
Disposable paper strips for carboxylate discrimination†
 a
and
Marco
Bonizzoni
*ab
a
and
Marco
Bonizzoni
*ab
Abstract
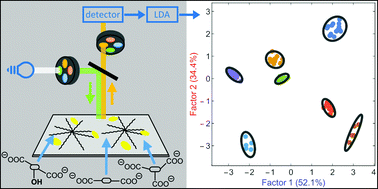
We describe a method for the differentiation of carboxylate anions on disposable paper supports (common printer paper, filter paper, chromatography paper), based on differential patterns of interactions between carboxylates and a fluorescent sensing system. The sensor was built from commercially available components, namely a polycationic fifth generation amine-terminated poly(amidoamine) dendrimer (PAMAM G5) and a small organic fluorophore (calcein) through non-covalent interactions. The assay's physical dimensions were chosen to conform to the microwell plate standard so detection could be carried out on widely available plate reader instrumentation. The sensing complex was first deposited in spots on a paper support to prepare the sensor strip; a carboxylate solution was then loaded on each spot. Nuanced changes in fluorescence were associated with carboxylate binding to the PAMAM dendrimer, characteristic of the structure and affinity of each carboxylate. Such signal changes, interpreted through Linear Discriminant Analysis (LDA), contained enough information to recognize and successfully discriminate most anions in the panel. Among the substrates we tested, chromatography paper was the most promising. The relationship between the structure of the carboxylates and the patterns giving rise to their differentiation was also discussed. Finally, the long-term stability (“shelf life”) of the pre-assembled [calcein·dendrimer] sensing system was found to be excellent when deposited on paper support.
- This article is part of the themed collection: The Mechanics of Supramolecular Chemistry
 Please wait while we load your content...
Please wait while we load your content...

