
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Acellular oxidative potential assay for screening of amorphous silica nanoparticles†
Dalibor
Breznan‡
 a,
Nazila
Nazemof‡
b,
Filip
Kunc
c,
Myriam
Hill
d,
Djordje
Vladisavljevic
d,
James
Gomes
b,
Linda J.
Johnston
c,
Renaud
Vincent
ae and
Prem
Kumarathasan
*ab
a,
Nazila
Nazemof‡
b,
Filip
Kunc
c,
Myriam
Hill
d,
Djordje
Vladisavljevic
d,
James
Gomes
b,
Linda J.
Johnston
c,
Renaud
Vincent
ae and
Prem
Kumarathasan
*ab
aEnvironmental Health Science and Research Bureau, Health Canada, Ottawa, ON K1A 0K9, Canada. E-mail: premkumari.kumarathasan@canada.ca; Fax: +613-946-2600; Tel: +613-218-4530
bInterdisciplinary School of Health Sciences, University of Ottawa, Ottawa, ON K1N 7K4, Canada
cMetrology Research Centre, National Research Council Canada, Ottawa, Ontario K1A 0R6, Canada
dNew Substances Assessment and Control Bureau, Health Canada, Ottawa, ON K1A 0K9, Canada
eDepartment of Biochemistry, Microbiology and Immunology, University of Ottawa, Ottawa, ON K1H 8M5, Canada
First published on 28th May 2020
Abstract
Silica nanoparticles (SiNPs) are used in a wide range of consumer products, engineering and medical applications, with likelihood of human exposure and potential health concerns. It is essential to generate toxicity information on SiNP forms and associated physicochemical determinants to conduct risk assessment on these new materials. To address this knowledge gap, we screened a panel of custom synthesized, well-characterized amorphous SiNPs pristine and surface-modified (–C3-COOH, –C11-COOH, –NH2, –PEG) of 5 different sizes: (15, 30, 50, 75, 100 nm) for their oxidative potential using an acellular assay. The assay is based on oxidation of dithiothreitol (DTT) by reactive oxygen species and can serve as a surrogate test for oxidative stress. These materials were characterized for size distribution, aggregation, crystallinity, surface area, surface modification, surface charge and metal content. Tests for association between oxidative potential of SiNPs and their physicochemical properties were carried out using analysis of variance and correlation analyses. These test results suggest that the size of amorphous SiNPs influenced their oxidative potential irrespective of the surface modification, with 15 nm exhibiting relatively higher oxidative potential compared to the other sizes. Furthermore, SiNP surface area, surface modification and agglomeration in solution also appeared to affect oxidative potential of these SiNPs. These findings indicate that physicochemical properties are critical in influencing the oxidative behaviour of amorphous SiNPs, with potential to trigger cellular oxidative stress and thus toxicity, when exposed. This information advances our understanding of potential toxicities of these amorphous SiNPs and supports risk assessment efforts and the design of safer forms of silica nanomaterials.
Introduction
Engineered nanoparticles (NPs) provide challenges for hazard identification and risk evaluation due to a lack of reliable physicochemical and toxicity data, creating difficulties for government agencies to establish effective safety evaluation guidelines. Among the various engineered nanomaterials, amorphous silica nanoparticles (SiNPs) have received relatively more attention and have been frequently used as additives to cosmetics, drugs, printer toners, varnishes and food.1–4 Due to their biocompatibility, easy surface functionalization and resistance to biodegradation in the cellular environment, silica nanoparticles are currently being synthesized on a large scale for biomedical and biotechnology applications such as cancer therapy, in gene carriers, drug delivery and enzyme immobilization.5–10The growing use of nanomaterials including SiNPs raises concerns in terms of their potential harmful effects to human health and the environment and places them at a high priority for toxicity screening and risk assessment by regulatory authorities.11,12 However, toxicological studies on SiNPs are far behind the pace of their production and applications. There are emerging reports on the analyses of the toxicological properties of SiNPs.12–16 The physicochemical properties of SiNPs highly depend on the synthetic method for their production and can influence the toxicity of these materials.1,17 Unique physicochemical properties of engineered nanomaterials such as particle size (surface area and size distribution), aggregation/agglomeration, chemical composition (purity, crystallinity, electronic properties) and surface structure (surface reactivity, surface groups, inorganic or organic coating) can determine the fate of NP–cell interactions.
Formation of reactive oxygen species (ROS) has been suggested as one of the mechanisms by which SiNPs could exert toxicity.18–20 The excessive production of ROS-mediated oxidative stress triggered by exposures to environmental pollutants such as complex mixtures of air pollution particles containing ultrafine and nano-scale components can be associated with disruption in the normal mechanism of cellular signaling leading to various disease processes including cardiovascular disease, COPD, diabetes, cancer, Alzheimer's disease.21–23 The assessment of oxidative potential as a surrogate for ROS generation has been proposed as a useful metric to measure the capacity of particulate matter (PM) to oxidise target molecules in biological systems.24 It is an attractive measure because it integrates various biologically relevant properties, including size, surface and chemical composition.25 Both cellular26–28 and acellular29–32 methods have been developed to measure the oxidative potential of NPs.
Methodological approaches for redox activity are based either on direct measurement of ROS in cellular environment using high performance liquid chromatography (HPLC), electron spin resonance spectroscopy (ESR) and fluorescence-based methods33 or indirect measurement using particle-induced depletion of the reductants (e.g. DTT) or the antioxidants (e.g. vitamin C, glutathione and uric acid)34,35 in acellular tests. Acellular methods require a less controlled environment, are less costly and can be relatively rapid for high-throughput identification of NP hazard. Furthermore, in vitro acellular screening for oxidative potential of nanomaterials can be of value in terms of predicting relative reactivities and thus potencies of these particles when exposed to cells.
Among all available acellular methods, the dithiothreitol (DTT) assay is one of the commonly used assays to study oxidative potential associated with micron-sized environmental particles, such as ambient air particulate matter with complex matrices.30 This assay can serve as an initial screening step to identify the oxidative potential of particles prior to conducting more extensive cell or animal exposure studies. The DTT assay is based on the presence of redox-active chemical species in particles to oxidize DTT to its disulfide form. In general, the redox active species, such as transition metals, conjugated unsaturated organic species in complex air pollutant matrices can donate electron to dissolved molecular oxygen, forming superoxide, a ROS, which in turn can form other ROS, such as hydrogen peroxide which in the presence of metals, such as iron can lead to the formation of hydroxyl radical (˙OH). One of the limitations noted for this assay was its inability to discriminate between ROS, for instance, its inability to measure specifically ˙OH generation.33 The DTT assay is a fast and inexpensive assay and therefore is a candidate assay for high-throughput screening for oxidative potential.36,37 Nevertheless, the engineered nanomaterials, such as pure amorphous silica nanoparticles do not contain redox active metals or organic species to participate in redox-cycling reactions. Therefore it can be challenging to apply DTT assay to measure oxidative potential of such particles and thus the sensitivity of the assay can be questionable. Yet, the simplicity, cost and speed of the DTT assay are attractive as a first pass screening tool for oxidative potential measurement of nanomaterials compared to in vitro or in vivo exposure studies and thus warrants feasibility testing with these materials.
The objective of this study was to test the applicability of the cell-free assay based on DTT oxidation to screen various sizes (15, 30, 50, 75, 100 nm) of pristine and surface-modified SiNPs (–C3-COOH, –C11-COOH, –NH2, –PEG) for their oxidative potential, and to identify the physicochemical properties of SiNPs that can influence their oxidative potential by testing for associations between them. The analytical approach is presented in Fig. 1. To the best of our knowledge, this study is the first report for a comprehensive screening of nanoforms of amorphous SiNPs using the acellular DTT assay.
 | ||
| Fig. 1 Schematic drawing of the general analytical approach. | ||
Experimental
Materials
Dithiothreitol (DTT), 5,5′-dithiobis-(2-nitrobenzoic acid) (DTNB), 1 M phosphate buffer, di-tert-butyl peroxide (DTBP), tert-butyl hydroperoxide (TBHP), Fe(II) sulphate hydrate and ultrapure water were obtained from Sigma Aldrich (Oakville, ON, Canada). Dulbecco's Modified Eagle's Medium (phenol red-free) and with phenol red, fetal bovine serum (FBS) and phosphate buffered-saline (10×) were purchased from Fisher Scientific (Nepean, ON, Canada). Custom synthesized well-characterized pristine amorphous silica nanoparticles (SiNPs) of different sizes (15, 30, 50, 75, 100 nm) and the corresponding surface modified –C3-COOH, –C11-COOH, –(OH)2Si(OCH2CH2)nOCH3 (n = 9–12) (–PEG) and –(OH)2Si(CH2)2CH2NH2 (–NH2) variants were purchased from Advanced Quantum Materials Inc. (AQM, Edmonton, AB, Canada).For comparative purposes, reference particles were included in the experiments. Micron-sized standard reference materials (SRM-1879 silicon dioxide, SiO2 and SRM-154b titanium dioxide, TiO2) were obtained from the National Institute of Standards and Technology (Gaithersburg, MD, USA). Ottawa ambient air EHC-6802 particles were included to represent an urban particulate matter standard. The preparation and characterization of EHC-6802 particles has been previously described.38 A reference nanoparticle, 12 nm-sized amorphous silica was also included in the experiment (cat. # 718483; Sigma Aldrich, Oakville, ON, Canada).
Characterization of physicochemical properties
The pristine SiNPs were synthesized using modified-Stöber sol–gel based process that included a calcination step. The same batch of a specific size of the pristine SiNPs was used to prepare the corresponding surface modified counterparts. The physicochemical properties of the particles were analyzed by transmission electron microscopy (TEM) for size distribution, X-ray diffraction (XRD) to assess the crystallinity, Fourier transform infrared spectroscopy (FT-IR) for functional groups and thermogravimetric analysis (TGA) for functional group loss, by AQM. In addition, Brunauer–Emmett–Teller (BET) analyses for surface area, quantitative nuclear magnetic resonance (qNMR) analyses for surface modifications, dynamic light scattering (DLS) and zeta potential for agglomeration size and surface charge in aqueous media and inductively coupled plasma mass spectrometry (ICP-MS/AES) for elemental analysis were conducted as part of this study.Electron microscopy
Bright field transmission electron microscopy (TEM) images were taken with a JEOL 2010 TEM (with LaB6 electron gun) using accelerating voltage of 200 kV. TEM samples were prepared by depositing a drop of ethanol or toluene solution (ca. 0.1 mg mL−1) of the pristine or functionalized SiO2 nanoparticle suspensions onto carbon coated copper grid (obtained from Electron Microscopy Inc.). The nanoparticle sizes were determined upon averaging the dimensions of at least 50–100 particles chosen manually using ImageJ software (version 1.45).X-ray powder diffraction (XRD)
XRD data were obtained using an Inel MPD Multi Purpose Diffractometer equipped with a CPS 120 curved position sensitive X-ray detector and copper Kα (8.047 KeV energy) radiation source. The samples were made by depositing the SiNPs on a Si (111) wafer.Fourier transform infrared (FT-IR) spectroscopy
FT-IR spectroscopic analyses were performed using a Thermo Nicolet Magna 750 IR Spectrometer. Samples for FT-IR analysis were prepared by drop coating a methanol solution of the pristine and surface-functionalized SiNPs onto an electronic-grade Si-wafer (N-type, 100 surface, 100 mm thickness and 10 ohm cm resistivity).Thermogravimetric analysis (TGA)
TGA was performed using a Mettler Toledo Star TGA/DSC system. Pristine and surface-functionalized SiNP samples were placed in a Pt pan and heated under Ar atmosphere from 35 to 700 °C at a rate of 10 °C min−1 or 25 °C min−1 as indicated.Quantitative nuclear magnetic resonance (qNMR)
Functional group contents of SiNPs were quantified by dissolution of the NPs in basic solution, followed by solution NMR using a Bruker Avance 400 Mz spectrometer, according to a published protocol.39 SiNPs (4–10 mg) were treated with sodium deuteroxide solution in D2O (0.65 mL, 0.4 M) and dispersed by sonication in an ultrasonic bath for 10 min. Samples were then placed in a heated mixer and shaken at 45 °C for 16 hours, after which they were brought to room temperature; maleic acid calibrant solution in D2O (20 μL, 98.62 mM) was added and the sample was vortexed. These samples were transferred into a 5 mm NMR tube and the NMR measurement was conducted within 24 hours. Triplicate analyses were performed. Due to the high-throughput requirements, NMR analyses were carried out using a zg30 pulse program instead of zg90 as typically used for quantitative NMR experiments. This step introduces a difference of ≤5% in the quantification, but shortens the experiment duration by a factor of 4. Spectral width was 20 ppm with 6.175 ppm transmitter frequency offset. 32 scans with 2 dummy scans were recorded at 6 s relaxation delay. Maximal receiver gain was set prior to each measurement. All acquired spectra were phase corrected manually and baseline correction was done by 5th order polynomial fit. Baseline-resolved diagnostic signals of the functional moiety were identified, integrated, and the average integral was compared with the integral of the calibrant, maleic acid.ICP-MS/AES
Metal content of pristine SiNP samples was analyzed using inductively coupled plasma-mass spectrometry/atomic emission spectroscopy (ICP-MS/AES, Varian Vista-Pro, Mulgrave, Australia) after acid-digestion following the previously reported procedure.40 In brief, the various SiNPs were digested in 50% HNO3 for 8 h at 80 °C and after filtration via a 0.22 mm filter and the filtrates were analyzed by ICP-MS/AES.41 A reagent blank was analyzed by ICP-MS/AES and the blank values were subtracted from the sample analysis results to obtain the actual metal concentrations in these samples. All analyses were done in duplicate.Brunauer–Emmett–Teller analysis (BET)
The BET method with nitrogen absorption was utilized to obtain the specific surface areas for the SiNPs. ASAP 2020 instrument (Micrometrics, Norcross, GA, USA) was used for the gas-adsorption based SiNP surface area analyses. Prior to degassing, the SiNP samples contained in the sample tube were weighed. The samples were degassed under vacuum to a pressure of 10 mm per Hg at 80 °C, followed by a second degassing step at a pressure of 300 μm per Hg for 90 minutes. The temperature was increased at 100 °C min−1 to 120 °C at which time the degassing was continued for 12 hours. The SiNP samples were cooled to 20 °C and complete degassing was verified. The specific surface area was determined by the multipoint BET method.Particle preparations
All the amorphous SiNP variants and reference particles (EHC-6802 urban dust particles and micron-sized SiO2 and TiO2) were re-suspended in ultrapure water (Sigma-Aldrich, Oakville, ON, Canada) to a concentration of 500 μg mL−1. The suspensions were vortexed for 30 s and sonicated (6 cycles of 30 s on/off with 50–60% amplitude) on ice by using a probe sonicator (120 W, 20 KHz; Model CL-18, Fisher Scientific, Ottawa, ON, Canada). Sonicated samples were vortexed for 30 s and agitated by a rocking shaker. The suspended particles were employed to conduct the oxidative potential analyses.Dynamic light scattering (DLS) and zeta potential
The particle stocks were vortexed for 30 s and sonicated in a water bath sonicator (100 W, 20 kHz; Branson Ultrasonics, Danbury, CT, USA) for 20 min prior to sample preparation as follows; A 10 μl aliquot of aqueous suspensions of pristine or surface-modified SiNP variants (500 μg mL−1) was combined with 100 μL of 0.5 M potassium phosphate buffer (pH 7.4) and a final volume of 500 μL was made up with ultrapure water to obtain 10 μg mL−1 SiNP concentration for hydrodynamic diameter measurement by DLS. The samples were mixed using Eppendorf Thermomixer R dry-block shaker (Eppendorf, Mississauga, ON) for 10 min at 37 °C. Subsequently, the samples were vortexed for 10 s and transferred into cuvettes for measurements taken immediately and after 20 min incubation at room temperature (RT). Identical sample preparation was used for the zeta potential analyses. The DLS (z-average and polydispersity index) and zeta potential values were obtained using the Zetasizer Nano ZS (Malvern Instruments, UK) as reported earlier.42 All measurements were conducted in triplicate.Oxidative potential analyses
The procedure reported by Janssen et al. was followed to perform the dithiothreitol (DTT) assay after optimization with minor modifications.25 Briefly, 10 μl of pristine or surface-modified SiNP suspension in ultrapure water at 500 μg mL−1 was incubated in the dark room with gentle agitation in a thermomixer at 550 rpm (Eppendorf Thermomixer R) with 100 μl of 0.5 M potassium phosphate buffer (pH 7.4) and 340 μl of ultrapure water for 10 min at 37 °C. Then, 50 μl of 1 mM DTT was added to the incubation vial and vortexed for 10 s. A 100 μl aliquot of the DTT reaction mixture was withdrawn and transferred to a vial containing 10 μl of 1.5 mM 5,5′-dithiobis(2-nitrobenzoic acid) (DTNB) to quench the oxidation reaction at different time periods (3, 10, 20, 60 min). The reaction between the remainder of the reduced DTT and DTNB is fast and forms 2-nitro-5-thiobenzoic acid (TNB), which is stable in the final solution for at least 2 h at RT. This mixture was centrifuged at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000g for 40 s and the supernatant was transferred to 96-well plates. The absorbance was measured at 412 nm with a POLARstar Omega plate reader spectrophotometer (BMG Lab tech, Ortenberg, Germany).
000g for 40 s and the supernatant was transferred to 96-well plates. The absorbance was measured at 412 nm with a POLARstar Omega plate reader spectrophotometer (BMG Lab tech, Ortenberg, Germany).
Since both DTT and TNB are sensitive to light, care was taken to exclude light as much as possible by working in a dark room and sealing the 96-well plate with aluminium foil during the experiment. Initially, the reaction conditions were optimized using the reference particles, as well as pristine and COOH-modified SiNPs. The time period of 20 min was chosen as the optimal period for the DTT oxidation by these SiNPs and was used to screen the oxidative potential of all SiNP variants. A subset of SiNP variants (15 and 75 nm pristine and surface-modified SiNPs) were then subjected to free radical initiators DTBP or TBHP prior to conducting the DTT assay to assess the extent of oxidative potential amplification, if possible. Here, 100 μM TBHP or DTBP was initially reacted at RT with 20 μM Fe(II) sulfate hydrate and phosphate buffer (0.5 M; pH 7.4) and incubated for 15 min at 37 °C with continuous agitation using a thermomixer (550 rpm). Ten μL of the SiNP suspension (500 μg mL−1) in water was added to the incubated mixture and continuously shaken. After 10 min, DTT assay was carried out as described earlier. All analyses were conducted in duplicate.
Statistical analyses
The presented data for the DTT assay were normalized to the corresponding blanks and to the sum of the plate responses per each experiment. The experimental data were assessed for statistical significance by Kruskal–Wallis method or a two-way analysis of variance (ANOVA), as appropriate, using SigmaPlot v12.5 (Systat Software, San Jose, CA, USA). Where the assumptions of normality and equal variance were not met, the data were rank-transformed prior to conducting the ANOVA. For the Kruskal–Wallis method, Dunn's test was applied for the pairwise multiple comparisons (α = 0.05). For the two-way ANOVA, the multiple comparisons analysis was conducted using the Holm–Sidak post hoc test (α = 0.05). Nanoparticle size (size) and surface modification (mod) were applied as factors in the two-way ANOVA. Pearson product moment correlation was used to test for associations between oxidative potential values for the different SiNP forms and their physicochemical properties.Results and discussion
Physicochemical characterization
In this study, a set of custom synthesized amorphous SiNPs including pristine and surface-modified particles of various sizes were assessed for their in vitro acellular oxidative properties. Since the assessment of physicochemical properties of SiNPs is critical to understand nanomaterial reactivity and their relative potencies in biological systems, the SiNP variants were characterized for key physicochemical parameters. These measurements included TEM of pristine and surface-modified SiNP variants for size analyses of dry, primary particles and the BET method to determine the surface area of the SiNPs. Agglomeration behaviour of the SiNPs in solution-state was assessed using DLS method, while the electrophoretic mobility of the SiNPs in aqueous suspension was determined by zeta potential analysis.The TEM analysis results for the pristine and surface-modified SiNPs (Fig. 2 and S1†) showed that among the pristine SiNPs, in dry form, the 15 nm-sized particles showed a higher tendency to agglomerate than the SiNPs of other sizes. Also, as the size of pristine SiNPs increased, the particles were spherical with uniform particle size distribution. Similar behaviour was observed with surface-coated SiNP particles with 15 nm size being agglomerated across all surface modifications compared to the other sizes. In contrast, the 100 nm surface-coated SiNPs were spherical and well separated in dry-state. Furthermore, BET measurements for the SiNPs are summarized in Table 1. The BET-derived surface area data was compared to surface area estimates determined from TEM size. A correlation analysis between the BET and TEM surface areas revealed a lack of significant association (r = 0.232, p = 0.263).
 | ||
| Fig. 2 TEM images for pristine SiNPs of different sizes. (a) 15 nm, (b) 30 nm, (c) 50 nm, (d) 75 nm and (e) 100 nm pristine SiNPs. | ||
| SiNP ID | TEM diameter (nm) | DLS diameter (nm) | PDI (DLS) | TEM SA (m2 g−1) | BET SSA (m2 g−1) | Agglom (DLS/TEM) ratio | Agglom SA ratio | Zeta potential (mV) | Total metals (ppm) | Transition metals (ppm) |
|---|---|---|---|---|---|---|---|---|---|---|
| TEM mean diameter was obtained using transmission electron microscopy; DLS hydrodynamic diameter (Z-average) and polydispersity index (PDI) and zeta potential measurements were determined for SiNPs in 0.5 M K3PO4 buffer (pH 7.4); TEM surface area (SA) was derived from the mean TEM diameter; BET specific surface area was determined using the Brunauer–Emmett–Teller method; DLS/TEM ratio is a surrogate estimate of the agglomeration state of the SiNPs in the buffer; agglomeration SA ratio (TEM SA × (DLS/TEM ratio)) is an estimate for surface area of the SiNP agglomerates in buffer available for their potential activity; total metal content of the SiNPs was measured using the ICP-MS/AES analysis; transition metals represent the sum of transition elements in the SiNPs; dash line (−) indicates data not available. TEM diameter and TEM-derived surface area for C3- and C11-COOH-modified SiNPs indicated by a star (*) were not separately determined and were assumed to be equivalent to their pristine counterparts from which the modified materials were formed. | ||||||||||
| 15-P | 17.2 ± 3.5 | 657.4 ± 172.4 | 0.545 | 156.6 | 29.6 | 38.2 | 5982.1 | −15.9 ± 1.2 | 848.24 ± 48.17 | 785.15 |
| 30-P | 28.8 ± 3.5 | 1128.0 ± 423.8 | 0.720 | 94.5 | 97.2 | 39.2 | 3704.4 | −13.4 ± 2.8 | 615.55 ± 8.44 | 541.30 |
| 50-P | 47.9 ± 3.2 | 835.2 ± 108.9 | 0.684 | 56.9 | 75.2 | 17.4 | 990.1 | −12.0 ± 2.1 | 902.74 ± 19.11 | 828.03 |
| 75-P | 78.2 ± 5.5 | 260.1 ± 30.2 | 0.292 | 34.5 | 17.0 | 3.3 | 113.9 | −16.6 ± 2.3 | 476.36 ± 4.55 | 419.10 |
| 100-P | 101.1 ± 5.2 | 274.6 ± 38.1 | 0.329 | 26.9 | 33.0 | 2.7 | 72.6 | −18.6 ± 0.8 | 401.62 ± 10.96 | 359.94 |
| 15-C3 | 17.2 ± 3.5* | 530.6 ± 49.4 | 0.507 | 156.6* | 162.0 | 30.9 | 4838.9 | −17.8 ± 0.7 | — | — |
| 30-C3 | 28.8 ± 3.5* | 499.2 ± 73.9 | 0.477 | 94.5* | 117.0 | 17.3 | 1634.9 | −24.7 ± 1.3 | — | — |
| 50-C3 | 47.9 ± 3.2* | 473.2 ± 69.2 | 0.497 | 56.9* | 67.2 | 9.9 | 563.3 | −27.8 ± 1.7 | — | — |
| 75-C3 | 78.2 ± 5.5* | 215.1 ± 15.5 | 0.382 | 34.5* | 39.0 | 2.8 | 96.6 | −32.2 ± 2.3 | — | — |
| 100-C3 | 101.1 ± 5.2* | 144.7 ± 3.2 | 0.198 | 26.9* | 37.8 | 1.4 | 37.7 | −29.9 ± 2.5 | — | — |
| 15-C11 | 17.2 ± 3.5* | 445.3 ± 82.0 | 0.444 | 156.6* | 20.1 | 25.9 | 4055.9 | −21.4 ± 4.5 | — | — |
| 30-C11 | 28.8 ± 3.5* | 593.7 ± 240.0 | 0.629 | 94.5* | 78.9 | 20.6 | 1946.7 | −34.8 ± 2.8 | — | — |
| 50-C11 | 47.9 ± 3.2* | 875.9 ± 195.7 | 0.791 | 56.9* | 51.5 | 18.3 | 1041.3 | −21.7 ± 1.6 | — | — |
| 75-C11 | 78.2 ± 5.5* | 188.0 ± 5.9 | 0.359 | 34.5* | 30.6 | 2.4 | 82.8 | −35.8 ± 2.4 | — | — |
| 100-C11 | 101.1 ± 5.2* | 161.2 ± 2.9 | 0.232 | 26.9* | 34.0 | 1.6 | 43.0 | −32.0 ± 2.9 | — | — |
| 15-NH2 | 13.7 ± 1.4 | 822.4 ± 177.5 | 0.723 | 164.3 | 9.2 | 60.0 | 9858.0 | −6.7 ± 1.7 | — | — |
| 30-NH2 | 30.4 ± 2.7 | 786.1 ± 288.2 | 0.618 | 74.6 | 11.0 | 25.9 | 1932.1 | −14.9 ± 2.6 | — | — |
| 50-NH2 | 51.3 ± 3.7 | 437.3 ± 62.2 | 0.487 | 43.7 | 19.7 | 8.5 | 371.5 | −8.5 ± 3.3 | — | — |
| 75-NH2 | 78.0 ± 5.4 | 1158.1 ± 146.7 | 0.768 | 28.8 | 38.2 | 14.9 | 429.1 | −15.9 ± 0.7 | — | — |
| 100-NH2 | 101.1 ± 6.0 | 396.7 ± 50.2 | 0.471 | 22.4 | 35.6 | 3.9 | 87.4 | −17.5 ± 3.9 | — | — |
| 15-PEG | 13.7 ± 1.4 | 528.3 ± 54.2 | 0.471 | 164.3 | 19.6 | 38.6 | 6342.0 | −2.1 ± 0.5 | — | — |
| 30-PEG | 34.0 ± 3.4 | 624.5 ± 132.4 | 0.575 | 66.1 | 13.2 | 18.4 | 1216.2 | −6.0 ± 5.8 | — | — |
| 50-PEG | 52.6 ± 5.8 | 345.0 ± 45.0 | 0.466 | 42.8 | 35.2 | 6.6 | 282.5 | −3.7 ± 2.4 | — | — |
| 75-PEG | 76.5 ± 5.3 | 282.8 ± 54.7 | 0.331 | 28.8 | 30.3 | 3.7 | 106.6 | −0.5 ± 0.3 | — | — |
| 100-PEG | 104.6 ± 6.0 | 178.7 ± 3.2 | 0.209 | 21.3 | 22.5 | 1.7 | 36.2 | −1.7 ± 2.8 | — | — |
The average hydrodynamic diameters (nm) of the different SiNPs in 0.5 M potassium phosphate buffer, polydispersity indices (PDI) and the surface charge values (zeta potential) are provided in Table 1. In general, hydrodynamic sizes of the particles were higher for the smaller particles (≤50 nm sized SiNPs) relative to the larger 75 and 100 nm variants. This observation indicates increased agglomeration of the smaller SiNPs in aqueous media. Also, the hydrodynamic size value within each nano-size SiNP group was larger for the pristine SiNPs compared to their corresponding surface-modified variants for ≤50 nm sized SiNPs. In terms of surface-modified SiNPs, their size in the dry-state appeared to be the primary determinant of agglomeration of these amorphous SiNPs, although, the –NH2 modification on the surface of SiNPs led to increased agglomeration compared to PEGylation. All SiNPs exhibited negative surface charge values. Surface charge was not significantly associated with other physicochemical characteristics. A correlation analysis between DLS size in aqueous media and TEM size of SiNPs showed a significant inverse relationship (r = −0.557, p = 0.004). It was also interesting to note that for samples which showed marked agglomeration in aqueous solution (15, 30 nm size SiNPs), the agreement between the BET and TEM estimates for specific surface area was poor. In addition, there was a lack of association between DLS size and BET surface area (r = 0.221, p = 0.289) suggesting that the surface area available for nitrogen absorption for dry-state samples may not correlate well with the available surface area for the same SiNPs in an aqueous environment under the conditions of the biological assays.
Structural analysis of SiNPs assessed by XRD confirmed the amorphous state of all of the pristine SiNPs with a typical pattern of broad reflection at 2 theta = 23° (Fig. S2†). Functionalization by any of the surface functional groups did not alter the amorphous nature of the nanoparticles (data not shown). Similarly, surface functionality (e.g. organic moieties such as short and long chain carboxylic acids groups, NH2 and PEG) was confirmed using the FT-IR methodology. Functional group analyses by FT-IR (Fig. 3) demonstrated the different stretching vibrational frequencies for Si–O, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, –CH2, –OH, NH2 at (e.g. for the 15 nm size). The FT-IR spectra of all samples are dominated by ν (Si–O) vibrational feature (∼1050 cm−1 for symmetric and ∼800 cm−1 for asymmetric stretch) and δ (Si–O–H) bending modes (∼1600 cm−1), respectively. The FT-IR spectra (Fig. 3(b)) of all C3-COOH-functionalized SiO2 nanoparticles exhibit distinct ν (OH), ν (CHx), and ν (CO) stretching mode indicate the presence of alkyl chain and –COOH on the surface. The FT-IR spectra of all C11-COOH-functionalized SiO2 nanoparticles exhibit distinct ν (OH), ν (CHx), and ν (CO) stretching mode indicate the presence of alkyl chain and –COOH on the surface (Fig. 3(c)). The spectra of all NH2-SiO2 samples are dominated by ν (Si–O) vibrational stretching (∼1050 cm−1 for asymmetric and ∼800 cm−1 for symmetric stretch) and δ (Si–O–H) bending modes (∼1600 cm−1), respectively (Fig. 3(d)). The spectra exhibit distinct ν (CH2) stretching (∼2920, 2870 cm−1) and δ (CH2) bending (∼1460 cm−1) modes indicating the presence of methylene groups of surface-bound 3-aminopropyl groups. The FT-IR spectrum exhibits two weak vibrational bands at ∼3360 and ∼3295 cm−1, which can be attributed to symmetric and asymmetric NH2 stretches. In addition, a medium intensity bending mode is observed at ∼1577 cm−1 for NH2 bending vibration. Like NH2-SiO2, spectra of all PEG-functionalized SiO2 samples are dominated by ν (Si–O) vibrational stretching (∼1050 cm−1 for asymmetric and ∼800 cm−1 for symmetric stretch) and δ (Si–O–H) bending modes (∼1600 cm−1), respectively (Fig. 3(e)). The spectra exhibit distinct ν (CH2) stretching (∼2920, 2870 cm−1) and δ(CH2) bending (∼1460 cm−1) modes indicate the presence of methylene groups of PEG chain. In addition, a shoulder at ∼1220 cm−1 is observed for all samples, which may be attributed to the ν (C–O) stretching mode in surface-bound PEG.
O, –CH2, –OH, NH2 at (e.g. for the 15 nm size). The FT-IR spectra of all samples are dominated by ν (Si–O) vibrational feature (∼1050 cm−1 for symmetric and ∼800 cm−1 for asymmetric stretch) and δ (Si–O–H) bending modes (∼1600 cm−1), respectively. The FT-IR spectra (Fig. 3(b)) of all C3-COOH-functionalized SiO2 nanoparticles exhibit distinct ν (OH), ν (CHx), and ν (CO) stretching mode indicate the presence of alkyl chain and –COOH on the surface. The FT-IR spectra of all C11-COOH-functionalized SiO2 nanoparticles exhibit distinct ν (OH), ν (CHx), and ν (CO) stretching mode indicate the presence of alkyl chain and –COOH on the surface (Fig. 3(c)). The spectra of all NH2-SiO2 samples are dominated by ν (Si–O) vibrational stretching (∼1050 cm−1 for asymmetric and ∼800 cm−1 for symmetric stretch) and δ (Si–O–H) bending modes (∼1600 cm−1), respectively (Fig. 3(d)). The spectra exhibit distinct ν (CH2) stretching (∼2920, 2870 cm−1) and δ (CH2) bending (∼1460 cm−1) modes indicating the presence of methylene groups of surface-bound 3-aminopropyl groups. The FT-IR spectrum exhibits two weak vibrational bands at ∼3360 and ∼3295 cm−1, which can be attributed to symmetric and asymmetric NH2 stretches. In addition, a medium intensity bending mode is observed at ∼1577 cm−1 for NH2 bending vibration. Like NH2-SiO2, spectra of all PEG-functionalized SiO2 samples are dominated by ν (Si–O) vibrational stretching (∼1050 cm−1 for asymmetric and ∼800 cm−1 for symmetric stretch) and δ (Si–O–H) bending modes (∼1600 cm−1), respectively (Fig. 3(e)). The spectra exhibit distinct ν (CH2) stretching (∼2920, 2870 cm−1) and δ(CH2) bending (∼1460 cm−1) modes indicate the presence of methylene groups of PEG chain. In addition, a shoulder at ∼1220 cm−1 is observed for all samples, which may be attributed to the ν (C–O) stretching mode in surface-bound PEG.
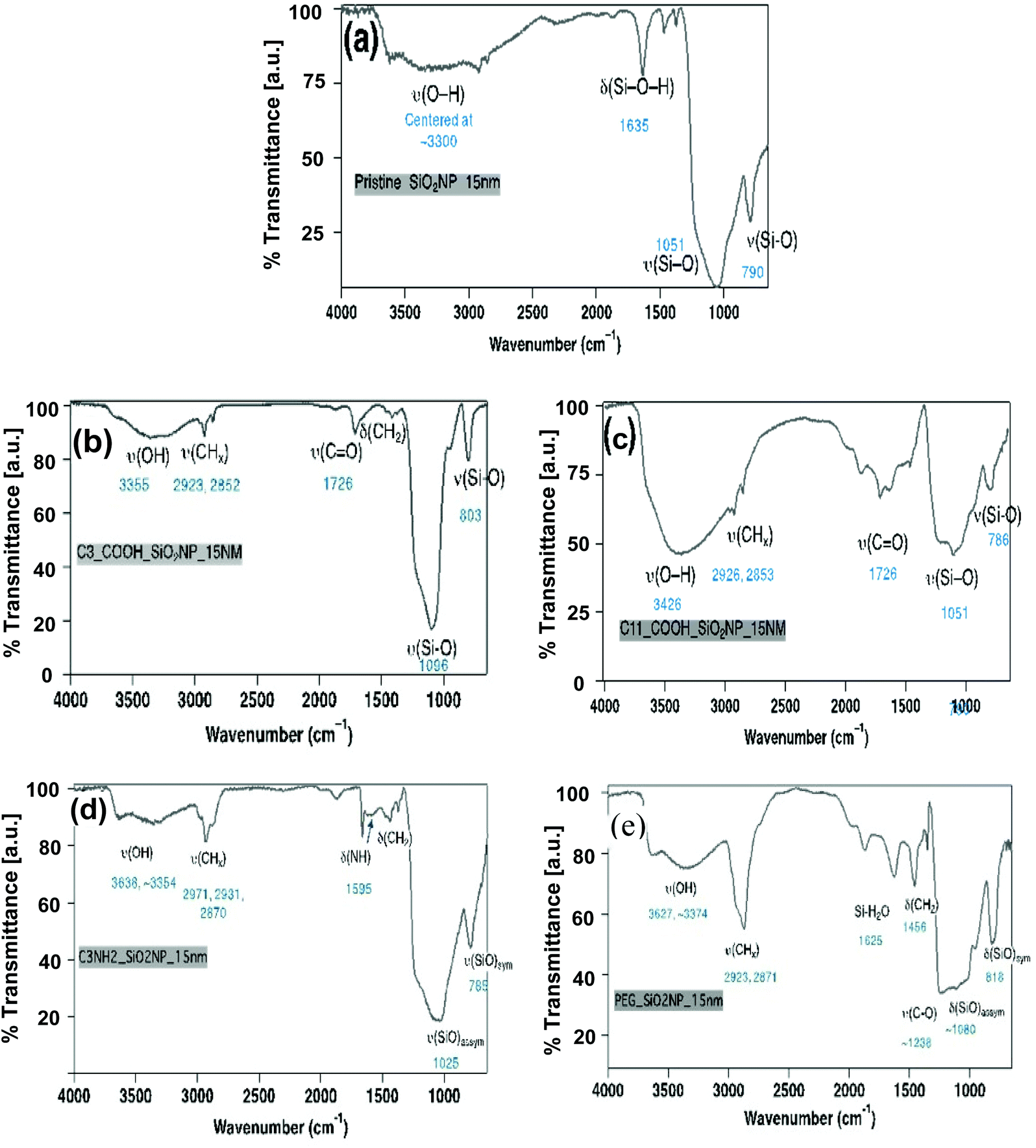 | ||
| Fig. 3 Functional group (FT-IR spectra) analysis for 15 nm-sized SiNPs; (a) pristine, (b) C3-COOH, (c) C11-COOH, (d) NH2, (e) PEG-modified SiNPs. | ||
The total metal content and the sum of transition metals for all five pristine SiNPs used in this work are shown in Table 1. The sum of all elements measured for each SiNP was below 1000 ppm (1 mg g−1), indicating that all SiNPs were more than 99.9% pure. However, there were some differences in total amount of trace elements present between the SiNPs. Surface modification did not appear to alter the amount of metals present in the SiNPs (30 nm surface-modified SiNPs were sampled; data not shown). The detailed elemental composition is indicated in Table S1.† There were differences in amounts of specific elements between the various pristine SiNPs, as well, the SiNPs appeared to be enriched in aluminium and titanium (>100 ppm), as well as boron, magnesium, calcium, chromium and zirconium (>10 ppm; Table S1†). Chemical analyses data on pristine SiNPs revealed differences in the levels of contaminant metals, implying perhaps differences in the batches of source materials used in the synthesis of these nanomaterials or the additives used in synthesis (Table S1†). For instance, there were increased levels of alkaline earth metals, transition metals, and metalloids namely, barium, strontium, titanium, copper, zirconium, cerium, aluminum, lead and boron in the ≤50 nm sized SiNPs compared to the 75 and 100 nm variants. These metals are known to contribute to oxidative stress reactions and similarly to toxicity effects in biological systems.43–47
Surface density of the organic functional groups on surface-modified SiNP variants was determined using TGA and qNMR. Thermogravimetric weight losses under inert argon atmospheres for the pristine SiNPs are illustrated in Fig. 4. The mass loss profiles were relatively simple with initial loss of water molecules adsorbed on the surface, for the pristine SiNPs at temperatures ∼250 °C (Fig. 4). There was a variable mass loss at higher temperature, which based on previous literature48 may be due to condensation of surface hydroxyl groups. As well, potential contaminants introduced during the synthesis process and possibly also loss of matrix components could contribute to the observed mass loss. TGA profiles for the surface modified SiNPs showed loss of water at low temperature, followed by mass loss between ∼250 and 700 °C that is tentatively assigned to surface functional groups (Fig. 4). Moreover, the mass loss between 250 and 700 °C (i.e., eliminating the water contribution), an assumed silica density of 2.2 g cm−3, and a surface area calculated from the mean TEM diameter were used to derive an estimate of surface coverage. The surface coverage estimates are presented in Table 2 and show that NH2-modified SiNPs have markedly higher surface density values relative to PEG-modified SiNPs. The C3-COOH and C11-COOH-modified SiNPs featured intermediate levels of surface coverage (Table 2). Note that these estimates ignore any mass loss due to matrix components or condensation of surface silanols in the same temperature range as the loss of functional group.
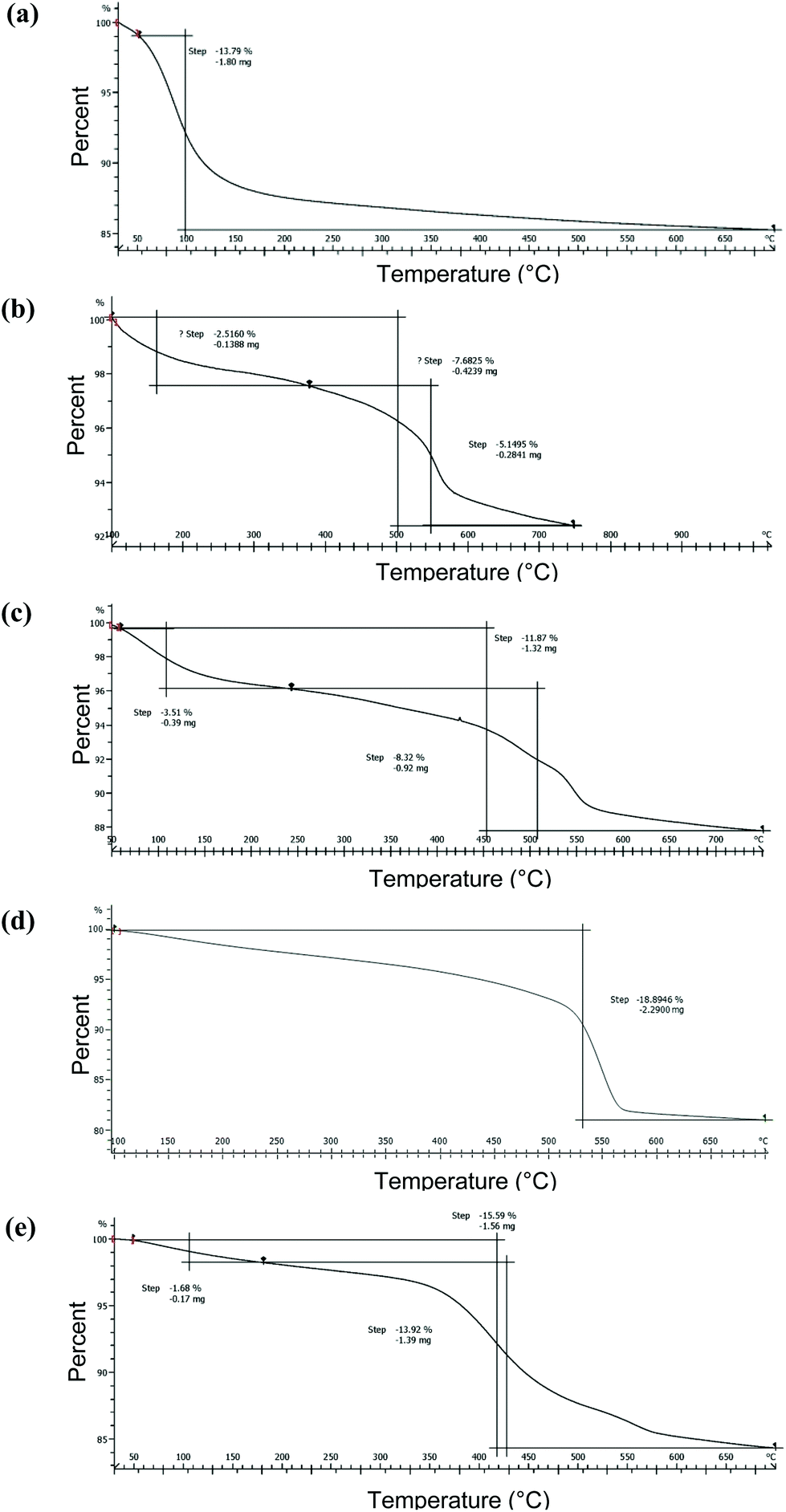 | ||
| Fig. 4 TGA analysis of 15 nm-sized SiNPs; (a) pristine, (b) C3-COOH, (c) C11-COOH, (d) NH2, (e) PEG-modified SiNPs. | ||
| SiNP ID | Mass loss from TGA (wt%) | TGA (μmol g−1) | Surface density TGA (μmol m−2) | qNMR (μmol g−1) | Fraction of monolayer coverage (qNMR) |
|---|---|---|---|---|---|
| The TGA mass loss between ∼250 and 700 °C was assumed to be due to only the organic functional group and was used to calculate the functional group content (μmol g−1) and surface density. qNMR was also used to estimate the functional group content (μmol g−1) and to calculate the corresponding fraction of monolayer coverage, assuming that a full monolayer corresponds to 1.3 molecules per nm2 of the SiNP surface. | |||||
| 15-C3 | 5.15 | 351 | 3.6 | 522 ± 55 | 1.5 |
| 30-C3 | 4.04 | 267 | 7.0 | 401 ± 8 | 1.9 |
| 50-C3 | 11.12 | 679 | 15.1 | 370 ± 6 | 2.9 |
| 75-C3 | 3.52 | 239 | 12.8 | 124 ± 3 | 1.6 |
| 100-C3 | 1.72 | 113 | 9.0 | 139 ± 7 | 2.3 |
| 15-C11 | 8.32 | 325 | 2.1 | 196 ± 13 | 0.6 |
| 30-C11 | 9.76 | 378 | 4.0 | 375 ± 1 | 1.8 |
| 50-C11 | 13.72 | 532 | 9.4 | 221 ± 11 | 1.8 |
| 75-C11 | 6.55 | 256 | 7.3 | 129 ± 5 | 1.7 |
| 100-C11 | 3.57 | 140 | 5.2 | 129 ± 1 | 2.2 |
| 15-NH2 | 18.9 | 1232 | 7.5 | 757 ± 137 | 1.7 |
| 30-NH2 | 9.98 | 619 | 8.3 | 1360 ± 110 | 6.8 |
| 50-NH2 | 9.07 | 620 | 14.2 | 1310 ± 10 | 12.1 |
| 75-NH2 | 9.97 | 667 | 21.8 | 269 ± 6 | 3.6 |
| 100-NH2 | 5.12 | 320 | 14.3 | 458 ± 83 | 7.7 |
| 15-PEG | 13.92 | 180 | 1.1 | 196 ± 5 | 0.4 |
| 30-PEG | 12.66 | 171 | 2.6 | 235 ± 14 | 1.3 |
| 50-PEG | 7.42 | 107 | 2.5 | 121 ± 4 | 1.1 |
| 75-PEG | 7.55 | 103 | 3.6 | 106 ± 3 | 1.4 |
| 100-PEG | 4.78 | 64 | 3.0 | 70 ± 2 | 1.2 |
Quantitative analysis of the functional group content was carried out using a previously reported method that relies on dissolution of silica NPs in base, followed by measurement of the 1H NMR spectrum and quantification of the released functional group by comparison to an internal standard.39 Representative spectra are shown in Fig. 5 for the four functionalized 15 nm SiNPs. For C3-COOH and NH2-modified SiNPs the three methylene signals (a, b, c) were well-separated from the impurity signals due to residual ethanol and all three were used for integration. For C11-COOH-modified SiNPs, peak “d” corresponds to the interior methylene groups of the undecyl chain and was not included in the integration, due to the interference from the trace ethanol signal. Peak “d” was also excluded from the integration for the polyethylene glycol (PEG)-modified silica NPs since the PEG chain length is not known. The functional group content measured by qNMR is provided in Table 2. Comparison of NMR and TGA data indicates that the two methods give similar results for PEG-functionalized SiNPs, with the exception of the 30 nm samples. This finding agrees well with the previous study in which it was observed that SiNPs functionalized with a large molecular weight group such as PEG revealed a similarity by NMR and TGA, due to the increased mass loss for a large functional group in the TGA.48 The agreement was poor for the other samples, with TGA giving estimates that are both lower and higher than the NMR values. Note that the structural identification of the functional groups by NMR significantly increases the confidence in this method for quantification of the functional group content. Across all particles assessed, the correlation between qNMR and TGA methods in quantification of functional groups on the surface of SiNPs revealed a significant association (r = 0.607, p = 0.005) between the two techniques. However, detailed analyses within each surface modification revealed a strong association between the two methods for PEG-modified SiNPs (r = 0.960, p = 0.01) only. While the higher temperature losses observed in the thermograms for the surface-modified SiNPs are attributable to functional group loss, absolute determination of functional group loss can still be confounded by mass loss due to other components in this temperature regimen, as observed for their pristine counterparts. This complication explains the lack of correlation between qNMR and TGA data for the majority of the individual surface-modified SiNPs (–C3-COOH, –C11-COOH, –NH2). Thus it is challenging to use TGA results as a measure of the organic functional group contents associated with surface modifications, unless a reliable correction method can be applied for the mass loss that corresponds to surface groups other than the intended surface modifications.48
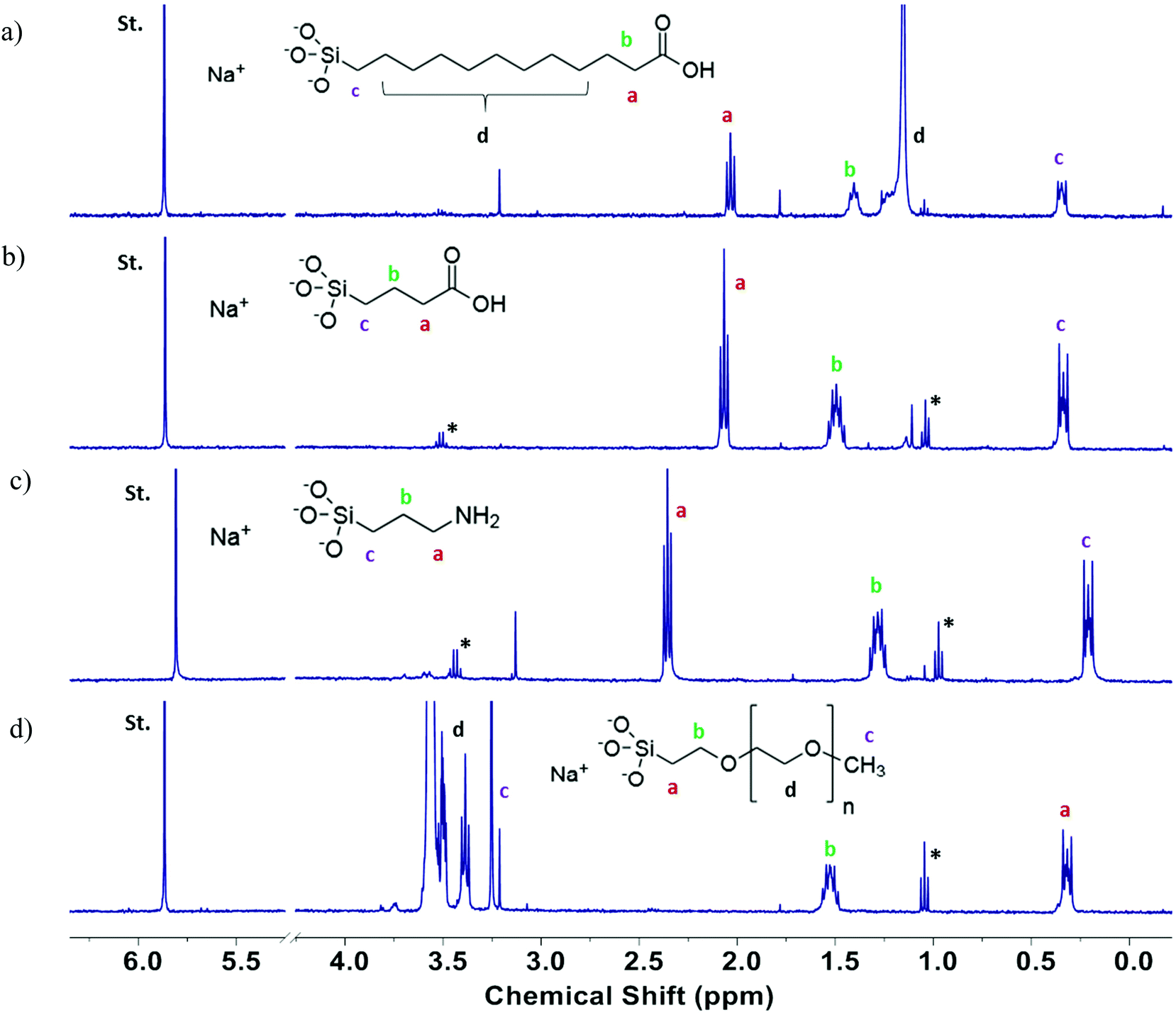 | ||
| Fig. 5 Determination of functional group content of SiNPs by dissolution qNMR. The H2O signal at 4.7 ppm has been removed and the spectra are expanded for display purposes. The letters indicate the carbon atom in the functional group structure corresponding to the peak in the NMR spectrum. The star indicates the presence of an EtOH peak. (a) 15 nm C11-COOH; (b) 15 nm C3-COOH; (c) 15 nm NH2; (d) 15 nm PEG. | ||
Oxidative potential
There are reports on the application of the DTT oxidation assay to quantify the oxidative potential of ambient air pollution particles consisting of complex matrices containing transition metals and various organics that can participate in redox-cycling mechanisms.49–51 On the contrary, engineered nanomaterials are prepared for intended use and are relatively pure compared to air particles and the applicability of the DTT assay for oxidative potential screening requires exploration. Here, we applied the DTT assay to assess oxidative potential of synthesized SiNPs that do not contain redox-cycling species in its chemical composition. Nevertheless, our findings revealed that all reference particles (EHC-6802, TiO2, SiO2, and SiNP 12 nm) and SiNPs used in this study were able to oxidize DTT in aqueous media under the experimental conditions applied, at various time points (3–60 min; Fig. S3†). In addition, some variability in the 0–3 min time point was observed across the particles, indicating that the DTT oxidation reaction is rapid. Therefore, the reactivity at 3 minutes was selected to represent the initial reaction of DTT and SiNPs, prior to the quenching with DTNB. Here, DTNB reacts with the free “thiol” groups present in unreacted DTT. Based on the time course, data for 20 min is reported subsequently, since the extent of the DTT oxidation at this time point for all SiNPs, appeared to be the most stable across all particles and suitable for further analysis. Time-related changes in the oxidation of DTT by the various SiNPs exhibited an apparent decrease in the DTT oxidation at 60 min compared to that at 20 min for most particles (Fig. S3†). The apparent decrease in the DTT oxidation at 60 min (longer time period) may be attributed to a masking effect caused by alternate reactions of DTT that result in reaction products with a free “thiol” group which in turn can react with DTNB (instead of disulfide-containing product) and absorb at the wavelength measured for DTT-DTNB product.The relative oxidative potential values of the amorphous SiNPs revealed significant-size related changes (2-way ANOVA: p < 0.05; Fig. 6(a)) at 20 min (but not at 60 min of reaction time). Note that the lower the DTNB absorbance, the higher will be DTT oxidation to the disulfide product. All tested SiNPs indicated generally low redox reactivity in the DTT-based assay. The DTT oxidation results indicated that 15 and 30 nm size pristine SiNPs were relatively more reactive (p < 0.05) compared to the larger size pristine SiNP variants and that surface modifications did not show any deviations from this effect. The 15 nm SiNPs, regardless of modification type, were significantly more reactive in comparison to the 30 nm SiNPs (two-way ANOVA, size main effect, p = 0.002; 15 nm versus 30 nm, p < 0.001). It was also interesting to note that both amorphous 12 nm SiNP, which is a reference material, and the test pristine 15 nm SiNP exhibited similar levels of oxidation of DTT. Among the reference particles, EHC-6802 had the highest activity in DTT depletion after a 20 min reaction, while TiO2 had the least activity (Kruskal–Wallis test, p = 0.006; EHC-6802 versus TiO2, p < 0.05; Fig. 6(a)). Low redox activity of TiO2 suspension with the acellular assay DCFH-DA has also previously been reported.52 The difference between EHC-6802 and the other reference particles used in this work may be related to the fact that EHC-6802 is an environmental particle comprised of a complex mixture of organics and metals, whereas the other reference particles (SiO2 and TiO2) have a much simpler composition.
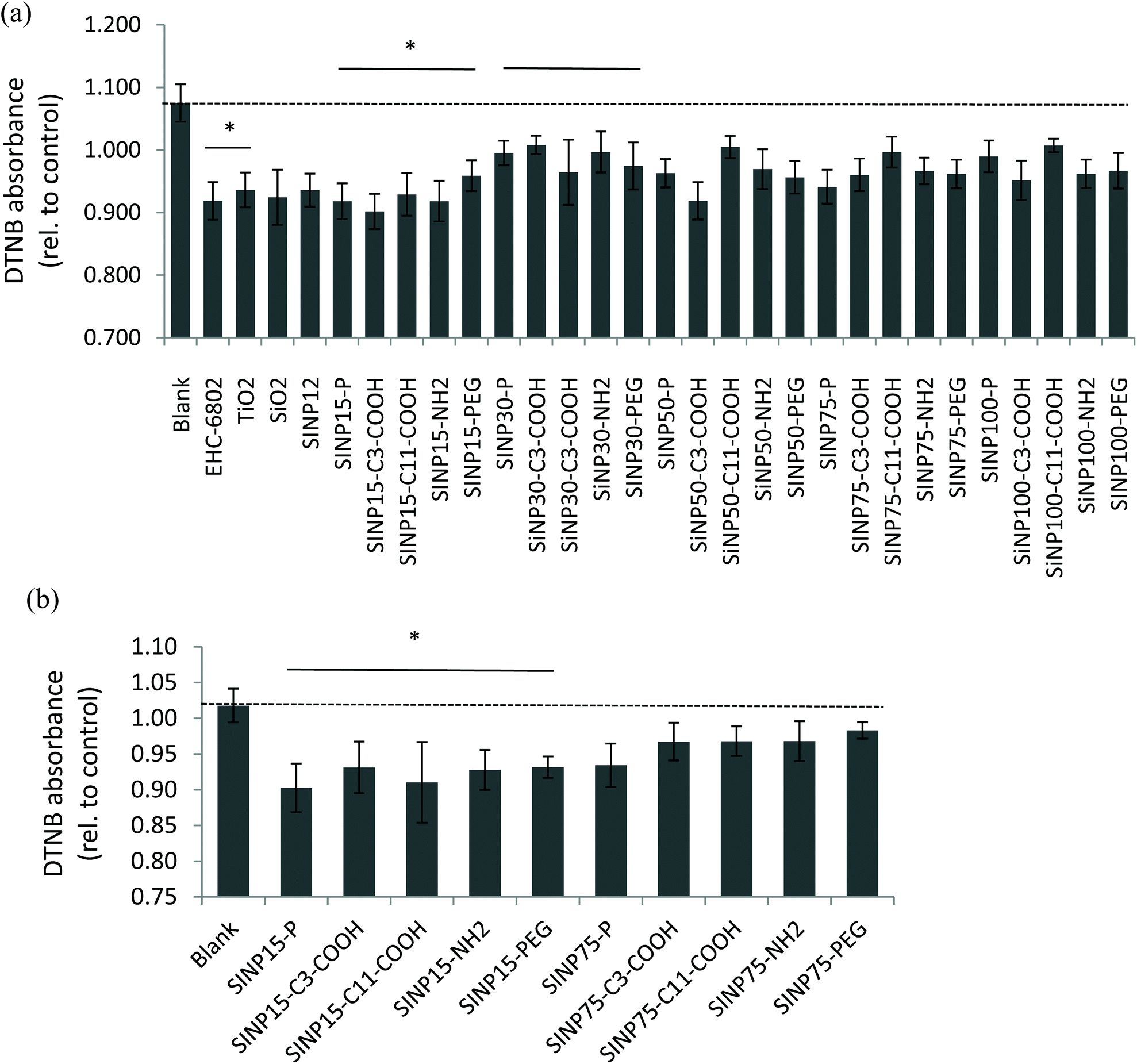 | ||
| Fig. 6 Intrinsic oxidative capacity of SiNPs and reference particles based on DTT depletion (a) at selected 20 min time point, and (b) the oxidation of DTT ± 100 μM tert-butyl peroxide and 20 μM Fe2+ by 15 and 75 nm SiNPs after 20 min co-incubation. The increased DTT substrate depletion is indicated by lower values of the DTNB absorbance (below 1.0). | ||
A modified protocol was tested to evaluate whether the presence of the free radical initiator TBHP in the presence of Fe2+ in test media could enhance the oxidative potential of these nanoparticles. This test was only conducted with the pristine and surface-modified 15 and 75 nm SiNPs and showed that the 15 nm pristine SiNP was relatively more oxidatively active compared to the 75 nm variant (Fig. 6(b); two-way ANOVA, size main effect, p < 0.001; 15 nm versus 75 nm, p < 0.001). Interestingly, this test also confirmed dominant size-related oxidative potential of these SiNPs.
Increasing evidence suggests that physicochemical properties of nanomaterials are important drivers in perturbation of biological functions.1,12,40 Therefore, potential linkages between the physicochemical properties of the SiNPs (summarised in Tables 1 and 2) with their redox potential as measured by the DTT oxidation assay (presented in Fig. 6(b)) were explored through a Pearson correlation analysis (Table 3). When only pristine SiNPs were included in the correlation analysis, associations with physicochemical properties were less clear (data not shown), perhaps due to small number of observations (sample size, n). The correlation analysis including all pristine and surface-modified SiNPs revealed significant positive association of the DTT oxidation activity of the SiNPs with their TEM surface area (r = 0.490, p = 0.013, Table 3). TEM surface area increased as the SiNP size decreased as illustrated in Table 1, and thus the positive association of TEM surface area with DTT oxidation activity suggests nanoparticles’ size-related effect on oxidative behaviour. This is consistent with the ANOVA results (Fig. 6), where smaller size SiNP particles (e.g. 15 nm) exhibited significant increase in oxidative potential. Similarly, correlation tests also indicated that agglomeration SA ratio (r = 0.480, p = 0.015) was positively associated with oxidative potential. Agglomeration SA ratio represents an approximation of the agglomerated SiNP surface area in buffer available for potential reactivity (see footnote in Table 1). Agglomeration SA ratio is also a size- and surface modification-dependent parameter (Table 1) thus suggesting impact of size and surface modification-related effects on the oxidative potential values of these SiNPs. In addition, a marginal association (0.1 > p > 0.05) was also observed between the DTT oxidation activity and the DLS/TEM ratio, which represents an estimate of the agglomeration state of the SiNPs (r = 0.339, p = 0.098). It was interesting to note that the DTT oxidative capacity did not correlate with metal contents suggesting that the trace amounts of contaminant metals do not contribute significantly to the oxidative potential of these nanomaterials. Interestingly, correlation tests on DTT oxidation activity versus functional group content measured by NMR (Table 2) within the different surface-modified SiNP groups revealed a significant negative association for C11-COOH-modified silica nanoparticles only (DTT oxidation versus fraction of monolayer coverage; r = −0.879, p ≤ 0.05) and marginal associations (0.1 > p > 0.05) for DTT oxidation and the remaining functional group-related measures (Table 3), suggesting the effect of surface modification. These associations may be attributed to the longer alkyl chain length of this C11-COOH-SiNP, which facilitates internal stable hydrogen bonding between the –COOH group and the “O” bonded to “Si”. This could potentially impede the hydrogen-abstraction from the –SH group in DTT and thus negatively affect the DTT oxidation. This is supported by the relatively increased hydrogen bonding in the C11-COOH-SiNP revealed by the –OH stretching vibration (e.g.Fig. 3; ∼3500 cm−1) compared to the other SiNPs. The observed correlations indicate that physicochemical parameters such as size, surface area, surface modifications and agglomeration behaviour of these SiNPs may be key determinants of their DTT oxidative potential.
| Acellular DTT activity versus | Correlation coefficient (r) | P-Value |
|---|---|---|
| All of the physicochemical variables were correlated with 1-DTT activity values.a Correlations including all SiNPs.b Correlations including C11-COOH-modified SiNPs only; statistically significant correlations (p < 0.05) are shown in bold; marginally significant correlations (0.1 > p > 0.05) are italicized; non-significant correlations are not shown. | ||
| TEM SA (m2 g−1)a | 0.490 | 0.013 |
| Agglomeration (DLS/TEM) ratioa | 0.339 | 0.098 |
| Agglomeration SA ratioa | 0.480 | 0.015 |
| Surface density (μmol m−2)b | −0.828 | 0.083 |
| Molecules per nm2b | −0.867 | 0.057 |
| Fraction of monolayer coverage (1.3 molecules per nm2)b | −0.879 | 0.050 |
Our observations are in line with the previous reports on amorphous silica nanomaterial exposure-related oxidative stress and physicochemical determinants of their reactivity in cells.18,53–55 SiNPs have been shown to participate in ROS formation in biological systems. For instance, SiNPs (1–100 nm) are known to exhibit toxicity in vitro (immortalized mammalian cell lines), with associated formation of ROS and subsequent oxidative stress1 and toxicity of SiNPs were shown to be associated with their physicochemical properties such as size. Also, Duan et al. have reported on the formation of ROS in human umbilical vein endothelial cells (HUVEC) exposed to pristine SiNPs (62 nm), as well as the induction of apoptosis and cytotoxicity.56 Athinarayanan et al. have exposed human lung fibroblasts (WI38 cell line) to SiNPs (10–20 nm) and showed ROS production and cytotoxicity in these cells.57 We have also previously shown that exposures of A549 cells to amorphous SiNP 12 nm from another source (obtained commercially) can induce release of ROS, changes in lipid metabolism and changes in cytosolic Ca2+, as suggested by pathway analysis.40 These SiNPs were highly cytotoxic across multiple cell lines.
These findings suggest that the DTT oxidation results could provide insight into the reactivity of amorphous SiNPs at a cellular level. Thus, this assay can serve as a first-tier bioanalytical tool to scan for potential biological reactivity.
Conclusion
Smaller-sized amorphous pristine SiNPs (15 nm) exhibit greater potential to oxidize DTT, as well as to agglomerate compared to relatively larger-sized SiNPs. Also, physicochemical properties including TEM-derived surface area, impacted by size of SiNPs and agglomeration SA ratio, impacted by SiNP size and surface modification were positively correlated with the oxidative potential of these amorphous SiNP variants, while surface coverage of organics was negatively correlated with the DTT oxidation only for the C11-COOH-modified SiNPs, also exhibiting the effect of surface modification. This study reveals the capacity of the DTT assay to serve as an initial acellular screening tool to predict SiNP reactivities, and future work on these SiNPs in biological systems can unravel the relationship between their oxidative potential and related cellular toxicity characteristics.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
Chemicals Management Plan (CMP) and Nanotechnology Section nano research fund from NSACB, Health Canada. The panel of the amorphous SiNPs was synthesized by Applied Quantum Materials Inc. in Edmonton, AB, Canada. We are grateful to Dr Nimal DeSilva at the Department of Earth and Environmental Sciences, University of Ottawa for conducting the ICP-MS/AES analysis. We are grateful to Drs Dharani Das and Yong-Lai Feng at Health Canada, for critical reviews of the manuscript. We thank Bing Wang, Health Canada and Daniel Hong (CO-OP student, University of Waterloo) for DLS technical assistance, Monica Sourial (Capstone course field placement student, Carleton University) for her assistance with the DTT assay and Floyd Toll, NRC for assistance with BET measurements.References
- D. Napierska, L. C. Thomassen, D. Lison, J. A. Martens and P. H. Hoet, Part. Fibre Toxicol., 2010, 7, 39 CrossRef CAS PubMed.
- L. R. Khot, S. Sankaran, J. M. Maja, R. Ehsani and E. W. Schuster, Crop Prot., 2012, 35, 64–70 CrossRef CAS.
- M. R. Kasaai, J. Nanotechnol., 2015, 2015, 852394 Search PubMed.
- A. Brinch, S. F. Hansen, N. B. Hartmann and A. Baun, Nanomaterials, 2016, 6, 33 Search PubMed.
- V. Vijayanathan, T. Thomas and T. J. Thomas, Biochemistry, 2002, 41, 14085–14094 CrossRef CAS PubMed.
- L. R. Hirsch, R. J. Stafford, J. A. Bankson, S. R. Sershen, B. Rivera, R. E. Price, J. D. Hazle, N. J. Halas and J. L. West, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 13549–13554 CrossRef CAS PubMed.
- M. Benezra, O. Penate-Medina, P. B. Zanzonico, D. Schaer, H. Ow, A. Burns, E. DeStanchina, V. Longo, E. Herz, S. Iyer, J. Wolchok, S. M. Larson, U. Wiesner and M. S. Bradbury, J. Clin. Invest., 2011, 121, 2768–2780 CrossRef CAS PubMed.
- A. Bitar, N. M. Ahmad, H. Fessi and A. Elaissari, Drug Discovery Today, 2012, 17, 1147–1154 CrossRef CAS PubMed.
- L. Tang and J. Cheng, Nano Today, 2013, 8, 290–312 CrossRef CAS PubMed.
- J. Chen, Z. Guo, H. Tian and X. Chen, Mol. Ther. – Methods Clin. Dev., 2016, 3, 1602 Search PubMed.
- WHO, Nanotechnology and human health: Scientific evidence and risk governance, Report of the WHO expert meeting 10–11 December 2012, Bonn, Germany, Copenhagen, WHO Regional Office for Europe, 2013.
- S. Murugadoss, D. Lison, L. Godderis, S. Van Den Brule, J. Mast, F. Brassinne, N. Sebaihi and P. H. Hoet, Arch. Toxicol., 2017, 91, 2967–3010 Search PubMed.
- J. Y. Kim, J. H. Park, M. Kim, H. Jeong, J. Hong, R. S. Chuck and C. Y. Park, Sci. Rep., 2017, 7, 14566 Search PubMed.
- L. Chen, J. Liu, Y. Zhang, G. Zhang, Y. Kang, A. Chen, X. Feng and L. Shao, Nanomedicine, 2018, 13, 1939–1962 CrossRef PubMed.
- Z. Du, S. Chen, G. Cui, Y. Yang, E. Zhang, Q. Wang, M. F. Lavin, A. J. Yeo, C. Bo, Y. Zhang, C. Li, X. Liu, X. Yang, C. Peng and H. Shao, Int. J. Mol. Med., 2019, 43, 1229–1240 CAS.
- K. Lee, J. Lee, M. Kwak, Y. L. Cho, B. Hwang, M. J. Cho, N. G. Lee, J. Park, S. H. Lee, J. G. Park, Y. G. Kim, J. S. Kim, T. S. Han, H. S. Cho, Y. J. Park, S. J. Lee, H. G. Lee, W. K. Kim, I. C. Jeung, N. W. Song, K. H. Bae and J. K. Min, J. Nanobiotechnol., 2019, 17, 24 CrossRef PubMed.
- H. Zhang, D. R. Dunphy, X. Jiang, H. Meng, B. Sun, D. Tarn, M. Xue, X. Wang, S. Lin, Z. Ji, R. Li, F. L. Garcia, J. Yang, M. L. Kirk, T. Xia, J. I. Zink, A. Nel and C. J. Brinker, J. Am. Chem. Soc., 2012, 134, 15790–15804 CrossRef CAS PubMed.
- E. J. Park and K. Park, Toxicol. Lett., 2009, 184, 18–25 CrossRef CAS PubMed.
- F. Wang, F. Gao, M. Lan, H. Yuan, Y. Huang and J. Liu, Toxicol. In Vitro, 2009, 23, 808–815 Search PubMed.
- Y. Ye, J. Liu, M. Chen, L. Sun and M. Lan, Environ. Toxicol. Pharmacol., 2010, 29, 131–137 CrossRef CAS PubMed.
- C. Terzano, F. Di Stefano, V. Conti, E. Graziani and A. Petroianni, Eur. Rev. Med. Pharmacol. Sci., 2010, 14, 809–821 CAS.
- M. Lodovici and E. Bigagli, J. Toxicol., 2011, 2011, 487074 Search PubMed.
- A. Valavanidis, T. Vlachogianni, K. Fiotakis and S. Loridas, Int. J. Environ. Res. Public Health, 2013, 10, 3886–3907 CrossRef PubMed.
- P. J. Borm, F. Kelly, N. Künzli, R. P. Schins and K. Donaldson, Occup. Environ. Med., 2007, 64, 73–74 CrossRef PubMed.
- N. A. H. Janssen, A. Yang, M. Strak, M. Steenhof, B. Hellack, M. E. Gerlofs-Nijland, T. Kuhlbusch, F. Kelly, R. Harrison, B. Brunekreef, G. Hoek and F. Cassee, Sci. Total Environ., 2014, 472, 572–581 CrossRef CAS PubMed.
- V. Bonvallot, A. Baeza-Squiban, A. Baulig, S. Brulant, S. Boland, F. Muzeau, R. Barouki and F. Marano, Am. J. Respir. Cell Mol. Biol., 2001, 25, 515–521 CrossRef CAS PubMed.
- A. Kubátová, L. C. Dronen, M. J. Picklo and S. B. Hawthorne, Chem. Res. Toxicol., 2006, 19, 255–261 Search PubMed.
- T. Xia, M. Kovochich, J. Brant, M. Hotze, J. Sempf, T. Oberley, C. Sioutas, J. I. Yeh, M. R. Wiesner and A. E. Nel, Nano Lett., 2006, 6, 1794–1807 CrossRef CAS PubMed.
- T. Shi, R. P. F. Schins, A. M. Knaapen, T. Kuhlbusch, M. Pitz, J. Heinrich and P. J. A. Borm, J. Environ. Monit., 2003, 5, 550–556 RSC.
- A. K. Cho, C. Sioutas, A. H. Miguel, Y. Kumagai, D. A. Schmitz, M. Singh, A. Eiguren-Fernandez and J. R. Froines, Environ. Res., 2005, 99, 40–47 CrossRef CAS PubMed.
- I. S. Mudway, G. Fuller, D. Green, C. Dunster and F. J. Kelly, Report: Quantifying the London specific component of PM10 oxidative activity, University of London, Defra, UK, 2011.
- B. Zomer, L. Collé, A. Jedyńska, G. Pasterkamp, I. Kooter and H. Bloemen, Anal. Bioanal. Chem., 2011, 401, 2945–2954 Search PubMed.
- Q. Xiong, H. Yu, R. Wang, J. Wei and V. Verma, Environ. Sci. Technol., 2017, 51, 6507–6514 CrossRef CAS PubMed.
- I. S. Mudway, S. T. Duggan, C. Venkataraman, G. Habib, F. J. Kelly and J. Grigg, Part. Fibre Toxicol., 2005, 2, 6 CrossRef PubMed.
- J. G. Ayres, P. Borm, F. R. Cassee, V. Castranova, K. Donaldson, A. Ghio, R. M. Harrison, R. Hider, F. Kelly, I. M. Kooter, F. Marano, R. L. Maynard, I. Mudway, A. Nel, C. Sioutas, S. Smith, A. Baeza-Squiban, A. Cho, S. Duggan and J. Froines, Inhalation Toxicol., 2008, 20, 75–99 CrossRef CAS PubMed.
- P. J. O'Brien, Chem.-Biol. Interact., 1991, 80, 1–41 CrossRef.
- G. L. Squadrito, R. Cueto, B. Dellinger and W. A. Pryor, Free Radicals Biol. Med., 2001, 31, 1132–1138 CrossRef CAS PubMed.
- E. M. Thomson, A. Williams, C. L. Yauk and R. Vincent, Part. Fibre Toxicol., 2009, 6, 6 Search PubMed.
- F. Kunc, V. Balhara, A. Brinkmann, Y. Sun, D. M. Leek and L. J. Johnston, Anal. Chem., 2018, 90, 13322–13330 CrossRef CAS PubMed.
- D. Breznan, D. D. Das, J. S. O'Brien, C. MacKinnon-Roy, S. Nimesh, N. Q. Vuong, S. Bernatchez, N. DeSilva, M. Hill, P. Kumarathasan and R. Vincent, Nanotoxicology, 2017, 11, 223–235 CrossRef CAS PubMed.
- M. K. Kim and W. K. Jo, Int. Arch. Occup. Environ. Health, 2006, 80, 40–50 CrossRef CAS PubMed.
- D. D. Das, Y. Yang, J. S. O'Brien, D. Breznan, S. Nimesh, S. Bernatchez, M. Hill, A. Sayari, R. Vincent and P. Kumarathasan, J. Nanomater., 2014, 12, e176015 Search PubMed.
- K. Bhattacharya, M. Davoren, J. Boertz, R. P. Schins, E. Hoffmann and E. Dopp, Part. Fibre Toxicol., 2009, 6, 17 Search PubMed.
- K. Jomova and M. Valko, Toxicology, 2011, 283, 65–87 CrossRef CAS PubMed.
- E. Asadpour, H. R. Sadeghnia, A. Ghorbani and M. T. Boroushaki, Iran. J. Pharm. Res., 2014, 13, 1141–1148 CAS.
- E. Cheraghi, A. Golkar, K. Roshanaei and B. Alani, Int. J. Fertil. Steril., 2017, 11, 166–175 CAS.
- D. Schwotzer, M. Niehof, D. Schaudien, H. Kock, T. Hansen, C. Dasenbrock and O. Creutzenberg, J. Nanobiotechnol., 2018, 16, 16 CrossRef PubMed.
- F. Kunc, V. Balhara, Y. Sun, M. Daroszewska, Z. J. Jakubek, M. Hill, A. Brinkmann and L. J. Johnston, Analyst, 2019, 144, 5589–5599 RSC.
- Y. Kumagai, S. Koide, K. Taguchi, A. Endo, Y. Nakai, T. Yoshikawa and N. Shimojo, Chem. Res. Toxicol., 2002, 15, 483–489 Search PubMed.
- M. Y. Chung, R. A. Lazaro, D. Lim, J. Jackson, J. Lyon, D. Rendulic and A. S. Hasson, Environ. Sci. Technol., 2006, 40, 4880–4886 CrossRef CAS PubMed.
- Y. Wang, C. Arellanes, D. B. Curtis and S. E. Paulson, Environ. Sci. Technol., 2010, 44, 4070–4075 CrossRef CAS PubMed.
- L. K. Limbach, P. Wick, P. Manser, R. N. Grass, A. Bruinink and W. J. Stark, Environ. Sci. Technol., 2007, 41, 4158–4163 CrossRef CAS PubMed.
- H. Nabeshi, T. Yoshikawa, K. Matsuyama, Y. Nakazato, S. Tochigi, S. Kondoh, T. Hirai, T. Akase, K. Nagano, Y. Abe, Y. Yoshioka, H. Kamada, N. Itoh, S. Tsunoda and Y. Tsutsumi, Part. Fibre Toxicol., 2011, 8, 1 CrossRef CAS PubMed.
- M. Ahamed, Hum. Exp. Toxicol., 2013, 32, 186–195 CrossRef CAS PubMed.
- A. Nemmar, S. Beegam, P. Yuvaraju, J. Yasin, A. Shahin and B. H. Ali, Cell. Physiol. Biochem., 2014, 34, 255–265 CrossRef CAS PubMed.
- J. Duan, Y. Yu, Y. Li, Y. Yu and Z. Sun, Biomaterials, 2013, 34, 5853–5862 CrossRef CAS PubMed.
- J. Athinarayanan, V. S. Periasamy, M. A. Alsaif, A. A. Al-Warthan and A. A. Alshatwi, Cell Biol. Toxicol., 2014, 30, 89–100 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0an00380h |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |