
A nanobiosensor for the simple detection of small molecules using non-crosslinking aggregation of gold nanoparticles with G-quadruplexes†
 de
Masahiro
Fujita
*b
and
de
Masahiro
Fujita
*b
and
Abstract
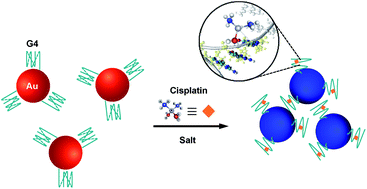
This work demonstrates a simple and specific colorimetric sensor for a hazardous small molecule, cisplatin, using a G-quadruplex (G4) DNA as a sensing probe and non-crosslinking aggregation of gold nanoparticles (AuNPs) as a signal enhancer. AuNPs functionalized with G4 strands dispersed stably in the colloidal system. The colloidal solution colour changed in a short time from red to purple-blue after cisplatin was added to the system. The cisplatin molecule can bind at the N7 guanine base of G4, resulting in the formation of a cisplatin–DNA adduct. The adduct formed does not act as a cross-linker between the particles, but probably causes a reduction in steric repulsion, associated with unfolding of the G4 strand. This induces non-crosslinking aggregation of particles. The plasmon absorption peak shift arising from the aggregation—that is, the degree of colour change—varied with the cisplatin concentration. This sensor showed a good analytical performance with linearity from 15.0 to 30.0 μM and a detection limit of 12.9 μM. Moreover, the response of the colour change to cisplatin was found to be faster than those for other analogues such as carboplatin and oxaliplatin, indicating that this system has a high specificity for cisplatin detection.
 Please wait while we load your content...
Please wait while we load your content...

