
Evaluation of the electroanalytical performance of carbon-on-gold films prepared by electron-beam evaporation†

Abstract
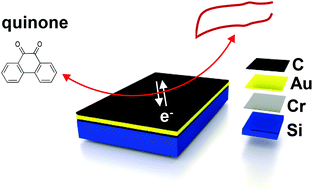
Carbon film electrodes can often be used without pretreatment, and their fabrication allows for flexibility in size and shape and for mass production. In this work, we are exploring layered structures comprised of thin films of carbon on gold (eC/Au) prepared by electron-beam evaporation. These extremely flat films are not pyrolyzed and are comprised of mainly amorphous carbon but still exhibit reasonable conductivity due to the underlying gold layer. eC/Au electrodes, without any pretreatment, yield similar heterogeneous electron-transfer rates for benchmark redox systems and significantly lower background current in comparison with polished glassy carbon. Interestingly, they show insignificant adsorption of quinones, which is uncommon for most carbon electrode materials. However, eC/Au is still prone to adsorption of airborne hydrocarbons when exposed to ambient air like most graphitic materials. With reproducibly fast electron transfer kinetics, low background current, negligible adsorption, and ultraflat surface, eC/Au films are a promising candidate for electrochemical and sensor applications.
- This article is part of the themed collection: Versatile Electrochemical Approaches
 Please wait while we load your content...
Please wait while we load your content...

