
Reaction parameter comparison and optimization of multiple displacement amplification
Mengting
Huang
,
Fang
Yang
,
Jiye
Fu
,
Pengfeng
Xiao
,
Jing
Tu
 * and
Zuhong
Lu
* and
Zuhong
Lu
State Key Laboratory of Bioelectronics, School of Biological Science and Medical Engineering, Southeast University, Nanjing, 210096, China. E-mail: jtu@seu.edu.cn; Tel: +86-025-83792396
First published on 22nd November 2019
Abstract
Multiple displacement amplification (MDA) has been widely used in biological and medical fields as a simple, stable and high-fidelity whole-genome amplification (WGA) method. From the reaction itself, many factors may influence the amplification quality of MDA. In this study, we analyzed MDA under different conditions of template input, amplification time, Mg2+ concentration, DNA denaturation methods and amplification temperature, and assessed their effects on the performance of MDA. We found that the use of different DNA denaturation methods before isothermal incubation can influence the amplification speed of MDA, which is represented as the reciprocal of the total time of the amplification reaction. Analyzed using next-generation sequencing, MDA coverage uniformity was found to be correlated with the amplification temperature. An appropriately high incubation temperature leads to more even coverage uniformity. Based on the results of this study, practical advice is offered to researchers to acquire better MDA performance.
Introduction
Multiple displacement amplification (MDA) is an isothermal DNA amplification method using ϕ29 DNA polymerase.1 After an initial random priming step, this enzyme facilitates DNA elongation via strand-displacement activity to form a hyper-branched DNA network, which results in sufficient DNA product after MDA.MDA has been widely used in biological and medical fields as a simple and stable whole-genome amplification (WGA) method with high fidelity. As it mainly depends on a moderate isothermal reaction, no thermal cycler is required. Furthermore, DNA can be amplified by ϕ29 DNA polymerase after brief alkaline treatment, which saves on labor for cell culture, bacterial collection and lysis, as well as DNA extraction.2 This characteristic greatly increases its application scenarios, such as clinical diagnosis,3 forensic investigation4 and metagenomics research.5 The “debranching” ability of ϕ29 polymerase enables amplification to be activated even without DNA denaturation, which also makes it more convenient.6 Another advantage of MDA is its consistent ability to attain high yields using various amounts of template DNA (100 fg to 10 ng).1 Trace DNA, such as DNA from a single sperm, can be amplified to generate sufficient product of up to 13–35 μg using MDA.7 The proofreading character of ϕ29 DNA polymerase by its exonuclease domain gives MDA high fidelity in the amplification process.8 It has been reported that the error rate of MDA is 5 × 10−6, which is 100-fold lower than that of Taq DNA polymerase.9 This feature makes it more suitable for single-nucleotide polymorphism (SNP) allele detection10 and single cell sequencing.11 Although MDA exhibits the above advantages, it must be admitted that MDA has some limitations that need to be considered. The uneven amplification in MDA causes bias and influences the result of Copy Number Variation (CNV) detection12 and also leads to a relatively high Allele Dropout (ADO) rate.13 Furthermore, the cell-to-cell reproducibility of MDA is lower than that of DOP-PCR and MALBAC,14 which makes it hard to determine CNV by increasing the number of cells sequenced.13
Although there have been many studies attempting to improve the MDA performance by changing the volume of the reaction,15,16 using microfluidic devices17–20 or changing the primers,21,22 all of these methods require an extra experimental appliance or new reagents. In the reaction itself, many factors may influence the performance of MDA, such as: (i) template input; (ii) amplification time; (iii) Mg2+ concentration; (iv) DNA denaturation methods; (v) amplification temperature. DNA denaturation methods that are often used in MDA include alkaline denaturation, heat denaturation and non-denaturation. Dean et al. found that heat denaturation tends to damage the DNA target.1 Paez et al. demonstrated that alkaline denaturation prior to MDA improved the call rate of SNPs and reduced the ADO rates compared to non-denaturation.23 However, Spits et al. observed that no significant differences were found between samples amplified with or without a 95 °C denaturation step in terms of amplification rates, ADO, preferential amplification (PA) rates and DNA yield.24 To our knowledge, no study has been performed to compare those three denaturation methods simultaneously in terms of their influence on the amplification time required to complete the reaction and reach the amplification plateau.
In this study, we analyzed MDA under different conditions of the five factors mentioned above and assessed their effects on the performance of MDA. As several previous studies1,4 have demonstrated how template input and amplification time influence the amplification yield, we also made comparisons with their work. Using next-generation sequencing, the coverage uniformity of MDA under different amplification temperatures was calculated and compared. Practical advice is offered to researchers for choosing suitable experimental conditions to acquire optimum MDA performance.
Materials and methods
In this study, all glass and plastic consumables were properly sterilized, dried and UV-treated before use. A dedicated set of pipettes were cleaned with ethanol (Sigma Aldrich) every time before experiments. All reagents were vortexed and spun briefly before use. Nuclease-free water (Ambion) was used in all experiments. Room temperature was kept at 25 °C using an air conditioner during experiments.Genomic DNA and MDA reaction buffer preparation
Genomic DNA of GM12878 cells (Coriell Institute) was harvested using a QIAamp DNA Mini Kit (Qiagen), and then quantitated using a Qubit dsDNA High Sensitivity Kit (ThermoFisher Scientific).MDA reaction buffer was prepared on ice and preserved at −20 °C before formal experiments, containing 37.5 mM Tris–HCl (pH 7.5) (HyClone), 50 mM KCl (Sigma), 20 mM (NH4)2SO4 (Sigma), 10 mM MgCl2 (Ambion), 4 mM DTT (Qiagen), 4 mM dNTP (ThermoFisher), 50 μM random thiophosphate-modified hexamer primers (Genescript) and 0.5 M Tre (Sigma). All the concentrations shown here are the final concentrations in 50 μL reaction volume.
The default conditions of MDA
The default conditions of template input, amplification time, Mg2+ concentration, DNA denaturation method and amplification temperature were 6 pg (equivalent to DNA content in single human cell), 10 hours, 10 mM, alkaline lysis and 30 °C, respectively. If the parameters are not specified, MDA was conducted as the following procedure.Purified DNA in 2.5 μL elution buffer was firstly denatured with an equal volume of alkaline lysis solution (400 mM KOH (Sigma), 10 mM EDTA (Invitrogen)) and incubated for 3 min at room temperature. Then, 5 μL neutralization solution (300 mM Tris–HCl buffer (HyClone), 200 mM HCl (Sigma)) was added, and the complex was vortexed and spun briefly to obtain 10 μL of denatured DNA solution in total.
MDA reaction mix was prepared on ice, containing 30 μL reaction buffer, 4 μL ϕ29 DNA polymerase (104 U mL−1, NEB), 1 μL pyrophosphatase inorganic (yeast) (100 U mL−1, NEB) and 5 μL nuclease-free water. Denatured DNA in 10 μL solution was added to attain a total volume of 50 μL amplification solution. Reactions were incubated for 10 h at 30 °C and terminated by heating to 65 °C for 10 min using a thermal cycler (Applied Biosystems) with a heated lid. The DNA product was quantitated using a Qubit dsDNA High Sensitivity Kit.
MDA under different conditions of template input, amplification time and Mg2+ concentration
In the experiments of MDA from different template inputs, initial amounts were set to 6 pg, 60 pg and 600 pg purified DNA. Amplification time was set to 4 h, 6 h, 8 h, 10 h, 12 h and 14 h in the experiments of MDA with different incubation times. In the prepared solution of MDA reaction buffer, the concentration of Mg2+ was changed to 6 mM, 8 mM, 10 mM, 12 mM, 15 mM, 18 mM, 20 mM and 25 mM to test how Mg2+ influenced amplification performance.Different denaturation methods before MDA
The heat denaturation step was carried out as follows: 6 pg DNA was diluted with nuclease-free water to make the total volume up to 10 μL, which was equivalent to the final volume of alkaline denatured DNA solution. Then the diluted DNA template was incubated at 95 °C for 3 min and chilled to 4 °C in a thermo cycler with heated lid at 105 °C to prevent evaporation. DNA denatured by alkaline lysis was carried out as the default procedure in the materials and methods. As for the amplification conducted without denaturation, the same amount of DNA was diluted to 10 μL in volume and incubated at 4 °C for 3 min.To track the change of DNA content during amplification, we added 2× EvaGreen dye to the denatured DNA solution. We chose EvaGreen dye rather than SYBR Green I because it claims to be extremely stable both thermally and hydrolytically and can be used at much higher dye concentration to obtain a high resolution signal.25 The total volume of the reaction complex reduced proportionally to 10 μL in this study. In order to correct for the spectral baseline shift resulting from either instrument instability or solvent evaporation, ROX was also added. The MDA reaction was performed in a real-time PCR thermo cycler (Applied Biosystems 7500) and fluorescence intensities were collected every 15 min. Real-time isothermal amplification data were exported by the Applied Biosystems 7500 Real-Time PCR Software v2.3, and analyzed using a Python script. The relative fluorescence intensities were calculated as the ratio of the fluorescence of EvaGreen dye divided by the fluorescence of ROX dye.
Two independent repeated experiments of each denaturation method (heat, alkaline and no denaturation) were conducted to verify the results.
MDA under different amplification temperature
The incubation temperature was set to 15 °C, 20 °C, 25 °C, 30 °C, 35 °C, 40 °C and 45 °C, while three independent repeated experiments of each parameter were carried out to verify the results, and two of them were finally sequenced.The produced DNA was prepared to construct a sequencing library using the Next Ultra II Kit (NEB). The quality of the resulting libraries was checked using an Agilent 2100 Bioanalyzer system (Agilent). After barcoding and pooling in proportions, all the libraries were subsequently sequenced in the Illumina HiSeq 4000 platform with 150 bp pair-end reads.
The original sequencing data were firstly checked by FastQC26 to obtain the eligible reads and then mapped to the hg19 reference genome using the BWA-MEM alignment algorithm.27 Mapped reads were then sorted and piled up using SAMtools.28 The Lorenz curves were plotted according to the study of Zong et al.14 using a Perl script. The coefficient of variation (CV) was calculated by a Perl script based on the following equation:

![[D with combining macron]](https://www.rsc.org/images/entities/i_char_0044_0304.gif) represents the average depth of all the bins. The CV of the read depths along the genome versus bin size ranging from 1 bp to 10 Mb was plotted to provide more information on the average variation on diverse scales.29
represents the average depth of all the bins. The CV of the read depths along the genome versus bin size ranging from 1 bp to 10 Mb was plotted to provide more information on the average variation on diverse scales.29
Results and discussion
Template input
In order to investigate how the amount of template input affects MDA reaction, we conducted experiments with initial inputs of 6 pg, 60 pg and 600 pg of purified DNA, with the amplification time set as 10 h. As shown in Fig. 1, all the experiments obtained sufficient product yield, ranging from 28.85 μg to 29.73 μg DNA. The difference among yields from low template input (6–600 pg) was rather low. The efficiency of MDA was found to be quite stable and barely influenced by the template input from 6 pg to 600 pg, which is also in accordance with Dean et al.'s previous work.1 This verifies the stability of the MDA product, which is very useful when applied to metagenomics studies and single cell sequencing.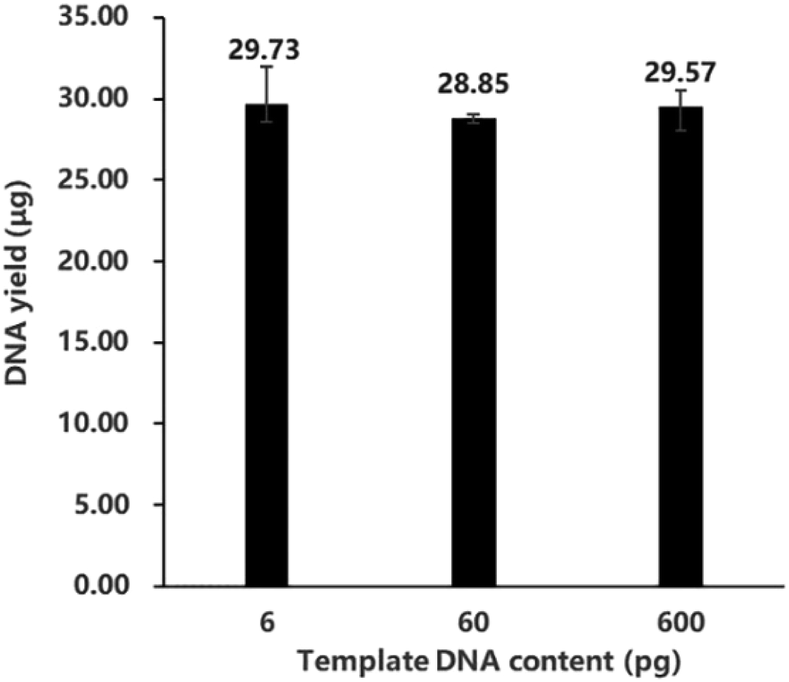 | ||
| Fig. 1 The DNA yields of MDA conducted with different template inputs: 6 pg, 60 pg and 600 pg DNA (n = 5). Total volume of every reaction system was 50 μL. The numbers on the column demonstrate the average yields of repeated experiments. The error bars display the highest and the lowest yields in the repeated experiments. | ||
Amplification time
Amplification time was set to 4 h, 6 h, 8 h, 10 h, 12 h and 14 h to explore the influence of different incubation times on MDA. Fig. 2A indicates that a short incubation time (4 h) led to a low yield. Although all the supplements were sufficient at this time, not all of the accessible template DNA was bound by ϕ29 DNA polymerase and primers for elongation. When the time was increased from 4 h to 8 h, the yield increased dramatically. From 4 h, the amplified DNA product had generated new templates and facilitated nonlinear amplification. With no shortage of reactants, the highest amplification speed could be obtained. The amplification speed here is represented as the reciprocal of the total time of the amplification reaction. As the incubation time was extended to 8 h and longer than 8 h, the yield reached a plateau at around 31 μg DNA (about 620 ng μL−1). It has been reported that MDA tends to be a self-limiting reaction, reaching a plateau at a DNA yield of about 700 ng μL−1.17 An equilibrium is reached between reactants and products. We also observed a slight decrease of yield after 8 h, which was due to the exonuclease activity of the polymerase enzyme to degrade the amplification product.4 | ||
| Fig. 2 MDA of different amplification times. (A) The DNA yields of MDA (50 μL volume) conducted for different amplification times, ranging from 4 h to 14 h (n = 5). (B) The change of relative fluorescence intensity versus amplification time. The MDA reaction was performed in a real-time PCR thermo cycler and fluorescence intensities of EvaGreen and ROX dye were collected every 0.25 h (15 min). The relative fluorescence intensities were calculated as the ratio of the fluorescence of EvaGreen dye divided by the fluorescence of ROX dye. | ||
As the relative fluorescence intensity increases with DNA concentration,25Fig. 2B demonstrates how the DNA concentration changed over time in one amplification reaction. The slope of the amplification curve can be considered as the instantaneous amplification speed. The instantaneous amplification speed of MDA increased firstly, decreased subsequently, and stopped finally when the yield reached a plateau. Fig. 2A is similar to the latter part of Fig. 2B, although the former demonstrates the yields of MDA at different incubation times and the latter represents the change of DNA concentration in one reaction. In fact, when the reaction process is demonstrated more comprehensively, in Fig. 2B, we can find that MDA was most efficient when the amplification proceeded for 4 h to 5 h. For a quick clinical diagnosis, product amplification for only 4–5 h is good enough to meet requirements.
Mg2+ concentration
Mg2+, whose role is as an enzyme activator and cofactor, can bind to two motifs of ϕ29 DNA polymerase (-D-NSLYP- and -K-NS(L/V)YG-) and promote its activity.30 However, high Mg2+ concentration often produces nonspecific products and induces replication errors from misincorporation with dNTPs.31 Consequently, appropriate Mg2+ concentration is vital to the MDA reaction.As shown in Fig. 3, when the concentration of Mg2+ was relatively low in the system (6–8 mM), MDA product was scarce. With the increase of Mg2+ ion concentration (10–15 mM), MDA reached its highest efficiency and exhibited a DNA yield of around 30 μg (about 600 ng μL−1). MDA had the highest yield (30.25 μg in average) when the Mg2+ concentration was 12 mM. However, when the concentration of Mg2+ was above 15 mM, the product yield declined sharply. The template could barely be amplified when the Mg2+ concentration reached 25 mM.
 | ||
| Fig. 3 DNA yields of MDA conducted with different Mg2+ concentrations, ranging from 6 mM to 25 mM (n = 4). Total volume of every reaction system was 50 μL. | ||
In summary, MDA cannot be well conducted under either too high or too low concentration of Mg2+. For those who self-prepare the amplification solution or want to improve the performance of MDA in the design of a commercial kit, when the dNTPs concentration in the system is 4 mM, the optimum concentration of Mg2+ to achieve a high yield is 10–15 mM.
DNA denaturation method
Alkaline lysis and heat lysis are two methods widely adopted by researchers for DNA denaturation and cell lysis before applying isothermal denaturation, due to the strong displacement ability of ϕ29 DNA polymerase.6 Different denaturation methods lead to various initial states of reactants, which may result in different amplification speeds in the initial stage of reaction.To investigate whether the denaturation method can influence the amplification speed of MDA, different denaturation treatments were conducted on the template DNA before incubation. As shown in Fig. 4, all the repeated experiments exhibit high consistency and distinguishable features under the different conditions of various denaturation methods. Whether and how DNA is denatured influence the amplification speed of MDA. Based on calculations, the average times taken to reach the highest instantaneous amplification speed were 225.0 min, 292.5 min and 247.5 min for MDA with alkaline denaturation, heat denaturation and no denaturation treatment, respectively (shown by the red squares in Fig. 4).
 | ||
| Fig. 4 The change of relative fluorescence intensity versus amplification time. The green, yellow and blue curves represent MDA with alkaline denaturation, heat denaturation and no denaturation, respectively. The results of two independent repeated experiments are displayed in different shades of the same color. The fluorescence intensities were collected every 0.25 h (15 min). The purple squares on the curves mark the earliest detection time. The red squares on the curves highlight the time when MDA reaches its highest instantaneous amplification speed. | ||
We considered the earliest time that relative fluorescence intensities exceeded 0.1 as the earliest detection time (shown by the purple squares in Fig. 4). As for alkaline denaturation, the earliest detection time was at 165 min and the average time to reach the highest instantaneous amplification speed was 225 min, which was the fastest among all the methods. After the introduction of the alkaline solution, dsDNA was denatured to ssDNA. The acidic solution added later neutralized the system to preserve enzyme activity but did not change the state of ssDNA. At the initial stage of the amplification, ssDNA was free to be bonded by hexamer primers at multiple sites, which led to a high startup rate and therefore a shortened time to reach the highest instantaneous amplification speed. MDA can be conducted without the necessity of DNA denaturation and results in sufficient yield. Although DNA remained double-stranded at the beginning of the reaction, the strong strand displacement by ϕ29 DNA polymerase facilitated the fracture of hydrogen bonds and allowed primers to bind as the reaction progressed. This resulted in a relatively longer earliest detection time (195 min) but the highest instantaneous amplification speed could be achieved at the average time of 247.5 min. As for MDA with heat denaturation, it had the slowest startup rate and needed the longest time to reach its highest instantaneous amplification speed. As heat denaturation tends to damage the DNA target,1 the amplification speed of MDA with heat denaturation was even slower than MDA without DNA denatured prior to incubation. The final yield of MDA with heat denaturation was also unstable. Moreover, heat denaturation may result in the mutation of the template DNA, which makes it more difficult to start up. Previous research has shown that more CG → TA artifactual mutations are induced with denaturation at elevated temperature than alkaline denaturation.32,33
Based on the previous work1,23,24 and this study, we suggest that if MDA is conducted from purified DNA, no denaturation is actually needed to obtain sufficient yield in a short time, which also saves on labor and reagents. If non-extracted DNA needs to be amplified, such as that from uncultured bacteria, alkaline denaturation is recommended as it provides cell lysis and can reach its highest instantaneous amplification speed in the shortest time. Alkaline denaturation also improved the call rate of SNPs and reduces the ADO rates.23 Heat denaturation is not suggested in any of the application scenarios because it leads to low startup speed and artifactual mutations.
Amplification temperature
In most MDA reactions, the isothermal incubation temperature is set to 30 °C. However, this temperature is far below the inactivation temperature of ϕ29 DNA polymerase in the final process (65 °C). As indicated in Fig. 5, the enzyme is capable of amplifying DNA under the temperature condition between 20 °C and 40 °C, and the optimum temperature for ϕ29 DNA polymerase to attain the maximum yield (29.45 μg) is around 30 °C. This result verifies why most protocols suggest that MDA should be conducted at 30 °C. It should be noticed that the temperature stabilities may vary from different batches of ϕ29 polymerases, but the tendency shown here almost remains consistent. | ||
| Fig. 5 The DNA yields of MDA at different incubation temperatures, ranging from 15 °C to 45 °C. The total volume of every reaction system was 50 μL. | ||
Nevertheless, we sequenced products of MDA under the temperature conditions of 20 °C, 25 °C, 30 °C and 35 °C, and found different results in terms of the coverage breadth and uniformity. As shown in Table 1, when equal amounts of sequencing data in every sample were randomly subsampled and analyzed, there were no remarkable differences or inclinations in the results of mapping rate, average sequencing depth and sequencing quality. The GC-content of the MDA product increased slightly with the incubation temperature, from 37.51% (on average) at 20 °C to 38.60% (on average) at 35 °C. We sequenced unamplified bulk GM12878 genomic DNA to a depth of ∼94× and the GC-content was found to be 40.77%. MDA was reported to have high coverage in the low GC-content region and low coverage in the high GC-content region, which means that it exhibits GC-content amplification bias.34 From the result of the sequencing data, products amplified by MDA of higher incubation temperature (35 °C) had less GC-content amplification bias than that of lower temperature. GC hydrogen bonds are more stable than AT hydrogen bonds. A possible explanation is that high temperature decreases the stability and facilitates the break of GC hydrogen bonds, reducing the GC-content amplification bias. Notably, within a certain temperature range (20–35 °C), the higher the incubation temperature was, the better the coverage of MDA product could be. The average coverage rate of samples processed at 35 °C was 56.23%, which was almost twice as high as that of the samples processed at 20 °C (27.32%). Meanwhile, we could see that the repeatability of samples amplified at 30 °C and 35 °C was relatively higher than that of the samples amplified at low temperature.
| Sample name | Incubation temperature (°C) | Reads count | Mapping rate (%) | Average depth (×) | Mapping quality | GC-content (%) | Coverage rate (%) |
|---|---|---|---|---|---|---|---|
| a The GC-content in unamplified bulk data of GM12878 cells is 40.77%. | |||||||
| D1-1 | 20 | 60![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000000 000000 |
98.93 | 2.8014 | 54.1412 | 37.77 | 34.82 |
| D1-2 | 60000000 |
98.82 | 2.8056 | 54.0683 | 37.25 | 19.82 | |
| D2-1 | 25 | 60000000 |
98.65 | 2.8069 | 54.3536 | 37.34 | 43.02 |
| D2-2 | 60000000 |
98.6 | 2.8103 | 54.6143 | 37.11 | 22.98 | |
| D3-1 | 30 | 60000000 |
97.45 | 2.7752 | 54.5535 | 37.49 | 59.57 |
| D3-2 | 60000000 |
97.5 | 2.7756 | 54.5038 | 37.49 | 49.83 | |
| D4-1 | 35 | 60000000 |
97.39 | 2.7096 | 54.5871 | 38.62 | 60.36 |
| D4-2 | 60000000 |
97.76 | 2.7304 | 54.3536 | 38.57 | 52.09 | |
As for the coverage uniformity, a Lorenz curve (bin size = 5 kb) was plotted, as shown in Fig. 6A, to provide the cumulative fraction of reads as a function of the cumulative fraction of the genome.35 A y = x line would represent perfectly even coverage and larger deviation means more bias. From Fig. 6A and B, we could find that MDA conducted at 35 °C outperforms MDA conducted at other temperature in uniformity of genome coverage. In order to comprehensively demonstrate the coverage uniformity on diverse scales, we calculated the CV of read depths along the genome versus bin size ranging from 1 bp to 10 Mb and plotted them in Fig. 6C and D. MDA incubated at 35 °C has the lowest CV value among all the experiments, indicating that an appropriately high incubation temperature can lead to more even coverage uniformity. Furthermore, Alsmadi et al.36 also found that elevated incubation temperatures could improve DNA amplification specificity and reduce template-independent DNA amplification. Some modifications of ϕ29 polymerase could also be found to enhance thermostability and enable enzymatic activity at higher temperatures compared to wild type enzymes.37 In applications where coverage uniformity in the amplification product is more important than yield, for example in CNV detection, higher temperature such as 35 °C are recommended for MDA rather than 30 °C in the traditional protocol to alleviate the impact of GC-content amplification bias and increase the coverage uniformity.
 | ||
| Fig. 6 Comparison of MDA conducted at different temperatures. (A and B) Lorenz curves at the variable 5 kb bin-size resolution. The cumulative fraction of mapped reads was plotted as a function of the cumulative fraction of the genome. (A and B) represent Lorenz curves of different temperatures in two independent repeated experiments. (C and D) Copy-number normalized coefficient of variation of read depths along the genome versus bin size ranging from 1 bp to 10 Mb. (C and D) represent CV plots in two independent repeated experiments. | ||
Conclusions
In this study, different conditions of template input, amplification time, Mg2+ concentration, DNA denaturation method and amplification temperature were investigated, to explore how these parameters influence the MDA reaction. The efficiency of MDA was found to be quite stable and barely influenced by the template input. When the dNTP concentration was 4 mM, the optimum concentration of Mg2+ to attain high MDA yield was 10–15 mM. Whether and how DNA was denatured influenced the amplification speed of MDA. Amplification of template DNA treated with alkaline denaturation exhibited a high start-up speed and was able to acquire sufficient yield in a short time, while amplification of undenatured DNA took second place. If non-extracted DNA needs to be amplified, alkaline denaturation is recommended as it provides cell lysis and can reach its highest instantaneous amplification speed in the shortest time. If MDA is conducted using purified DNA, amplifying without denaturation will save on labor and also have no negative effects on the amplification speed and final yield. Heat denaturation is not recommended because it leads to a low startup speed, unstable yield and artifactual mutations. MDA can be conducted at temperatures between 20 °C and 35 °C. The optimum temperature for MDA to attain the maximum yield is 30 °C. However, MDA conducted at 35 °C has higher coverage breadth and uniformity than that at 30 °C. In applications where coverage uniformity in the MDA product is more important than yield, a higher temperature such as 35 °C is recommended rather than 30 °C in the traditional protocol. Our study helps researchers in choosing suitable experimental conditions based on their requirements.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by project 61971125 of National Natural Science Foundation of China and the Fundamental Research Funds for the Central Universities of China.References
- F. B. Dean, S. Hosono, L. Fang, X. Wu, A. F. Faruqi, P. Bray-Ward, Z. Sun, Q. Zong, Y. Du, J. Du, M. Driscoll, W. Song, S. F. Kingsmore, M. Egholm and R. S. Lasken, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 5261–5266 CrossRef CAS PubMed.
- A. Taylor-Brown, D. Madden and A. Polkinghorne, Microb. Genomics, 2018, 4, e000145 Search PubMed.
- Y. Liu, J. Yao and M. Walther-Antonio, Biomicrofluidics, 2019, 13, 34109 CrossRef PubMed.
- B. Bruijns, A. Veciana, R. Tiggelaar and H. Gardeniers, Biosensors, 2019, 9, 85 CrossRef.
- P. Andersson, M. Klein, R. A. Lilliebridge and P. M. Giffard, Clin. Microbiol. Infect., 2013, 19, E405–E408 CrossRef CAS PubMed.
- E. Tenaglia, Y. Imaizumi, Y. Miyahara and C. Guiducci, Chem. Commun., 2018, 54, 2158–2161 RSC.
- Z. Jiang, X. Zhang, R. Deka and L. Jin, Nucleic Acids Res., 2005, 33, e91 CrossRef PubMed.
- C. Garmendia, A. Bernad, J. A. Esteban, L. Blanco and M. Salas, J. Biol. Chem., 1992, 267, 2594–2599 CAS.
- J. R. Nelson, Y. C. Cai, T. L. Giesler, J. W. Farchaus, S. T. Sundaram, M. Ortiz-Rivera, L. P. Hosta, P. L. Hewitt, J. A. Mamone, C. Palaniappan and C. W. Fuller, BioTechniques, 2002, 32, S44–S47 CrossRef.
- F. He, W. Zhou, R. Cai, T. Yan and X. Xu, J. Hum. Genet., 2018, 63, 407–416 CrossRef CAS.
- R. M. Bowers, N. C. Kyrpides, R. Stepanauskas, M. Harmon-Smith, D. Doud, T. B. K. Reddy, F. Schulz, J. Jarett, A. R. Rivers, E. A. Eloe-Fadrosh, S. G. Tringe, N. N. Ivanova, A. Copeland, A. Clum, E. D. Becraft, R. R. Malmstrom, B. Birren, M. Podar, P. Bork, G. M. Weinstock, G. M. Garrity, J. A. Dodsworth, S. Yooseph, G. Sutton, F. O. Glöckner, J. A. Gilbert, W. C. Nelson, S. J. Hallam, S. P. Jungbluth, T. J. G. Ettema, S. Tighe, K. T. Konstantinidis, W.-T. Liu, B. J. Baker, T. Rattei, J. A. Eisen, B. Hedlund, K. D. McMahon, N. Fierer, R. Knight, R. Finn, G. Cochrane, I. Karsch-Mizrachi, G. W. Tyson, C. Rinke, A. Lapidus, F. Meyer, P. Yilmaz, D. H. Parks, A. M. Eren, L. Schriml, J. F. Banfield, P. Hugenholtz and T. Woyke, Nat. Biotechnol., 2017, 35, 725–731 CrossRef CAS.
- T. J. Pugh, A. D. Delaney, N. Farnoud, S. Flibotte, M. Griffith, H. I. Li, H. Qian, P. Farinha, R. D. Gascoyne and M. A. Marra, Nucleic Acids Res., 2008, 36, e80 CrossRef CAS PubMed.
- L. Huang, F. Ma, A. Chapman, S. Lu and X. S. Xie, Annu. Rev. Genomics Hum. Genet., 2015, 16, 79–102 CrossRef CAS PubMed.
- C. Zong, S. Lu, A. R. Chapman and X. S. Xie, Science, 2012, 338, 1622–1626 CrossRef CAS.
- J. Gole, A. Gore, A. Richards, Y.-J. Chiu, H.-L. Fung, D. Bushman, H.-I. Chiang, J. Chun, Y.-H. Lo and K. Zhang, Nat. Biotechnol., 2013, 31, 1126–1132 CrossRef CAS PubMed.
- J. Li, N. Lu, X. Shi, Y. Qiao, L. Chen, M. Duan, Y. Hou, Q. Ge, Y. Tao, J. Tu and Z. Lu, Anal. Chem., 2017, 89, 10147–10152 CrossRef CAS PubMed.
- Y. Marcy, T. Ishoey, R. S. Lasken, T. B. Stockwell, B. P. Walenz, A. L. Halpern, K. Y. Beeson, S. M. D. Goldberg and S. R. Quake, PLoS Genet., 2007, 3, 1702–1708 CAS.
- Y. Nishikawa, M. Hosokawa, T. Maruyama, K. Yamagishi, T. Mori and H. Takeyama, PLoS One, 2015, 10, e0138733 CrossRef.
- A. M. Sidore, F. Lan, S. W. Lim and A. R. Abate, Nucleic Acids Res., 2016, 44, e66 CrossRef.
- Y. Fu, C. Li, S. Lu, W. Zhou, F. Tang, X. S. Xie and Y. Huang, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 11923–11928 CrossRef CAS.
- W. Wang, Y. Ren, Y. Lu, Y. Xu, S. D. Crosby, A. M. Di Bisceglie and X. Fan, BioTechniques, 2017, 63, 21–27 CrossRef CAS PubMed.
- O. Alsmadi, F. Alkayal, D. Monies and B. F. Meyer, BMC Res. Notes, 2009, 2, 48 CrossRef PubMed.
- J. G. Paez, M. Lin, R. Beroukhim, J. C. Lee, X. Zhao, D. J. Richter, S. Gabriel, P. Herman, H. Sasaki, D. Altshuler, C. Li, M. Meyerson and W. R. Sellers, Nucleic Acids Res., 2004, 32, e71 CrossRef.
- C. Spits, C. Le Caignec, M. de Rycke, L. van Haute, A. van Steirteghem, I. Liebaers and K. Sermon, Hum. Mutat., 2006, 27, 496–503 CrossRef CAS.
- F. Mao, W.-Y. Leung and X. Xin, BMC Biotechnol., 2007, 7, 76 CrossRef.
- J. Brown, M. Pirrung and L. A. McCue, Bioinformatics, 2017, 33, 3137–3139 CrossRef CAS.
- H. Li, arXiv preprint arXiv:1303.3997, 2013.
- H. Li, B. Handsaker, A. Wysoker, T. Fennell, J. Ruan, N. Homer, G. Marth, G. Abecasis and R. Durbin, Bioinformatics, 2009, 25, 2078–2079 CrossRef CAS.
- C. Chen, D. Xing, L. Tan, H. Li, G. Zhou, L. Huang and X. S. Xie, Science, 2017, 356, 189–194 CrossRef CAS.
- J. A. Cowan, Chem. Rev., 1998, 98, 1067–1088 CrossRef CAS.
- H. Khosravinia and K. P. Ramesha, Afr. J. Biotechnol., 2007, 6, 184–187 CAS.
- K. J. Fryxell and E. Zuckerkandl, Mol. Biol. Evol., 2000, 17, 1371–1383 CrossRef CAS.
- X. Dong, L. Zhang, B. Milholland, M. Lee, A. Y. Maslov, T. Wang and J. Vijg, Nat. Methods, 2017, 14, 491–493 CrossRef CAS.
- G. D. Evrony, E. Lee, B. K. Mehta, Y. Benjamini, R. M. Johnson, X. Cai, L. Yang, P. Haseley, H. S. Lehmann, P. J. Park and C. A. Walsh, Neuron, 2015, 85, 49–59 CrossRef CAS.
- B. J. O'Roak, L. Vives, W. Fu, J. D. Egertson, I. B. Stanaway, I. G. Phelps, G. Carvill, A. Kumar, C. Lee, K. Ankenman, J. Munson, J. B. Hiatt, E. H. Turner, R. Levy, D. R. O'Day, N. Krumm, B. P. Coe, B. K. Martin, E. Borenstein, D. A. Nickerson, H. C. Mefford, D. Doherty, J. M. Akey, R. Bernier, E. E. Eichler and J. Shendure, Science, 2012, 338, 1619–1622 CrossRef.
- O. Alsmadi, F. Alkayal, D. Monies and B. F. Meyer, BMC Res. Notes, 2009, 2, 48 CrossRef PubMed.
- R. Skirgaila and T. Povilaitis, US Pat., 20140322759A1, 2014.
| This journal is © The Royal Society of Chemistry 2020 |