
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn overview of therapeutic anticancer drug monitoring based on surface enhanced (resonance) Raman spectroscopy (SE(R)RS)
Chunchun
Li
,
Ziwei
Ye
,
Yikai
Xu
 * and
Steven E. J.
Bell
*
* and
Steven E. J.
Bell
*
School of Chemistry and Chemical Engineering, Queen's University Belfast, University Road, Belfast, BT7 1NN, UK. E-mail: yxu18@qub.ac.uk; S.Bell@qub.ac.uk
First published on 4th August 2020
Abstract
Therapeutic drug monitoring (TDM) is important for many therapeutic regimens and has particular relevance for anticancer drugs which often have serious effects and whose optimum dosage can vary significantly between different patients. Many of the features of surface enhanced (resonance) Raman spectroscopy (SE(R)RS) suggest it should be very suitable for TDM of anticancer drugs and some initial studies which explore the potential of SE(R)RS for TDM of anticancer drugs have been published. This review brings this work together in an attempt to draw some general observations about key aspects of the approach, including the nature of the substrate used, matrix interference effects and factors governing adsorption of the target molecules onto the enhancing surface. There is now sufficient evidence to suggest that none of these pose real difficulties in the context of TDM. However, some issues, particularly the need to carry out multiplex measurements for TDM of combination therapies, have yet to be addressed.
 Chunchun Li | Chunchun Li obtained her BSc in 2017 from East China University of Science and Technology in Applied Chemistry. Currently, she is a PhD student in chemistry under supervision of Prof. Steven E. J. Bell at Queen's University Belfast. Her research focuses on understanding molecular adsorption and related phenomenon on noble metal nanomaterial surfaces via SE(R)RS for the construction of nanomaterials with tailored surface properties. |
 Ziwei Ye | Dr Ziwei Ye received his BS degree in applied chemistry at East China University of Science and Technology in China. He proceeded to study in chemistry at Queen's University Belfast and obtained his PhD degree in 2019. He then continued to pursue his research in materials chemistry and currently holds a position as research assistant in Prof. Steven E. J. Bell's research group at Queen's University Belfast. His current research focuses on fabrication of multidimensional nanomaterials and their applications in photonics and photocatalysis. |
 Yikai Xu | Dr Yikai Xu is currently an independent Leverhulme Early Career Fellow at Queen's University Belfast. He obtained his BSc degree in Applied Chemistry at East China University of Science and Technology and completed his PhD research under the supervision of Prof. Steven E. J. Bell at QUB. Dr Xu is the recipient of the 2019 Kathleen Lonsdale Royal Irish Academy Prize for the most outstanding PhD research in chemical science in Ireland. His research interests are in the preparation and applications of plasmonic nanomaterials, particularly the preparation of hybrid materials containing plasmonic nanoparticle assemblies. |
 Steven Bell | Prof. Steven E. J. Bell received his PhD from Queen's University Belfast and worked at the Rutherford-Appleton Laboratory and the University of York before returning to QUB where he is a Professor of Physical Chemistry and Head of the School of Chemistry and Chemical Engineering. His research centres on nanomaterials and Raman spectroscopy. He has a particular interest in the application of Raman methods to real world problems including medical and security applications and was founder/director of a successful company manufacturing Raman spectrometers. |
Introduction
Many therapeutic anticancer drugs have serious side effects, causing damage to normal tissues and organs.1,2 This means that the treatment dosage needs to be optimized by balancing the need to provide the highest efficacy against minimising toxicity.3,4 For many drugs, the dosage is easy to adjust because there is a large difference between the effective dose and the levels giving toxicity. However, there are still a number of anticancer drugs for which the optimal dosage is hard to define due to their narrow therapeutic range. In addition, the response and toxicity of drugs in individual patients are quite different from aqueous environment so the various parameters of those drugs, pharmacokinetics and pharmacodynamics for example, are extremely valuable to study.5Therapeutic drug monitoring (TDM) is a key method in clinical chemistry and clinical pharmacology, which aims at accurately measuring the concentrations of medication in patient's blood.6 It has been applied in clinical practice since 1970s, primarily focusing on drugs with a narrow therapeutic window.7,8 Numerous factors will affect the appropriate therapeutic drug level, including personal factors such as age, gender, weight, and genetic make-up, as well as environmental factors such as diet, smoking habits and treatment history.8,9 Moreover, in most cases patients are treated with more than one anticancer drug simultaneously, so that TDM needs to be capable of carrying out multiplex measurements.10–14 This may be complex if there are significant drug–drug interaction.9 The most commonly applied platforms for TDM are immunoassays of various types and separation techniques combined with mass spectrometry (MS). In immunoassay, the detection of an analyte depends on the binding of the analyte with a specific binding molecule such as an antibody.15,16 Even though immunoassay provides a well-established and easy-operated platform technology, obvious shortfalls such as interference of the matrix, endogenous antibodies and drug metabolites etc. can limit its application in TDM. In addition, the low sensitivity of such a technique makes the detection of low concentration analytes difficult, while the need to have specific binding molecules for each drug target limits the generality.6,17 Compared to immunoassay, liquid chromatography (LC) combined with MS is a more robust platform and it has higher sensitivity for TDM. LC-MS avoids some of the interference effects which are found in immunoassay and it is able to detect multiple components in a complex mixed sample after chromatographic separation.6,18 However, LC-MS is time-consuming, expensive and laboratory-based. Apart from immunoassays and LC-MS, various spectroscopic methods have also been developed for TDM including NMR, Ultraviolet–visible (UV/vis), infrared (IR), fluorescence, X-ray photoelectron (XPS) and surface plasmon resonance (SPR) spectroscopies.19 Despite having some advantages, all these techniques have significant limitations associated with low sensitivity or limited accessibility.
SERS is an obvious potential alternative to existing methods for TDM. Many of its characteristics are exactly those which practical TDM requires. Most notably, the high sensitivity of SERS suggests that it will provide limits of detection and quantification that are in the range encountered in therapeutic applications.20,21 Moreover, this sensitivity can be increased if the excitation wavelength is chosen to fall within a strong absorption band of the target molecules, giving surface-enhanced resonance Raman spectroscopy (SERRS).22 Similarly, the ability of SE(R)RS to provide characteristic fingerprint spectra and to allow multiplex detection of several analytes simultaneously is a major advantage.23 Finally, these benefits can be realized in relatively low cost hand-held systems that are appropriate for bedside measurements. These considerations have led to several preliminary studies on monitoring anticancer drugs which have reported that it is possible to demonstrate some of these potential advantages in model systems which are similar to clinical applications. However, as is the case with many applications of SE(R)RS, a very wide range of drugs and experimental methods have been used in these studies so the overall picture is not clear.
The most notable variables in TDM SE(R)RS are the nature of the enhancing substrate used, excitation wavelength (SERS versus SERRS), data analysis method (univariate versus multivariate) and of course the chemical properties of the drug compounds studied, which is important since it determines which drugs will adsorb to the enhancing surface. The purpose of the review is to bring together information on the first generation of SE(R)RS studies and to draw out some general conclusions about the progress so far, as well as highlighting the areas which need to be developed further if SE(R)RS is to become established as a viable approach for TDM.
Methods for SE(R)RS therapeutic anticancer drug monitoring
The structures of the anticancer drugs which have been investigated by SE(R)RS in TDM-related studies are shown in Fig. 1, while Table 1 brings together some of the most important aspects of each of the studies. The first general observation from Fig. 1 is that the structures of the molecules which have been investigated are extremely diverse. From Table 1 the first notable aspect of the previous work listed is that both aggregated colloids and solid substrates have been used in these studies. Although it is often not explicitly stated, the rationale for using aggregated colloids in many of these studies is straightforward, aggregated colloids are extremely easy to prepare and offer intense SE(R)RS enhancement, which in some cases allowed the detection down to single molecule levels.24 As shown in Table 1, high signal enhancements have been achieved using even the most conventional types of aggregated colloids, such as aggregated citrate reduced Ag/Au colloids (CRSC/CRGC), which allowed anticancer drugs including methotrexate (2), mitoxantrone (5), 6-thioguanine (13) and doxorubicin (14) to be detected down to micro-molar levels.27–29,34,35,40–42 One common issue which arises in aggregated colloid systems is that colloid aggregation is a dynamic process. This means the plasmonic enhancement that aggregated colloids provide also changes over time, which can reduce the reproducibility of the SE(R)RS measurements. A popular approach to improve the reproducibility of SE(R)RS detection based on colloids is to use microfluidic systems. For example, Smith et al. have developed a flow cell system for mitoxantrone (5) detection in serum, which is simple and able to avoid interference from the biological matrix.35 In this work, a 632.8 nm excitation laser was selected to provide resonance enhancement and allow the SERRS signal of the drug to be recorded. This allowed mitoxantrone to be detected at concentrations as low as 0.04 nM. The same anticancer drug has also been successfully detected based on a microfluidic SERRS device with a 532 nm excitation laser.34 Similarly, Xu et al. have designed a SERS-microfluidic chip which can generate controllable aqueous micro-droplets in oil and used it for detection of 6-thioguanine (13) in human serum.40 Forming micro-droplets creates a stable and isolated micro-environment of colloid and drug, which increases the signal repeatability. A similar lab-on-a-chip SERS system has also been used for detection of methotrexate (2) in the therapeutic range (2.0–0.2 μM).29 Alternatively, it is possible to design novel colloidal aggregates with long-term stability or colloidal nanoparticles (NPs) with novel morphologies which make them SE(R)RS-active without the need for aggregation.45,46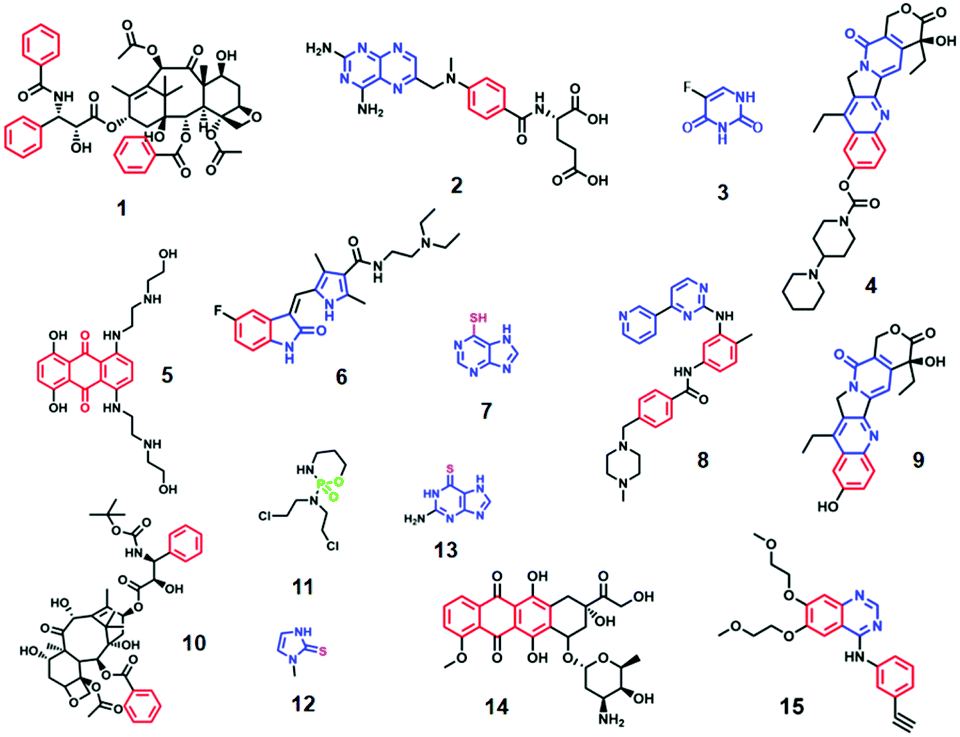 | ||
| Fig. 1 The structures of the anticancer drugs discussed in this review. | ||
| Name | Target cancer | Method | Bio-sample | LOD (linear quantification range) | Comments | Ref. | |
|---|---|---|---|---|---|---|---|
| 1 | Paclitaxel | Ovarian, breast and non-small cell lung cancer | Graphene oxide supported L-cysteine-functionalized star-like Au NP solid substrates; analyte droplet | Serum | 15 nM (15 nM–100 nM) | NPs surface modified | 25 |
| Microwave-treated Au-polystyrene beads solid film; analyte droplet | Plasma | 10 nM (10 nM–100 nM) | Multi-variate analysis | 26 | |||
| 2 | Methotrexate | Breast cancer, leukaemia, lung cancer, lymphoma, and osteosarcoma | LC-SERS combined system; Au aggregated colloid | Urine | 2.21 μM (n/a) | Portable Raman, binary analyte detection (drug and metabolite) | 27 |
| Drying analyte on dip-coated Au solid substrate | Serum | 0.1 μM (0.1 μM–20 μM) | Multivariate analysis | 28 | |||
| Microfluidic SERS system; Ag aggregated colloid | — | 0.17 μM (0.2 μM–2 μM) | — | 29 | |||
| 3 | 5-Fluorouracil (active metabolite of capecitabine) | Breast cancer, gastric cancer and colorectal cancer | HR-CSAS-SERS system; Ag aggregated colloid | Sweat | 2 μg mL−1 (2 μg mL–20 μg mL−1 | The drug was not mixed with sweat | 30 |
| 4 | Irinotecan | Colon cancer and small cell lung cancer | TLC-SERS system; Au/Ag colloids deposited on TLC spots | Serum | 2.73 μM (n/a) | Different capping agents tested; SERRS | 31 |
| Drying drug droplets on Klarite Au substrate | — | 60–70 ng (198 ng–4032 ng) | Binary drug detection; multivariate analysis | 32 | |||
| 5 | Mitoxantrone | Acute myeloid leukaemia, non-Hodgkin lymphoma, breast cancer and primary liver cancer | Flow cell; electron-beam evaporated Au on SiO2-polymer substrate | — | 61.03 ng mL−1 (0.061 μg mL−1–15.63 μg mL−1) | Binary drugs detection | 33 |
| Microfluidic system; Ag aggregated colloid | — | 500 nM (n/a) | SERRS | 34 | |||
| Flow cell system; Ag aggregated colloid | Plasma | 0.02 ng mL−1 (1 ng mL−1–43 ng mL−1) | SERRS | 35 | |||
| 6 | Sunitinib | Kidney cancer, gastrointestinal stromal tumours and pancreatic neuroendocrine tumours | Drying analyte on Klarite Au substrate | — | 18–26 ng (88 ng–1872 ng) | — | 32 |
| 7 | 6-Mercaptopurine | Acute lymphocytic leukaemia | Microfludidic system; using Ag NPs as bio-probe in cells | Living cells | 10 nM (10 nM–10 μM)* | Binary drug detection; linear quantification range not specified | 36 |
| β-Cyclodextrin functionalised Ag aggregated colloid | — | 2.4 nM (4 nM–20 nM) | NPs surface modified | 37 | |||
| 8 | Imatinib | Chronic myeloid leukaemia | Drying analyte on silicon nanopillar substrate coated with Ag | Plasma | 0.43 μM (n/a) | Portable Raman spectrometer; deproteinization; multivariate analysis | 38 |
| 9 | SN-38 (active metabolite of CPT-11) | Colorectal cancer | Drying analyte on Klarite Au substrate | — | 20–50 ng (107 ng–1092 ng) | Binary drug detection; multivariate analysis | 32 |
| 10 | Docetaxel | Breast cancer, non-small cell lung cancer, advanced stomach cancer, head and neck cancer and metastatic prostate cancer | Ag@ZnO NPs were immersed in analyte solution and then the mixture was dried onto an aluminium foil wafer | Plasma | 1 μg mL−1 (1 μg mL−1–1000 μg mL−1) | Multivariate analysis | 39 |
| 11 | Cyclophosphamide | Lymphomas, leukaemia, myeloma, lung cancer and breast cancer | GO-supported L-cysteine-functionalized star-like Au solid substrates; analyte droplet | Serum | 5 nM (5 nM–100 nM) | NPs surface modified; anti-fouling SAM | 25 |
| 12 | Methimazole | Hyperthyroidism (cancer complication) | Microfluidic system; using Ag NPs as bio-probe in cells | Living cells | 100 nM (100 nM–10 mM)* | Binary drug detection | 36 |
| 13 | 6-Thioguanine | Acute myeloid leukaemia | Droplet based microfluidic system; Au aggregated colloid | Serum | 0.66 μM (1 μM–100 μM)* | Deproteinization | 40 |
| 14 | Doxorubicin | Breast cancer, bladder cancer, Kaposi's sarcoma, lymphoma, and acute lymphocytic leukaemia | Ag aggregated colloid | Plasma | — | Multivariate analysis | 41 |
| Ag aggregated colloid | Plasma | 100 nM (100 nM–750 nM) | Multivariate analysis | 42 | |||
| Functionalised Au lithographic film | Plasma | 50 nM (0.05 μM–2 μM) | Mixed SAMs modified NPs; binary analytes detection (drug and metabolites) | 43 | |||
| 15 | Erlotinib | Non-small cell lung cancer and pancreatic cancer | Laser-ablated Au NP colloid; indirect competitor detection | Plasma | 300 nM (300 nM–3000 nM)* | NPs surface modified; click chemistry involved; using internal reference as competitor | 44 |
In contrast to aggregated colloids, solid enhancing substrates often exhibit significantly higher stability, which means that to facilitate the adsorption of weakly adsorbing analytes they can be soaked for long periods of time in the sample solution or a droplet of analyte can be directly dropped onto the substrate and Raman probed. For example, SERS signals of the anticancer drugs paclitaxel (1) and cyclophosphamide (11) have both been successfully obtained using this approach while the SERS signal of doxorubicin (14) has been observed in biological matrices using solid substrates within a flow-cell system.25,26,43 Alternatively, the analyte solution can be deposited and then allowed to dry directly onto the surface of the solid substrate to physically bring the analyte into close proximity with the enhancing surface. Meneghetti et al. have used this method to quantitatively detect doxorubicin (14), irinotecan (4) and SN-38 (9) on commercially available Klarite substrates while imatinib (8) spiked in plasma has also been measured with a limit of detection (LOD) of 0.43 μM.32,38
The approaches above can be extended by combining them with chromatographic separation. Bonifacio et al. have developed a TLC-SE(R)RS method for detecting the anti-neoplastic drug, irinotecan (4) in serum samples.31 In this work, the SE(R)RS signal of irinotecan was acquired by directly applying and then drying colloids on the relevant TLC spot. A pre-resonance effect was observed by using a 514 nm laser rather than a 785 nm laser. Moreover, Goodacre et al. have successfully demonstrated SERS detection of methotrexate (2), an anti-neoplastic drug, in urine samples, by coupling SERS with high-performance liquid chromatography (HPLC).27 By pre-separating methotrexate from its metabolite, 7-hydroxy methotrexate, by HPLC and then using aggregated colloid, the SERS signal of methotrexate was detected down to 2.21 μM using a portable Raman system.
Other less conventional approaches to anticancer drug detection have also been explored. For example, rather than using standard Au or Ag NPs, Chen et al. have used composite Ag@ZnO NPs to detect docetaxel (10) in plasma by mixing the particles with the analyte solution and then drying them onto an aluminium support.39 In addition, an example of indirect SERS detection of erlotinib (15), which uses the loss of the signal of a strongly scattering competitive molecule to signal the presence of the drug, has been presented by Meneghetti et al.44
Finally, SE(R)RS can, of course, be used alongside other synergistic techniques. For example, SERS has been used for identification of 5-fluorouracil (3) (the effective metabolite of capecitabine) while the concentration of capecitabine itself was measured using high-resolution continuum source absorption spectrometry which has high sensitivity for fluorine and low chlorine interference.30
Table 1 also shows the linear quantification ranges that have been achieved for the various anticancer drugs which have been studied using SE(R)RS with different types of enhancing substrates and sampling techniques. Achieving high accuracy and reproducibility in quantitative SE(R)RS analysis have been a longstanding challenge in SE(R)RS but in general, the majority of the methods discussed here demonstrated good linear quantification ranges of at least an order of magnitude with acceptable levels of uncertainty. In some cases multivariate methods have been used in analysis of the data and it seems likely that these will be used increasingly as SE(R)RS is applied for routine TDM where the desired output is the measured value of a concentration rather than a spectrum. However, in the current context, it is important to note that the clinically relevant concentration range varies widely between drugs and that only a small fraction of SE(R)RS studies to date have compared the quantification range they achieved for their target drug with the clinically relevant concentration range.25,27,29,32,35,38
Apart from the selection of an appropriate substrate, another equally important and general issue in SE(R)RS TDM of anticancer drugs is that the samples are typically in bio-fluids, which contain high concentrations of bio-molecules, particularly proteins, that interfere with the measurements by binding to the anticancer drugs and/or competing with the anticancer drugs for enhancing surface.47,48 For colloidal substrates the adsorption of protein contaminants also prevents colloid aggregation, which is often crucial for the formation of plasmonic hot-spots for effective SE(R)RS analysis.48
In general, the effect of protein in SE(R)RS is reduced using sample pre-treatment steps for deproteinization or by using enhancing surfaces with anti-fouling properties. Sample deproteinization typically involves treating the sample with alcohols, which separates the anticancer drugs from the proteins in solution and induces protein precipitation. This method has been successfully applied to SE(R)RS detection of irinotecan (4), 6-thioguanine (13), imatinib (8), erlotinib (15) and doxetaxel (10).31,38–40,44 It is also possible to remove proteins with adsorbent or filtering materials. For example, White et al. have successfully detected flucytosine, an antifungal agent, in serum using a vertical flow membrane system with inkjet-printed SERS sensors.49 In this work, layers of filter paper were utilized to filter out the protein and a portable Raman spectrometer used to obtain the SERS signals from the printed Au NPs. Quantitative detection of flucytosine in undiluted serum was achieved with a LOD of 10 μg mL−1. Moreover, Markin et al. have presented a pre-separation method for urine samples which used silica gel chromatography and allowed the detection of ceftriaxone, an antibiotic, down to 0.4 μg mL−1.50 Although these examples both focused on detection of non-anticancer drug molecules, the approach for protein separation is likely to be readily applicable for SE(R)RS TDM of anticancer drugs.
To create antifouling surfaces, Yu et al. have used a hierarchical surface modification strategy which involved pre-forming a self-assembled monolayer (SAM) of zwitterionic polymer brush on the surface of Au NPs before using them for SERS detection of doxorubicin (14).43 A similar approach was applied by De la Rosa et al. who used Au NPs functionalised with a zwitterionic amino acid L-cysteine SAM brush as the enhancing substrate, which allowed detection of paclitaxel (1) and cyclophosphamide (11) down to nano-molar level in serum.25
In general, a broad range of experimental methods have already been demonstrated for SE(R)RS detection of anticancer drugs. However, there is insufficient evidence to judge which method is optimum, or even if there exists a single optimum solution but the fact that so many methods are at least reasonably successful suggests that the availability of an appropriate enhancing substrate or need to reduce protein interference will not be the major limitations in implementing SE(R)RS as a method for TDM.
The adsorption of anticancer drugs on Au/Ag surfaces
The previous sections showed that many anticancer drugs can be detected by SE(R)RS. However, this in itself is not sufficient to demonstrate that SE(R)RS can be developed into a generally viable method for TDM of anticancer drugs, since there is a much larger number of anticancer drug molecules which have not been studied by SE(R)RS. Therefore, in this section we discuss whether the drugs which have been studied to date are representative of the large number of potential target analytes or if they are actually a special subset of anticancer drugs which are particularly favourable for SE(R)RS detection.SE(R)RS analysis of anticancer drugs requires the analyte to be in close proximity to the enhancing surface to experience sufficient signal enhancement. In most cases, this requires the drugs to be able to spontaneously adsorb onto the enhancing surface, which depends on both the molecular structure of the drug and the surface chemistry of the substrate material.51 It is therefore extremely important to discuss the types of interactions between the anticancer drugs and Ag/Au metal surfaces investigated so far, since this may serve as a general guideline to predict other potential anticancer drug candidates which could be detected via SE(R)RS.
Ag/Au NPs typically carry a layer of chemically bound capping agents on their surfaces. Therefore, to move into the enhancing electromagnetic field on the surface of the NPs the analyte must either replace the capping agents or adsorb onto them as a second layer, as illustrated in Fig. 2. To replace the initial capping agents on the surface of the enhancing metals the anticancer drugs need to have functional groups which adsorb strongly to the metal surface. The first molecule detected by SERS was pyridine and it is now well established that pyridine and various other types of nitrogen-containing heteroatomic functional groups have a strong affinity to Ag and Au surfaces.52–54 Indeed, ca. 66% of anticancer drugs which have been studied using SE(R)RS contain nitrogen-based hetero-aromatic rings. For example, Goodacre et al. have demonstrated that methotrexate (2), a pyrimidyl anticancer drug, can be detected using CRSC as enhancing substrate.27 Similarly, capecitabine (3), which also contains a pyrimidine group, has been found to spontaneously adsorb onto chloride-capped Ag NPs.30 Irinotecan (4), a pyridyl drug which is one of the most cytotoxic but effective drugs used for the treatment of metastatic colorectal cancer, could be quantitatively analyzed using both citrate-capped Au NPs and chloride-capped Ag NPs via SE(R)RS.31,32 Imatinib (8) which contains multiple nitrogen-based hetero-aromatic functional groups has also been successfully detected.38
 | ||
| Fig. 2 Schematic illustration of the two ways in which analytes may adsorb onto enhancing metal surface: chemical adsorption and physical adsorption. | ||
Another hetero atom which is well-established to be strongly adsorbing to Ag and Au surfaces is sulfur, with thiols/thioyls being the best-known.55,56 For example, 6-mercaptopurine (7), methimazole (12) and 6-thioguanine (13) has been detected down to 2.4 nM, 100 nM and 0.66 μM, respectively.36,40 It is notable that 20% of the anticancer drugs which have been studied by SE(R)RS contain sulfur while only 0.2% of anticancer drugs are sulfur containing.57
Apart from nitrogen-based hetero-aromatic and sulfur-based functional groups, amine functional groups are also commonly found in the group of anticancer drugs which have been studied by SE(R)RS. In fact, 13 of the 15 anticancer drugs which have been studied by SE(R)RS contain at least one amine group. These can be strongly binding, DFT calculations have shown that the adsorption strengths of amine groups are highly dependent on the overall structure of the molecule and the protonation state of the amine groups.58 A study showed that in 2016, the percentage of anticancer drugs containing R3N, R2NH or RNH2 groups was 38.6%, 40% and 6.8%, respectively.59
Besides sulfur and nitrogen compounds, phosphorous compounds can also form chemical bonds with Au, Ag.60 An example that is relevant here is cyclophosphamide (11) which is a non-aromatic anticancer drug which contains both amines and phosphoryl groups and could be detected down to 5 nM on graphene oxide supported L-cysteine-functionalized star-like Au NPs substrates, as demonstrated by Camacho-Villegas and De la Rosa.25
It is generally believed that polycyclic aromatic hydrocarbons (PAHs) do not possess strong affinity for Ag and Au nanosurfaces. However, we have recently shown that PAHs, such as naphthalene and fluoranthene, can adsorb directly onto the surface of CRGC by displacing citrate and chloride ions through strong π–metal interactions.61 Interestingly, the effect was only observed for CRGC but not CRSC. This is significant because, as shown in Fig. 1, most anticancer drugs that have been studied by SE(R)RS contain at least one aromatic ring. In fact, it has been reported that 74% of all known anticancer drugs contain at least one aromatic ring (including carbo-aromatic rings and hetero-aromatic rings) which could have important effects on the adsorption of aromatic anticancer drugs to Au and Ag substrates.57 The contribution of π–metal interactions to the adsorption of aromatic anticancer drugs is not clear in many cases since most of the aromatic drugs which have been studied by SE(R)RS also contain other surface binding functional groups. However, paclitaxel (1) and doxorubicin (14), both contain no well-defined conventional surface binding functional groups and would therefore be expected to preferentially adsorb on Au surfaces which allow π–metal interactions. Consistent with this expectation, all the existing SERS studies of paclitaxel (1) were obtained by using Au NPs as SERS enhancing substrates. Huang et al. successfully detected paclitaxel on bare Au at even 15 nM drug concentration, while work published by De la Rosa et al. showed detection on cysteine functionalised Au substrates with a LOD as low as 10 nM.25,26 However, SERS studies of doxorubicin (14) have been carried out on both Ag and Au substrates, suggesting that other moieties such as hydroxyl groups may also contribute to binding.41–43
Overall, SE(R)RS has been demonstrated for anticancer drugs containing several types of functional groups including hetero/carbo-aromatic rings (present in >70% anticancer drugs), various amine groups (present in >80% of anticancer drugs), sulphur-containing group (much less common, present in 0.2% of anticancer drugs) and phosphorous groups. The high proportion of these groups in anticancer drugs indicates that the drugs which have been investigated so far are unlikely to be especially favourable cases. Instead it is likely that most anticancer drugs possess sufficient affinity to Au and/or Ag surfaces to allow SE(R)RS detection.
Surface modification methods for SE(R)RS therapeutic anticancer drug monitoring
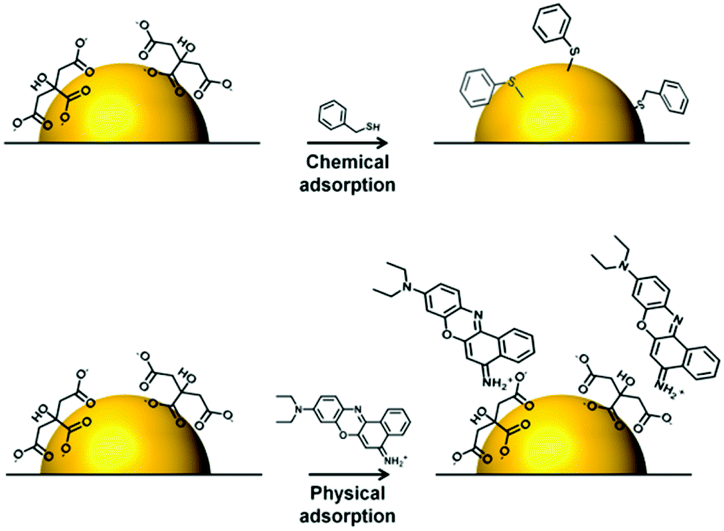
Within the SE(R)RS literature there are numerous examples of studies in which surface modification is used to promote the adsorption of target analytes of various types onto the enhancing surface, but the approach has been less widely adopted for anticancer drugs, presumably because their intrinsic tendency to adsorb to Ag and Au enhancing metals makes the need for them less pressing than for other weakly-adsorbing targets. However, surface-modification techniques may still be useful for improving the performance of SE(R)RS in TDM of anticancer drugs.For example, Tian et al. showed that using Ag NPs modified with β-cyclodextrin as the enhancing substrate increased not only the SERS signal intensity of 6-mercaptopurine (7) but also the speed of the analytical process and the linear quantification range of the drug, compared to as-prepared CRSC (Fig. 3).37 This signal improvement was attributed to the insertion of 6-mercaptopurine into the cavity of β-CD, which forced the drug molecules to stand vertically rather than lie flat on the surface of the enhancing substrate.
 | ||
| Fig. 3 Schematic illustration of the sensing mechanism when β-CD-AgNPs are used as the SERS-active substrate to detect 6-mercaptopurine.37 | ||
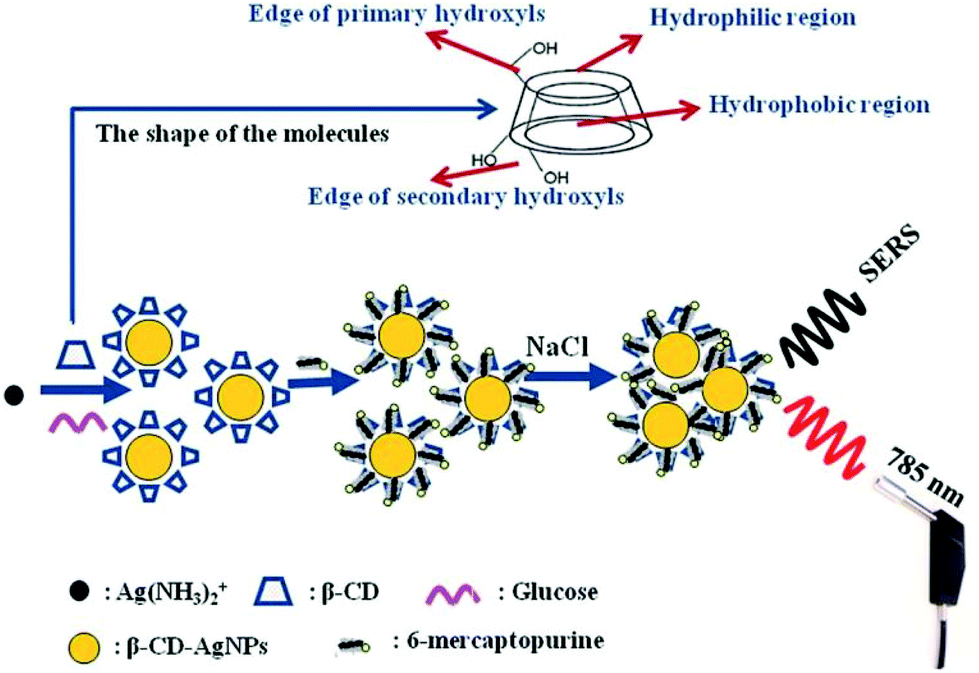
An alternative approach to using supramolecular hosts is to modify the surfaces with SAMs of thiol-terminated modifiers which are chosen to attract non-adsorbing analytes and/or to repel competing contaminants. As discussed in previous sections, SAMs of zwitterionic polymers have been used as antifouling layers in SERS TDM of doxorubicin (14) in plasma (Fig. 4).43 In addition to the zwitterionic polymer antifouling layer the mixed SAMs also contained different types of thiols which acted as either chemically-binding ‘reporters’ or physically attracting ‘attractors’ which allowed the detection of a variety of weakly/non adsorbing therapeutic drug molecules.
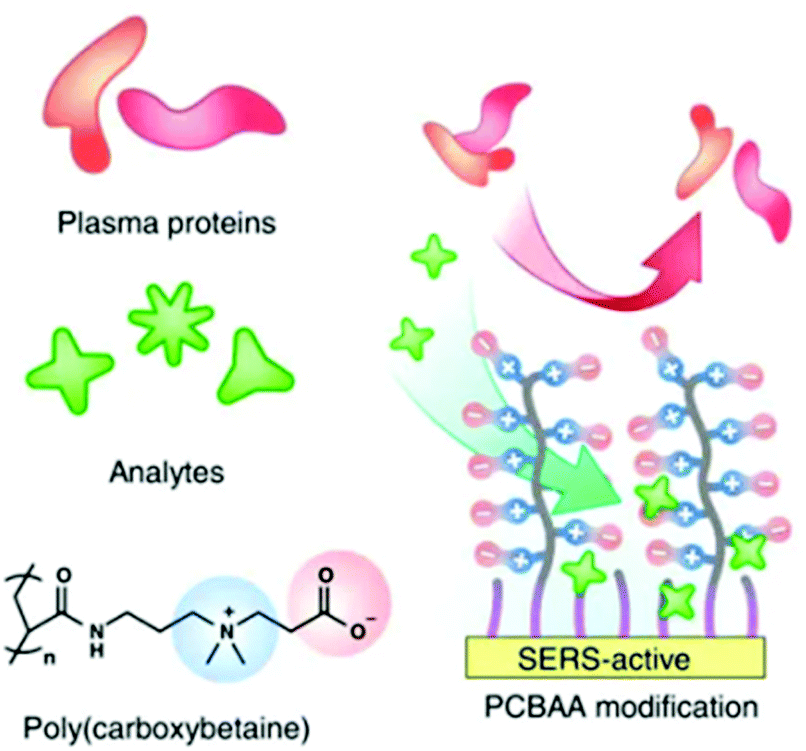 | ||
| Fig. 4 Schematic shows hierarchical pCBAA-based SERS-active substrate functionalized with zwitterionic non-fouling layer.43 Reprinted with permission from ref. 43. Copyright 2016 Springer Nature. | ||
Besides organic modifiers, the surface of Ag and Au enhancing metals can also be modified with other inorganic materials to form hybrid materials to increase the SERS signals of anticancer drugs. For example, graphene oxide (GO) supported Au NPs have been developed as SERS enhancing substrates for the detection of two anticancer drugs, paclitaxel (1) and cyclophosphamide (11).25 Using crystal violet as a probe molecule, the authors showed that the SERS signal of the probe molecule at 10−6 M on the hybrid enhancing substrate was ca. 2× stronger than the probe molecule at 10−4 M on pure Au enhancing colloids. In addition, the authors showed that the Au@GO substrates could be functionalised with L-cysteine modifiers to reduce protein adsorption (Fig. 5).
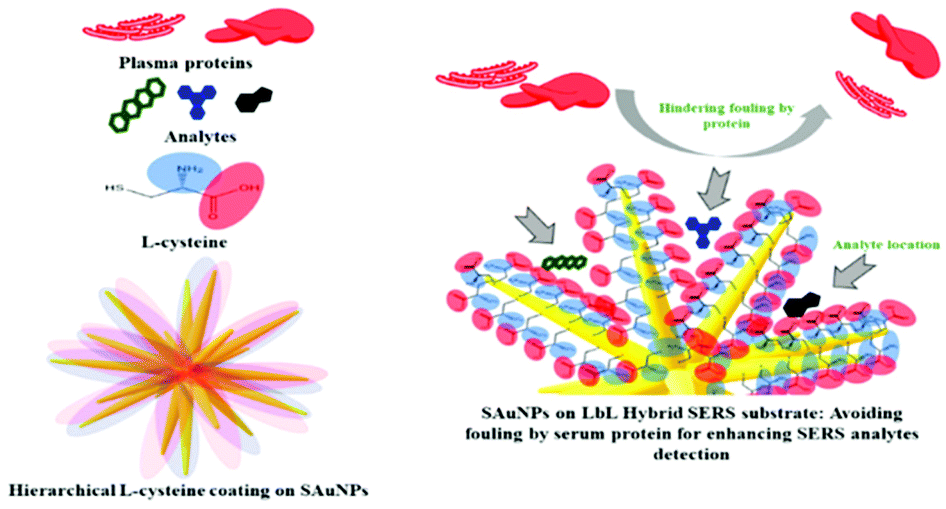 | ||
| Fig. 5 Schematic illustration of functionalization of AuNP SERS-substrate with a zwitterionic L-cysteine SAM, which acts as an anti-fouling brush.25 Reprinted with permission from ref. 25. Copyright 2019 American Chemical Society. | ||
Alternative methods for increasing sensitivity/selectivity of SERS measurements by adding other constituents to the enhancing material that might potentially be applied to anticancer drugs include substrates incorporating metal organic frameworks (MOFs), molecularly imprinted polymers (MIPs) and artificial peptides.62,63
Simultaneous SE(R)RS monitoring of multiple anticancer drugs in combination therapy
The studies discussed above suggest that SE(R)RS may well be an excellent method for anticancer drug monitoring. However, one significant omission is that the studies have not addressed the issue of making multiplex measurements. This is important because many anticancer drug regimens use at least two drugs simultaneously, since the combination may give synergistic effects and reduce the possibility of drug resistance developing. There are a small number of published studies which partly address the issue of multidrug detection, but full quantitative studies are still required. For example, Wang et al. have shown simultaneous SERS detection of two anticancer drugs in living cells.36In their study 6-mercaptopurine (7) and methimazole (12) were injected into a microfluidic chamber containing living cells which contained pre-incorporated Ag NPs. Both drugs gave their own distinctive SERS signal in HeLa and SKBR3 cells, respectively, as shown in Fig. 6. This demonstrated the possibility of achieving real-time label-free SERS detection of multiple drugs in vivo, which could be useful in studying the pharmacokinetics of drugs. However, no quantification of the drugs in this binary drug system was carried out.
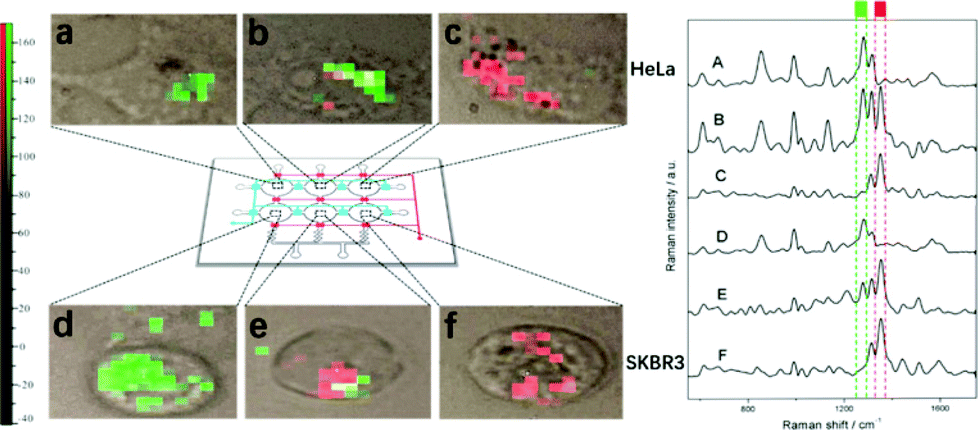 | ||
| Fig. 6 SERS image of 6 MP, MMI, and their mixture in HeLa and SKBR3 cells, respectively. The six images on the left (a, b, c, d, e, f) correspond to the mapping of a specific band of the six spectra on the right (A, B, C, D, E, F), respectively. The green blocks represent the band of 6 MP at 1289 cm−1 and red blocks represent the band of MMI at 1356 cm−1.36 Reprinted with permission from ref. 36. Copyright 2017 American Chemical Society. | ||
Cunningham et al. have reported SERS detection of a binary mixture of drugs, mitoxantrone (an anticancer drug (5)) and promethazine (an antihistamine) on Au.33 As shown in Fig. 7, both drugs gave detectable signals at four different total feedstock compositions. However, it is worth noticing that the difference in sensitivity for the two drugs meant that their strongest characteristic peaks (1037 cm−1 for promethazine and 1292 cm−1 for mitoxantrone) were only observed at similar intensity when the ratio between promethazine and mitoxantrone was ca. 3000![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. Even when the difference in Raman cross-section of the two analyte molecules are considered, this result obviously suggests that the amount of promethazine and mitoxantrone adsorbed on the surface of the enhancing substrate was very different from their ratio in the initial feedstock. This result is consistent with the results of competitive adsorption experiments of mixed thiols which have shown that differences in adsorption coefficients can lead to surface concentrations which are very different from the concentration of the analytes in the bulk solution.64 This is potentially an issue for multiplex SE(R)RS TDM since one drug within the therapeutic regimen may dominate the SE(R)RS signal to the extent that it may mask the SE(R)RS signals of the other components.
:1. Even when the difference in Raman cross-section of the two analyte molecules are considered, this result obviously suggests that the amount of promethazine and mitoxantrone adsorbed on the surface of the enhancing substrate was very different from their ratio in the initial feedstock. This result is consistent with the results of competitive adsorption experiments of mixed thiols which have shown that differences in adsorption coefficients can lead to surface concentrations which are very different from the concentration of the analytes in the bulk solution.64 This is potentially an issue for multiplex SE(R)RS TDM since one drug within the therapeutic regimen may dominate the SE(R)RS signal to the extent that it may mask the SE(R)RS signals of the other components.
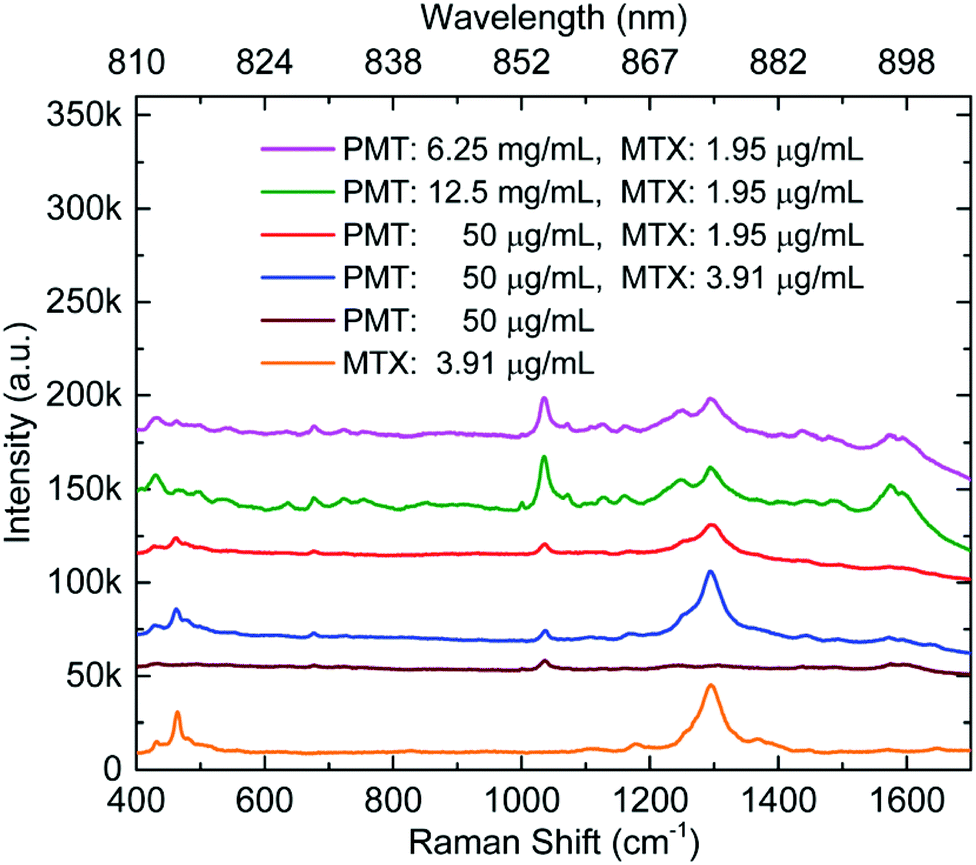 | ||
| Fig. 7 SERS spectra of promethazine and mitoxantrone mixtures, promethazine and mitoxantrone. Individual primary SERS intensity peaks for both promethazine and mitoxantrone compounds can be identified at 1037 and 1292 cm−1, respectively. Measurements were taken in sequence from the top to bottom.33 | ||
Other studies have investigated binary systems of drugs and their metabolites, for example, mixtures of the drug irinotecan (4) and its metabolite, SN-38 (9), which is the active compound. In experiments where mixed samples were dried onto a solid Au substrate it was found that the spectra were very similar although multivariate analysis could be used to separate them.32 In a second example, doxorubicin (14) and its metabolite, doxorubicinol were studied by SERS.43 When the molar ratio of the two components at fixed total concentration (2 μM) was altered, the resulting spectra showed a corresponding change in the relative intensities of bands characteristic of both species.
The studies above show that some of the issues which are associated with multiplex SE(R)RS in general are likely to be encountered in TDM of combinatorial anticancer chemotherapy. The most notable of these being the challenge in designing systems where the sensitivity of the analysis matches the concentration ranges of all the compounds which need to be monitored. This could mean that for mixtures where the therapeutic range of each drug is very different the sensitivity of the enhancing material to the higher concentration drugs would need to be suppressed. Conversely, in mixtures with similar therapeutic ranges, the challenge will be to ensure that the sensitivity to each of the drugs is approximately the same. It seems unlikely that these conditions will be met by simple unmodified surfaces, except by coincidence, so there is a clear need for approaches which allow the sensitivity of the assay to the various target compounds to be controlled, possibly by surface modification. This requirement does add an extra level of difficulty in designing SE(R)RS TDM methods for particular drug combinations and it may be the reason that up to this point there have been no quantitative studies of anticancer drug mixtures. However, the widespread use of combination therapies for different types of cancer mean that it is a challenge that will need to be met.
Conclusions
The SER(R)S studies of anticancer drugs published to date have demonstrated that limits of detection in the μM to nM range can be achieved for drugs with a very broad range of structures using a variety of enhancing substrates. As discussed above, this generality is associated with the structures of anticancer drugs which, despite their diversity, typically have at least one feature which helps to promote adsorption onto simple unmodified Au or Ag enhancing surfaces. Similarly, several methods for minimising the effects of interfering components within biofluid samples have been demonstrated, while more established protocols which are likely to be directly transferrable to SE(R)RS TDM of anticancer drugs are also available. These observations, coupled with the potential to carry out rapid bedside SE(R)RS testing using compact instruments, means that SE(R)RS does have real advantages compared to the techniques traditionally used for TDM, such as LC-MS.Apart from the normal issues that are associated with achieving reliable SE(R)RS quantification and are now well recognised,65 an additional challenge which is particularly relevant for SE(R)RS TDM of anticancer drugs is to develop SE(R)RS assays that can quantitatively monitor two or more drug compounds simultaneously. This is important because many therapeutic regimens in oncology involve the use of combinations of drugs. In principle, multiplex sample detection should be possible since each drug will give a characteristic finger print spectrum.23,66,67 However, successful multiplex detection will also require that the signals of each component can be detected at the required concentration range, which suggests that methods to control the sensitivity of each component will need to be developed. This will likely require combined research efforts in material fabrication and surface-modification to create novel enhancing substrates. In summary, there is now clear evidence that SE(R)RS has real advantages over established TDM approaches and that single drug analysis is already possible while multidrug monitoring remains an important challenge for the future.
Conflicts of interest
There are no conflicts to declare.Notes and references
- A. Remesh, Int. J. Basic Clin. Pharmacol., 2012, 1, 2 CrossRef.
- F. Kroschinsky, F. Stölzel, S. von Bonin, G. Beutel, M. Kochanek, M. Kiehl and P. Schellongowski, Crit. Care, 2017, 21, 89 CrossRef PubMed.
- E. Chatelut, Invest. New Drugs, 2003, 21, 141 CrossRef CAS PubMed.
- G. Powis, Drug Metab. Rev., 1983, 14, 1145 CrossRef CAS PubMed.
- D. Birkett, Aust. Prescr., 1997, 20, 9 CrossRef.
- A. Jaworska, S. Fornasaro, V. Sergo and A. Bonifacio, Biosensors, 2016, 6, 47 CrossRef PubMed.
- C. Neef, D. Touw and L. Stolk, Pharm. Med., 2008, 22, 235 CrossRef.
- J. Kang and M. Lee, Korean J. Intern. Med., 2009, 24, 1 CrossRef PubMed.
- E. Jaquenoud Sirot, J. W. van der Velden, K. Rentsch, C. Eap and P. Baumann, Drug Saf., 2006, 29, 735 CrossRef PubMed.
- N. Agrawal, J. Pallos, N. Slepko, B. Apostol, L. Bodai, L. Chang, A. Chiang, L. Thompson and J. Marsh, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 3777 CrossRef CAS PubMed.
- N. Ashizawa, Y. Tsuji, K. Kawago, Y. Higashi, M. Tashiro, M. Nogami, R. Gejo, M. Narukawa, T. Kimura and Y. Yamamoto, J. Infect. Chemother., 2016, 22, 331 CrossRef CAS PubMed.
- A. Vazquez, BMC Syst. Biol., 2013, 7, 31 CrossRef CAS PubMed.
- C. Stigliano, J. Key, M. Ramirez, S. Aryal and P. Decuzzi, Adv. Funct. Mater., 2015, 25, 3371 CrossRef CAS.
- J. Zhang, Y. Liang, X. Lin, X. Zhu, L. Yan, S. Li, X. Yang, G. Zhu, A. Rogach, P. Yu, P. Shi, L. Tu, C. Chang, X. Zhang, X. Chen, W. Zhang and C. Lee, ACS Nano, 2015, 9, 9741 CrossRef CAS PubMed.
- M. Broto, R. McCabe, R. Galve and M. Marco, Analyst, 2017, 142, 2404 RSC.
- G. Hempel, Methods of therapeutic drug monitoring including pharmacogenetics, 2nd edn, Elsevier, 2019 Search PubMed.
- M. Oellerich and A. Dasgupta, Personalized immunosuppression in transplantation, 1st edn, Elsevier, 2016 Search PubMed.
- F. Sime, M. Roberts, J. Roberts and T. Robertson, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2014, 960, 134 CrossRef CAS PubMed.
- J. Yi, K. Park, W. Kim, J. Rhee and W. Son, Arch. Pharmacal Res., 2018, 41, 355 CrossRef CAS PubMed.
- S. Dick, M. Konrad, W. Lee, H. McCabe, J. McCracken, T. Rahman, A. Stewart, Y. Xu and S. Bell, Adv. Mater., 2016, 28, 5705 CrossRef CAS PubMed.
- P. Stiles, J. Dieringer, N. Shah and R. Van Duyne, Annu. Rev. Anal. Chem., 2008, 1, 601 CrossRef CAS PubMed.
- T. Cotton, S. Schultz and R. Van Duyne, J. Am. Chem. Soc., 1982, 104, 6528 CrossRef CAS.
- J. Dougan and K. Faulds, Analyst, 2012, 137, 545 RSC.
- X. Qian and S. Nie, Chem. Soc. Rev., 2008, 37, 912 RSC.
- S. Panikar, G. Ramírez-García, S. Sidhik, T. Lopez-Luke, C. Rodriguez-Gonzalez, I. Ciapara, P. Castillo, T. Camacho-Villegas and E. De la Rosa, Anal. Chem., 2018, 91, 2100 CrossRef PubMed.
- C. Yuen, W. Zheng and Z. Huang, Biosens. Bioelectron., 2010, 26, 580 CrossRef CAS PubMed.
- A. Subaihi, D. Trivedi, K. Hollywood, J. Bluett, Y. Xu, H. Muhamadali, D. Ellis and R. Goodacre, Anal. Chem., 2017, 89, 6702 CrossRef CAS PubMed.
- S. Fornasaro, S. Marta, M. Rabusin, A. Bonifacio and V. Sergo, Faraday Discuss., 2016, 187, 485 RSC.
- I. Hidi, A. Mühlig, M. Jahn, F. Liebold, D. Cialla, K. Weber and J. Popp, Anal. Methods, 2014, 6, 3943 RSC.
- Y. Xing, H. Fuss, J. Lademann, M. Huang, H. Becker-Ross, S. Florek, A. Patzelt, M. Meinke, S. Jung and N. Esser, Spectrochim. Acta, Part B, 2018, 142, 91 CrossRef CAS.
- A. Vicario, V. Sergo, G. Toffoli and A. Bonifacio, Colloids Surf., B, 2015, 127, 41 CrossRef CAS PubMed.
- L. Litti, V. Amendola, G. Toffoli and M. Meneghetti, Anal. Bioanal. Chem., 2016, 408, 2123 CrossRef CAS PubMed.
- H. Wu and B. Cunningham, Nanoscale, 2014, 6, 5162 RSC.
- K. Ackermann, T. Henkel and J. Popp, ChemPhysChem, 2007, 8, 2665 CrossRef CAS PubMed.
- C. McLaughlin, D. MacMillan, C. McCardle and W. Smith, Anal. Chem., 2002, 74, 3160 CrossRef CAS PubMed.
- J. Fei, L. Wu, Y. Zhang, S. Zong, Z. Wang and Y. Cui, ACS Sens., 2017, 2, 773 CrossRef CAS PubMed.
- L. Yang, Y. Chen, H. Li, L. Luo, Y. Zhao, H. Zhang and Y. Tian, Anal. Methods, 2015, 7, 6520 RSC.
- S. Fornasaro, A. Bonifacio, E. Marangon, M. Buzzo, G. Toffoli, T. Rindzevicius, M. Schmidt and V. Sergo, Anal. Chem., 2018, 90, 12670 CrossRef CAS PubMed.
- J. Xu, T. Jiao, S. Arumugam, P. He, J. Zhu, H. Li and Q. Chen, Microchem. J., 2020, 155, 104373 CrossRef CAS.
- W. Zhang, Y. Wang, Y. Wang and Z. Xu, Sens. Actuators, B, 2019, 283, 532 CrossRef CAS.
- C. Eliasson, A. Lorén, K. Murty, M. Josefson, M. Käll, J. Abrahamsson and K. Abrahamsson, Spectrochim. Acta, Part A, 2001, 57, 1907 CrossRef CAS.
- A. Lorén, C. Eliasson, M. Josefson, K. Murty, M. Käll, J. Abrahamsson and K. Abrahamsson, J. Raman Spectrosc., 2001, 32, 971 CrossRef.
- F. Sun, H. Hung, A. Sinclair, P. Zhang, T. Bai, D. Galvan, P. Jain, B. Li, S. Jiang and Q. Yu, Nat. Commun., 2016, 7, 13437 CrossRef CAS PubMed.
- L. Litti, A. Ramundo, F. Biscaglia, G. Toffoli, M. Gobbo and M. Meneghetti, J. Colloid Interface Sci., 2019, 533, 621 CrossRef CAS PubMed.
- Z. Ye, C. Li, Q. Chen, Y. Xu and S. Bell, Angew. Chem., 2019, 131, 19230 CrossRef.
- L. Scarabelli, M. Coronado-Puchau, J. Giner-Casares, J. Langer and L. Liz-Marzán, ACS Nano, 2014, 8, 5833 CrossRef CAS PubMed.
- C. Tesseromatis and A. Alevizou, Eur. J. Drug Metab. Pharmacokinet., 2008, 33, 225 CrossRef CAS PubMed.
- S. Dominguez-Medina, J. Blankenburg, J. Olson, C. Landes and S. Link, ACS Sustainable Chem. Eng., 2013, 1, 833 CrossRef CAS PubMed.
- A. Berger, S. Restaino and I. White, Anal. Chim. Acta, 2017, 949, 59 CrossRef CAS PubMed.
- N. Markina, I. Goryacheva and A. Markin, Anal. Bioanal. Chem., 2018, 410, 2221 CrossRef CAS PubMed.
- S. Ding, E. You, Z. Tian and M. Moskovits, Chem. Soc. Rev., 2017, 46, 4042 RSC.
- M. Fleischmann, P. Hendra and A. McQuillan, Chem. Phys. Lett., 1974, 26, 163 CrossRef CAS.
- A. Kaczor, K. Malek and M. Baranska, J. Phys. Chem. C, 2010, 114, 3909 CrossRef CAS.
- A. Tadjeddine, A. Rille, O. Pluchery, P. Hébert, W. Zheng and T. Marin, Nucl. Instrum. Methods Phys. Res., Sect. A, 1999, 429, 481 CrossRef CAS.
- C. Vericat, M. Vela, G. Benitez, P. Carro and R. Salvarezza, Chem. Soc. Rev., 2010, 39, 1805 RSC.
- W. Pan, C. Durning and N. Turro, Langmuir, 1996, 12, 4469 CrossRef CAS.
- F. Mao, W. Ni, X. Xu, H. Wang, J. Wang, M. Ji and J. Li, Molecules, 2016, 21, 75 CrossRef PubMed.
- R. Hoft, M. Ford, A. McDonagh and M. Cortie, J. Phys. Chem. C, 2007, 111, 13886 CrossRef CAS.
- H. Singh, R. Kumar, S. Singh, K. Chaudhary, A. Gautam and G. Raghava, BMC Cancer, 2016, 16, 77 CrossRef PubMed.
- G. Niaura, A. Gaigalas and V. Vilker, J. Phys. Chem. B, 1997, 101, 9250 CrossRef CAS.
- Z. Ye, C. Li, Y. Xu and S. Bell, Analyst, 2019, 144, 448 RSC.
- G. Phan-Quang, N. Yang, H. Lee, H. Sim, C. Koh, Y. Kao, Z. Wong, E. Tan, Y. Miao, W. Fan, T. Liu, I. Phang and X. Ling, ACS Nano, 2019, 13, 12090 CrossRef CAS PubMed.
- S. Chen and Z. Zhang, Spectrochim. Acta, Part A, 2008, 70, 36 CrossRef PubMed.
- A. Stewart, S. Zheng, M. McCourt and S. Bell, ACS Nano, 2012, 6, 3718 CrossRef CAS PubMed.
- S. Bell, G. Charronb, E. Cortésc, J. Kneipp, M. Chapelle, J. Langerf, M. Procházkag, V. Tranh and S. Schlücker, Angew. Chem., 2020, 59, 5454 CrossRef CAS PubMed.
- S. Laing, K. Gracie and K. Faulds, Chem. Soc. Rev., 2015, 45, 1901 RSC.
- H. Kearns, R. Goodacre, L. Jamieson, D. Graham and K. Faulds, Anal. Chem., 2017, 89, 12666 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2020 |