem., 1992, 64, 143–168 CAS.
- I. Nazarenko, B. Lowe, M. Darfler, P. Ikonomi, D. Schuster and A. Rashtchian, Nucleic Acids Res., 2002, 30, e37 CrossRef PubMed.
- I. Nazarenko, R. Pires, B. Lowe, M. Obaidy and A. Rashtchian, Nucleic Acids Res., 2002, 30, 2089–2195 CrossRef CAS PubMed.
- V. J. Gadkar, D. M. Goldfarb, S. Gantt and P. A. G. Tilley, Sci. Rep., 2018, 8, 5548 CrossRef PubMed.
- S. Fukuta, H. Nagai, R. Suzuki, Y. Matsumoto, S. Kato, N. Saka, H. Horikawa and N. Miyake, Ann. Appl. Biol., 2017, 170, 170–178 CrossRef CAS.
- R. Suzuki, H. Tanaka, K. Suzuki, S. Fukuta, S. Kato and T. Ohno, J. Appl. Entomol., 2018, 142, 745–754 CrossRef CAS.
- R. L. Howard, D. J. French, J. A. Richardson, C. E. O'Neill, M. P. Andreou, T. Brown, D. Clark, I. N. Clarke, J. W. Holloway, P. Marsh and P. G. Debenham, Mol. Cell. Probes, 2015, 29, 92–98 CrossRef CAS PubMed.
- Y. Mori, T. Hirano and T. Notomi, BMC Biotechnol., 2006, 6, 3 CrossRef PubMed.
- I. Takayama, M. Nakauchi, H. Takahashi, K. Oba, S. Semba, A. Kaida, H. Kubo, S. Saito, S. Nagata, T. Odagiri and T. Kageyama, J. Virol. Methods, 2019, 267, 53–58 CrossRef CAS PubMed.
- H. Tani, T. Teramura, K. Adachi, S. Tsuneda, S. Kurata, K. Nakamura, T. Kanagawa and N. Noda, Anal. Chem., 2007, 79, 5608–5613 CrossRef CAS PubMed.
- Y. Kouguchi, T. Fujiwara, M. Teramoto and M. Kuramoto, Mol. Cell. Probes, 2010, 24, 190–195 CrossRef CAS PubMed.
- Y. Ayukawa, S. Hanyuda, N. Fujita, K. Komatsu and T. Arie, Sci. Rep., 2017, 7, 4253 CrossRef PubMed.
- U. Waiwijit, D. Phokaratkul, J. Kampeera, T. Lomas, A. Wisitsoraat, W. Kiatpathomchai and A. Tuantranont, J. Biotechnol., 2015, 212, 44–49 CrossRef CAS PubMed.
- N. A. Tanner, Y. Zhang and T. C. Evans, BioTechniques, 2012, 53, 81–89 CrossRef CAS PubMed.
- M. Mashooq, D. Kumar, A. K. Niranjan, R. K. Agarwal and R. Rathore, J. Microbiol. Methods, 2016, 126, 24–29 CrossRef CAS PubMed.
- D. Kumar, T. K. S. Chauhan, R. K. Agarwal, K. Dhama, P. P. Goswami, A. K. Mariappan, A. K. Tiwari and B. P. Mishra, Arch. Virol., 2017, 162, 979–985 CrossRef CAS PubMed.
- I. A. Nanayakkara and I. M. White, Analyst, 2019, 144, 3878–3885 RSC.
- I. A. Bhat, M. Mashooq, D. Kumar, R. Varshney and R. Rathore, J. Appl. Microbiol., 2018, 126(5), 1332–1339 CrossRef PubMed.
- C. S. Ball, Y. K. Light, C.-Y. Koh, S. S. Wheeler, L. L. Coffey and R. J. Meagher, Anal. Chem., 2016, 88, 3562–3568 CrossRef CAS PubMed.
- A. Priye, S. W. Bird, Y. K. Light, C. S. Ball, O. A. Negrete and R. J. Meagher, Sci. Rep., 2017, 7, 44778 CrossRef CAS PubMed.
- A. Priye, C. S. Ball and R. J. Meagher, Anal. Chem., 2018, 90, 12385–12389 CrossRef CAS PubMed.
- C. S. Ball, R. F. Renzi, A. Priye and R. J. Meagher, Lab Chip, 2016, 16, 4436–4444 RSC.
- K. A. Curtis, D. L. Rudolph, I. Nejad, J. Singleton, A. Beddoe, B. Weigl, P. LaBarre and S. M. Owen, PLoS One, 2012, 7, e31432 CrossRef CAS PubMed.
- A. Bektaş, Food Analytical Methods, 2018, 11, 686–692 CrossRef.
- K. A. Curtis, D. L. Rudolph and S. M. Owen, J. Med. Virol., 2009, 81, 966–972 CrossRef CAS PubMed.
- K. A. Curtis, D. L. Rudolph, D. Morrison, D. Guelig, S. Diesburg, D. McAdams, R. A. Burton, P. LaBarre and M. Owen, J. Virol. Methods, 2016, 237, 132–137 CrossRef CAS PubMed.
- D. L. Rudolph, V. Sullivan, S. M. Owen and K. A. Curtis, PLoS One, 2015, 10, e0126609 CrossRef PubMed.
- C. R. Phaneuf, B. Mangadu, H. M. Tran, Y. K. Light, A. Sinha, F. W. Charbonier, T. P. Eckles, A. K. Singh and C.-Y. Koh, Biosens. Bioelectron., 2018, 120, 93–101 CrossRef CAS PubMed.
- Y. S. Jiang, S. Bhadra, B. Li, Y. R. Wu, J. N. Milligan and A. D. Ellington, Anal. Chem., 2015, 87, 3314–3320 CrossRef CAS PubMed.
- S. Bhadra, Y. S. Jiang, M. R. Kumar, R. F. Johnson, L. E. Hensley and A. D. Ellington, PLoS One, 2015, 10, e0123126 CrossRef PubMed.
- Y. S. Jiang, T. E. Riedel, J. A. Popoola, B. R. Morrow, S. Cai, A. D. Ellington and S. Bhadra, Water Res., 2018, 131, 186–195 CrossRef CAS PubMed.
- S. Bhadra, T. E. Riedel, M. A. Saldaña, S. Hegde, N. Pederson, G. L. Hughes and A. D. Ellington, PLoS Neglected Trop. Dis., 2018, 12, e0006671 CrossRef PubMed.
- D.-C. Nyan and K. L. Swinson, Sci. Rep., 2015, 5, 17925 CrossRef CAS PubMed.
- W. Liu, S. Huang, N. Liu, D. Dong, Z. Yang, Y. Tang, W. Ma, X. He, D. Ao, Y. Xu, D. Zou and L. Huang, Sci. Rep., 2017, 7, 40125 CrossRef CAS PubMed.
- L. Wan, T. Chen, J. Gao, C. Dong, A. H.-H. Wong, Y. Jia, P.-I. Mak, C.-X. Deng and R. P. Martins, Sci. Rep., 2017, 7, 14586 CrossRef PubMed.
- P. Bakthavathsalam, G. Longatte, S. O. Jensen, M. Manefield and J. J. Gooding, Sens. Actuators, B, 2018, 268, 255–263 CrossRef CAS.
- J. Xu, Y. Hu, J. Guo, Y. Yang, J. Qiu, X. Li and Z. Xin, Food Analytical Methods, 2019, 12, 422–430 CrossRef.
- Y. Xu, Chem. Soc. Rev., 2011, 40, 2719–2740 RSC.
- P. Travascio, A. J. Bennet, D. Y. Wang and D. Sen, Chem. Biol., 1999, 6, 779–787 CrossRef CAS PubMed.
- P.-H. Chou, Y.-C. Lin, P.-H. Teng, C.-L. Chen and P.-Y. Lee, J. Virol. Methods, 2011, 173, 67–74 CrossRef CAS PubMed.
- R. Komura, T. Kawakami, K. Nakajima, H. Suzuki and C. Nakashima, J. Gen. Plant Pathol., 2018, 84, 247–253 CrossRef CAS.
- R. Kubota, A. M. Alvarez, W. W. Su and D. M. Jenkins, Biol. Eng. Trans., 2011, 4, 81–100 CAS.
- R. Kubota, P. LaBarre, J. Singleton, A. Beddoe, B. H. Weigl, A. M. Alvarez and D. M. Jenkins, Biol. Eng. Trans., 2011, 4, 69–80 CAS.
- R. Kubota, P. LaBarre, B. H. Weigl, Y. Li, P. Haydock and D. M. Jenkins, Chin. Sci. Bull., 2013, 58, 1162–1168 CrossRef CAS PubMed.
- D. M. Jenkins, R. Kubota, J. Dong, Y. Li and D. Higashiguchi, Biosens. Bioelectron., 2011, 30, 255–260 CrossRef CAS PubMed.
- R. Kubota and D. M. Jenkins, Int. J. Mol. Sci., 2015, 16, 4786–4799 CrossRef CAS.
- L. Becherer, M. Bakheit, S. Frischmann, S. Stinco, N. Borst, R. Zengerle and F. von Stetten, Anal. Chem., 2018, 90, 4741–4748 CrossRef CAS PubMed.
- L. Becherer, S. Knauf, M. Marks, S. Lueert, S. Frischmann, N. Borst, F. von Stetten, S. Bieb, Y. Adu-Sarkodie, K. Asiedu, O. Mitja and M. Bakheit, Emerging Infect. Dis., 2020, 26, 282–288 Search PubMed.
- Y. Wang, Y. Wang, R. Lan, H. Xu, A. Ma, D. Li, H. Dai, X. Yuan, J. Xu and C. Ye, J. Mol. Diagn., 2015, 17, 392–401 CrossRef CAS PubMed.
- Y. Wang, D. Li, Y. Wang, K. Li and C. Ye, Molecules, 2016, 21, E111 CrossRef PubMed.
- O. Higgins, E. Clancy, M. Cormican, T. W. Boo, R. Cunney and T. J. Smith, Int. J. Mol. Sci., 2018, 19, 524 CrossRef PubMed.
- C. Liang, Y. Chu, S. Cheng, H. Wu, T. Kajiyama, H. Kambara and G. Zhou, Anal. Chem., 2012, 84, 3758–3763 CrossRef CAS PubMed.
- J. Chen, X. Xu, Z. Huang, Y. Luo, L. Tang and J.-H. Jiang, Chem. Commun., 2018, 54, 291–294 RSC.
- L. Cao, X. Cui, J. Hu, Z. Li, J. R. Choi, Q. Yang, M. Lin, L. Ying Hui and F. Xu, Biosens. Bioelectron., 2017, 90, 459–474 CrossRef CAS PubMed.
- A. S. Basu, SLAS Technol., 2017, 22, 369–386 Search PubMed.
- P.-L. Quan, M. Sauzade and E. Brouzes, Sensors, 2018, 18, 1271 CrossRef.
- M. S. Draz and X. Lu, Theranostics, 2016, 6, 522–532 CrossRef CAS PubMed.
- G. A. S. Minero, C. Nogueira, G. Rizzi, B. Tian, J. Fock, M. Donolato, M. Strömberg and M. F. Hansen, Analyst, 2017, 142, 3441–3450 RSC.
- B. Tian, J. Ma, T. Zardán Gómez de la Torre, Á. Bálint, M. Donolato, M. F. Hansen, P. Svedlindh and M. Strömberg, ACS Sens., 2016, 1, 1228–1234 CrossRef CAS.
- Y. Ge, Q. Zhou, K. Zhao, Y. Chi, B. Liu, X. Min, Z. Shi, B. Zou and L. Cui, Int. J. Nanomed., 2017, 12, 2645–2656 CrossRef CAS PubMed.
- Y. Zhou, J. Xiao, X. Ma, Q. Wang and Y. Zhang, Appl. Microbiol. Biotechnol., 2018, 102, 5299–5308 CrossRef CAS PubMed.
- S. Wachiralurpan, T. Sriyapai, S. Areekit, P. Sriyapai, S. Augkarawaritsawong, S. Santiwatanakul and K. Chansiri, Front. Chem., 2018, 6, 90 CrossRef.
- R. Kumvongpin, P. Jearanaikool, C. Wilailuckana, N. Sae-Ung, P. Prasongdee, S. Daduang, M. Wongsena, P. Boonsiri, W. Kiatpathomchai, S. S. Swangvaree, A. Sandee and J. Daduang, J. Virol. Methods, 2016, 234, 90–95 CrossRef CAS.
- R. Suebsing, P. Prombun and W. Kiatpathomchai, Lett. Appl. Microbiol., 2013, 56, 428–435 CrossRef CAS PubMed.
- W. Jaroenram, N. Arunrut and W. Kiatpathomchai, J. Virol. Methods, 2012, 186, 36–42 CrossRef CAS.
- N. Arunrut, J. Kampeera, S. Sirithammajak, P. Sanguanrut, P. Proespraiwong, R. Suebsing and W. Kiatpathomchai, PLoS One, 2016, 11, e0151769 CrossRef PubMed.
- N. Arunrut, J. Kampeera, R. Suebsing and W. Kiatpathomchai, J. Virol. Methods, 2013, 193, 542–547 CrossRef CAS PubMed.
- K. Watthanapanpituck, W. Kiatpathomchai, E. Chu and N. Panvisavas, J. Leg. Med., 2014, 128, 923–931 CrossRef.
- F. F. Carlos, B. Veigas, A. S. Matias, G. Doria, O. Flores and P. V. Baptista, Biotechnology Reports, 2017, 16, 21–25 CrossRef PubMed.
- Y. Lu, X. Ma, J. Wang, N. Sheng, T. Dong, Q. Song, J. Rui, B. Zou and G. Zhou, Biosens. Bioelectron., 2017, 90, 388–393 CrossRef CAS PubMed.
- Y. Chen, N. Cheng, Y. Xu, K. Huang, Y. Luo and W. Xu, Biosens. Bioelectron., 2016, 81, 317–323 CrossRef CAS PubMed.
- A. B. Nurul Najian, E. A. R. Engku Nur Syafirah, N. Ismail, M. Mohamed and C. Y. Yean, Anal. Chim. Acta, 2016, 903, 142–148 CrossRef CAS PubMed.
- J. H. Jung, S. J. Oh, Y. T. Kim, S. Y. Kim, W.-J. Kim, J. Jung and T. S. Seo, Anal. Chim. Acta, 2015, 853, 541–547 CrossRef CAS PubMed.
- Y. Wang, H. Li, Y. Wang, L. Zhang, J. Xu and C. Ye, Front. Microbiol., 2017, 8, 192 CrossRef.
- J. Yu, F. Wang, X. Zhan, X. Wang, F. Zuo, Y. Wei, J. Qi and Y. Liu, Anal. Bioanal. Chem., 2019, 411, 647–658 CrossRef CAS.
- E. A. Phillips, T. J. Moehling, S. Bhadra, A. D. Ellington and J. C. Linnes, Anal. Chem., 2018, 90, 6580–6586 CrossRef CAS.
- P. C. Foo, Y. Y. Chan, M. Mohamed, W. K. Wong, A. B. Nurul Najian and B. H. Lim, Anal. Chim. Acta, 2017, 966, 71–80 CrossRef CAS.
- H. Y. Yin, T. J. Fang and H. W. Wen, Lett. Appl. Microbiol., 2016, 63, 16–24 CrossRef CAS PubMed.
- P. Prompamorn, P. Sithigorngul, S. Rukpratanporn, S. Longyant, P. Sridulyakul and P. Chaivisuthangkura, Lett. Appl. Microbiol., 2011, 52, 344–351 CrossRef CAS PubMed.
- W. Jaroenram, W. Kiatpathomchai and T. W. Flegel, Mol. Cell. Probes, 2009, 23, 65–70 CrossRef CAS PubMed.
- J. R. Choi, J. Hu, Y. Gong, S. Feng, A. Wan, W. Abu Bakar, B. Pingguan-Murphy and F. Xu, Analyst, 2016, 141, 2930–2939 RSC.
- L. A. Rigano, F. Malamud, I. G. Orce, M. P. Filippone, M. R. Marano, A. M. Do Amaral, A. P. Castagnaro and A. A. Vojnov, BMC Microbiol., 2014, 14, 86 CrossRef PubMed.
- L. A. Rigano, M. R. Marano, A. P. Castagnaro, A. M. Do Amaral and A. A. Vojnov, BMC Microbiol., 2010, 10, 176 CrossRef.
- Y. Ge, B. Wu, X. Qi, K. Zhao, X. Guo, Y. Zhu, Y. Qi, Z. Shi, M. Zhou, H. Wang and L. Cui, PLoS One, 2013, 8, e69941 CrossRef CAS PubMed.
- K. Roskos, A. I. Hickerson, H.-W. Lu, T. M. Ferguson, D. N. Shinde, Y. Klaue and A. Niemz, PLoS One, 2013, 8, e69355 CrossRef CAS PubMed.
- Y.-L. Sun, C.-H. Yen and C.-F. Tu, J. Vet. Med. Sci., 2014, 76, 509–516 CrossRef CAS PubMed.
- Z. Liu, Q. Zhang, N.-N. Yang, M.-G. Xu, J.-F. Xu, M.-L. Jing, W.-X. Wu, Y.-D. Lu, F. Shi and C.-F. Chen, J. Microbiol. Biotechnol., 2019, 29, 454–464 CrossRef PubMed.
- T. Kaewphinit, N. Arunrut, W. Kiatpathomchai, S. Santiwatanakul, P. Jaratsing and K. Chansiri, BioMed Res. Int., 2013, 2013, 926230 Search PubMed.
- Z. K. Njiru, Diagn. Microbiol. Infect. Dis., 2011, 69, 205–209 CrossRef CAS.
- J. Zhang, J. Cao, M. Zhu, M. Xu and F. Shi, World J. Microbiol. Biotechnol., 2019, 35, 31 CrossRef PubMed.
- D. Liu, W. He, M. Jiang, B. Zhao, X. Ou, C. Liu, H. Xia, Y. Zhou, S. Wang, Y. Song, Y. Zheng, Q. Chen, J. Fan, G. He and Y. Zhao, AMB Express, 2019, 9, 11 CrossRef PubMed.
- R. A. Waters, V. L. Fowler, B. Armson, N. Nelson, J. Gloster, D. J. Paton and D. P. King, PLoS One, 2014, 9, e105630 CrossRef PubMed.
- B. H. Park, S. J. Oh, J. H. Jung, G. Choi, J. H. Seo, D. H. Kim, E. Y. Lee and T. S. Seo, Biosens. Bioelectron., 2017, 91, 334–340 CrossRef CAS PubMed.
- A. B. Nurul Najian, P. C. Foo, N. Ismail, L. Kim-Fatt and C. Y. Yean, Mol. Cell. Probes, 2019, 44, 63–68 CrossRef CAS PubMed.
- J. Connolly and T. G. St Pierre, J. Magn. Magn. Mater., 2001, 225, 156–160 CrossRef CAS.
- B. Tian, Z. Qiu, J. Ma, T. La Zardán Gómez de Torre, C. Johansson, P. Svedlindh and M. Strömberg, Biosens. Bioelectron., 2016, 86, 420–425 CrossRef CAS PubMed.
- H. Ravan and R. Yazdanparast, Anal. Biochem., 2013, 439, 102–108 CrossRef CAS PubMed.
- H. Ravan and R. Yazdanparast, Anal. Chim. Acta, 2012, 733, 64–70 CrossRef CAS.
- P. Prakrankamanant, C. Leelayuwat, C. Promptmas, T. Limpaiboon, S. Wanram, P. Prasongdee, C. Pientong, J. Daduang and P. Jearanaikoon, Biosens. Bioelectron., 2013, 40, 252–257 CrossRef CAS.
- K. Imai, N. Tarumoto, K. Misawa, L. Runtuwene, J. Sakai, K. Hayashida, Y. Eshita, R. Maeda, J. Tuda, T. Murakami, S. Maesaki, Y. Suzuki, J. Yamagishi and T. Maeda, BMC Infect. Dis., 2017, 17(1), 621 CrossRef.
- K. Imai, N. Tarumoto, K. Amo, M. Takahashi, N. Sakamoto, A. Kosaka, Y. Kato, K. Mikita, J. Sakai, T. Murakami, Y. Suzuki, S. Maesaki and T. Maeda, Parasitol. Int., 2018, 67, 34–37 CrossRef CAS.
- J. Yamagishi, L. R. Runtuwene, K. Hayashida, A. E. Mongan, L. A. N. Thi, L. N. Thuy, C. N. Nhat, K. Limkittikul, C. Sirivichayakul, N. Sathirapongsasuti, M. Frith, W. Makalowski, Y. Eshita, S. Sugano and Y. Suzuki, Sci. Rep., 2017, 7, 3510 CrossRef.
- K. Imai, N. Tarumoto, L. R. Runtuwene, J. Sakai, K. Hayashida, Y. Eshita, R. Maeda, J. Tuda, H. Ohno, T. Murakami, S. Maesaki, Y. Suzuki, J. Yamagishi and T. Maeda, Malar. J., 2018, 17, 217 CrossRef.
- A. S. Patterson, D. M. Heithoff, B. S. Ferguson, H. T. Soh, M. J. Mahan and K. W. Plaxco, Appl. Environ. Microbiol., 2013, 79, 2302–2311 CrossRef CAS.
- N. Nakamura, K. Ito, M. Takahashi, K. Hashimoto, M. Kawamoto, M. Yamanaka, A. Taniguchi, N. Kamatani and N. Gemma, Anal. Chem., 2007, 79, 9484–9493 CrossRef CAS.
- N. Nakamura, K. Ito, M. Takahashi, S. Hongo, K. Hashimoto, M. Kawamoto, A. Taniguchi, N. Kamatani and N. Gemma, Clin. Biochem., 2009, 42, 1158–1161 CrossRef CAS.
- K. Hashimoto and Y. Ishimori, Lab Chip, 2001, 1, 61–63 RSC.
- W. Sun, P. Qin, H. Gao, G. Li and K. Jiao, Biosens. Bioelectron., 2010, 25, 1264–1270 CrossRef CAS PubMed.
- G. V. Soraya, J. Chan, T. C. Nguyen, D. H. Huynh, C. D. Abeyrathne, G. Chana, M. Todaro, E. Skafidas and P. Kwan, Biosens. Bioelectron., 2018, 111, 174–183 CrossRef CAS PubMed.
- H. J. Chun, S. Kim, Y. D. Han, K. R. Kim, J.-H. Kim, H. Yoon and H. C. Yoon, ACS Sens., 2018, 3, 2261–2268 CrossRef CAS PubMed.
- Y. D. Han, H.-S. Kim, Y. M. Park, H. J. Chun, J.-H. Kim and H. C. Yoon, ACS Appl. Mater. Interfaces, 2016, 8, 10767–10774 CrossRef CAS PubMed.
- K. Nawattanapaiboon, W. Kiatpathomchai, P. Santanirand, A. Vongsakulyanon, R. Amarit, A. Somboonkaew, B. Sutapun and T. Srikhirin, Biosens. Bioelectron., 2015, 74, 335–340 CrossRef CAS PubMed.
- H. H. Nguyen, J. Park, S. Kang and M. Kim, Sensors, 2015, 15, 10481–10510 CrossRef CAS PubMed.
- H. Šípová, M. Piliarik, M. Vala, K. Chadt, P. Adam, M. Bocková, K. Hegnerová and J. Homola, Procedia Eng., 2011, 25, 148–151 CrossRef.
- S. D. Soelberg, T. Chinowsky, G. Geiss, C. B. Spinelli, R. Stevens, S. Near, P. Kauffman, S. Yee and C. E. Furlong, J. Ind. Microbiol. Biotechnol., 2005, 32, 669–674 CrossRef CAS.
- X. Zhi, M. Deng, H. Yang, G. Gao, K. Wang, H. Fu, Y. Zhang, D. Chen and D. Cui, Biosens. Bioelectron., 2014, 54, 372–377 CrossRef CAS PubMed.
- M. Bartosik, L. Jirakova, M. Anton, B. Vojtesek and R. Hrstka, Anal. Chim. Acta, 2018, 1042, 37–43 CrossRef CAS.
- M. Bartosik and L. Jirakova, Curr. Opin. Electrochem., 2019, 14, 96–103 CrossRef CAS.
- A. K. L. Yang, H. Lu, S. Y. Wu, H. C. Kwok, H. P. Ho, S. Yu, A. K. L. Cheung and S. K. Kong, Anal. Chim. Acta, 2013, 782, 46–53 CrossRef CAS PubMed.
- Space systems - Definition of the Technology Readiness Levels (TRLs) and their criteria of assessment, 2014, ISO 16290:2014-12 Search PubMed.
- Clinical Laboratory Improvement Amendments (CLIA), available at: https://www.cms.gov/Regulations-and-Guidance/Legislation/CLIA/index.html.
- CLIA Categorizations, available at: https://www.fda.gov/medical-devices/ivd-regulatory-assistance/clia-categorizations.
- L. He and H.-s. Xu, Aquaculture, 2011, 311, 94–99 CrossRef CAS.
- T. M. Squires, R. J. Messinger and S. R. Manalis, Nat. Biotechnol., 2008, 26, 417–426 CrossRef CAS PubMed.
- J. Dostálek, H. Vaisocherová and J. Homola, Sens. Actuators, B, 2005, 108, 758–764 CrossRef.
- J. Homola, H. B. Lu, G. G. Nenninger, J. Dostálek and S. S. Yee, Sens. Actuators, B, 2001, 76, 403–410 CrossRef CAS.
- G. G. Nenninger, J. B. Clendenning, C. E. Furlong and S. S. Yee, Sens. Actuators, B, 1998, 51, 38–45 CrossRef CAS.
- P. K. Drain, E. P. Hyle, F. Noubary, K. A. Freedberg, D. Wilson, W. R. Bishai, W. Rodriguez and I. V. Bassett, Lancet Infect. Dis., 2014, 14, 239–249 CrossRef PubMed.
- D. Mabey, R. W. Peeling, A. Ustianowski and M. D. Perkins, Nat. Rev. Microbiol., 2004, 2, 231–240 CrossRef CAS.
- N. Phares, R. J. White and K. W. Plaxco, Anal. Chem., 2009, 81, 1095–1100 CrossRef CAS PubMed.
- L. Civit, A. Fragoso and C. K. O'Sullivan, Electrochem. Commun., 2010, 12, 1045–1048 CrossRef CAS.
- Y. Liu, Q. Liu, S. Chen, F. Cheng, H. Wang and W. Peng, Sci. Rep., 2015, 5, 12864 CrossRef CAS PubMed.
- Z. Wei, X. Xiao, J. Wang and H. Wang, Sensors, 2017, 17, 2500 CrossRef PubMed.
- J. Jiang, X. Wang, R. Chao, Y. Ren, C. Hu, Z. Xu and G. L. Liu, Sens. Actuators, B, 2014, 193, 653–659 CrossRef CAS.
- D. Zhang, J. Jiang, J. Chen, Q. Zhang, Y. Lu, Y. Yao, S. Li, G. Logan Liu and Q. Liu, Biosens. Bioelectron., 2015, 70, 81–88 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2020 |