
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Native detection of protein O-GlcNAcylation by gel electrophoresis†
Chuan
Fu
 and
Daan M. F.
van Aalten
*
and
Daan M. F.
van Aalten
*
Centre for Gene Regulation & Expression, School of Life Sciences, University of Dundee, Dundee, UK. E-mail: dmfvanaalten@dundee.ac.uk
First published on 2nd September 2020
Abstract
O-GlcNAcylation is an abundant and dynamic protein posttranslational modification (PTM), with crucial roles in metazoans. Studies of this modification are hampered by the lack of convenient methods for detecting native O-GlcNAcylation. Here, we describe a novel gel-based approach, Separation of O-GlcNAcylated Proteins by Polyacrylamide Gel Electrophoresis (SOPAGE), which enables detection of O-GlcNAc levels and dynamics.
Introduction
Glycosylation of serine or threonine on nuclear and cytoplasmic proteins with O-linked β-N-acetyl-D-glucosamine (O-GlcNAc) is an abundant, reversible and dynamic intracellular post-translational modification (PTM) that is essential for metazoan life.1–4 The cellular O-GlcNAcylation cycle is regulated by a pair of enzymes, O-GlcNAc transferase (OGT) and O-GlcNAcase (OGA). Addition or removal of O-GlcNAc moieties onto over 1000 proteins has been implicated in numerous cellular processes, including transcription, cell cycle progression, signal transduction, energy metabolism and protein quality control.5–7 It also shows a degree of interplay with regulatory protein phosphorylation, and serves as a link between nutrient availability and cellular responses.8O-GlcNAc is also essential for embryogenesis and neuronal development,7,9 and abnormal protein O-GlcNAcylation is implicated in diabetes,10 tumorigenesis,11 Alzheimer's disease (AD)12 and X-linked intellectual disability (XLID).13–15Although O-GlcNAc was discovered more than 30 years ago, there are only a few methods for monitoring its dynamics or stoichiometry.16,17 Typically, O-GlcNAcylation does not give rise to a detectable change of mobility on standard polyacrylamide gel electrophoresis (PAGE) due to the neutral charge of O-GlcNAc residue and the minor addition of mass. Initially, protein O-GlcNAc levels are quantified by radioactive labeling,18 high-pH anion exchange chromatography (HPAEC),19 or lectin weak affinity chromatography (LWAC).20,21 However, these methods are not routinely accessible, time-consuming or requiring large quantities of samples. Western blotting using O-GlcNAc specific antibodies (either pan-specific22 or site-specific Abs23) can assess the relative change in protein O-GlcNAc levels between samples, yet lacks the ability to provide information on accurate quantification. Quantitative mass spectrometry provides a solution; however, either isotope-labeled peptides are required as internal standards for MS1-based quantification,24 or precursor-fragment ions couples and the fragmentation behavior of peptides derived from proteins of interest must be pre-determined to facilitate Multiple Reaction Monitoring Mass Spectrometry (MRM-MS).25 Both these MS-based approaches require specific reagents for each site to be monitored and are labor-intensive, limiting their universal applicability. Recently, a mass-tag-based derivatization method achieved the spatial separation of O-GlcNAc protein in gels, facilitating quantification.26 The O-GlcNAc is first labeled with a β-1,4-galactosyltransferase mutant (Y289L) and azido-UDP-ketogalactose, and then reacted with alkyne-containing polyethylene glycol (PEG) via click chemistry. Here, accurate quantification relies on the completeness of the requisite enzymatic and chemical reactions, which can be affected by the diverse nature of the protein substrates. Besides, wheat germ agglutinin (WGA)-based affinity electrophoresis has recently been shown to retard glycosylated protein in gels.27 However, WGA non-specifically interacts with other types of glycosylation and has low affinity for O-GlcNAc (approx. 10 mM for free O-GlcNAc28), resulting in smeared bands. Functional dissection of O-GlcNAc would benefit from a more universal method for the one-step direct separation of O-GlcNAc subpopulations by gel electrophoresis but is still challenging.
Recently, we described an inactive mutant of the O-GlcNAcase derived from Clostridium perfringens (CpOGAD298N), which was capable of binding O-GlcNAc peptides and proteins specifically and with micromolar affinities.29,30CpOGAD298N can also act in a manner similar to O-GlcNAc pan-specific antibodies, contributing to the development of an O-GlcNAc enrichment and detection method at the level of the whole proteome.6,31
Here, we describe the synthesis of acrylamide-pendant inactive CpOGAD298N (AI-OGA) and its co-polymerization into polyacrylamide gels. Using AI-OGA, we developed an approach termed Separation of O-GlcNAcylated Proteins by Polyacrylamide Gel Electrophoresis (SOPAGE). We demonstrate that SOPAGE allows for the spatial separation of native O-GlcNAc proteins from their unmodified counterparts, with a mobility shift. This simple gel-based approach worked for either single- or multi-site O-GlcNAc model proteins, enabling the native detection of O-GlcNAcylation and determination of steady-state kinetics of OGT on native protein substrates.
Results and discussion
Immobilized AI-OGA reduces O-GlcNAc protein electrophoretic mobility
The principle of SOPAGE was outlined in Fig. 1a. The acryl-group was chosen for functionalization of the inactive CpOGAD298N mutant to prepare AI-OGA, because of its small size, lack of charge, stability in the absence of initiators (e.g., TEMED), and compatibility with other components used in PAGE. Synthesized AI-OGA was expected to copolymerize with acrylamide and bis-acrylamide during gel casting, forming a monolithic stationary phase. As such, O-GlcNAc proteins was anticipated to show reduced electrophoretic mobility as a result of continuous association-dissociation with immobilized AI-OGA.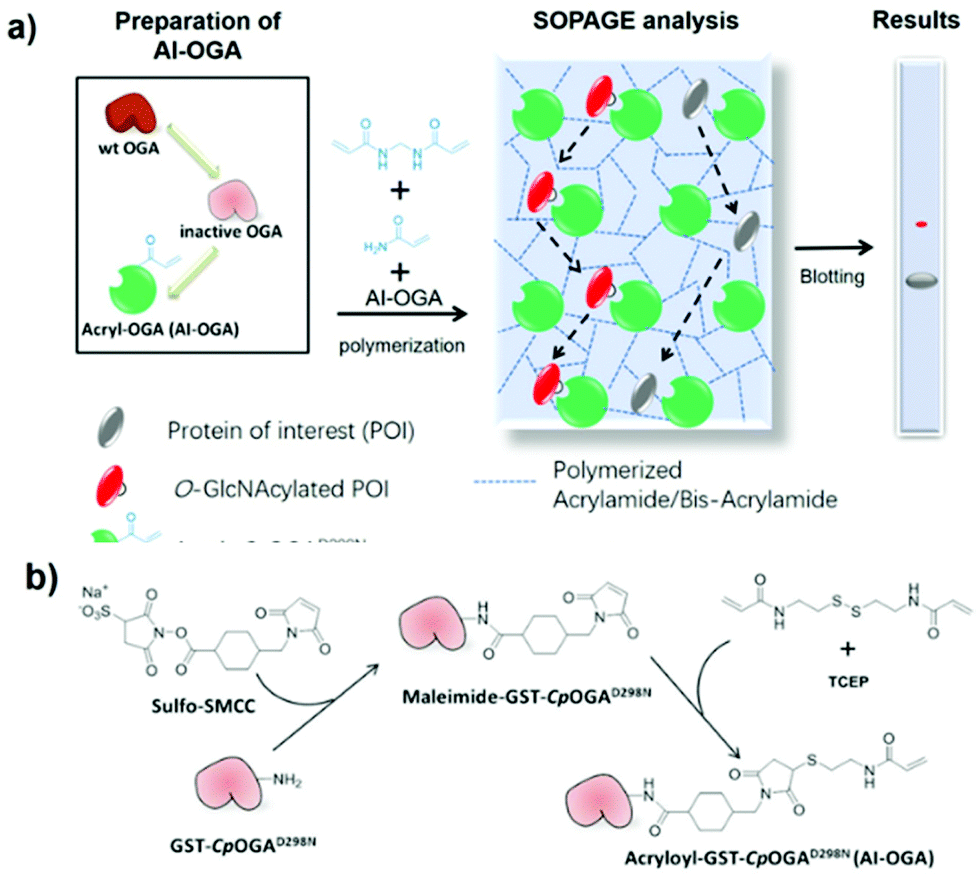 | ||
| Fig. 1 Schematic of the SOPAGE method (a) and synthesis of AI-OGA (b). | ||
AI-OGA was synthesized in a two-step reaction (Fig. 1b). Briefly, recombinant GST-CpOGAD298N expressed in E. coli (Fig. S1†) was first reacted with sulfo-SMCC in HEPES buffer (pH 7.5) to install maleimide groups onto the ε-amino group of lysine residues. Thereafter, excessive sulfo-SMCC was removed with NAP-5 column, and the buffer was changed to MES buffer (pH 5.2) to facilitate the subsequent reaction. Meanwhile, the disulfide bond in N,N′-bis(acryloyl)cystamine was reduced with TCEP in a parallel tube, and the resulting product with free thiol was added to maleimide-GST-CpOGAD298N to yield AI-OGA.
The resulting AI-OGA was characterized by function-based assays and mass spectrometry. First, we casted a two-layer gel with AI-OGA added to the upper-layer during polymerization (Fig. S2†). After electrophoresis, the gel was stained with Coomassie Blue to visualize the protein. As shown in Fig. 2a, the addition of AI-OGA did not affect gel polymerization, giving a monolithic gel sheet. Electrophoresis did not blur the boundary between the two layers (Fig. 2a), suggesting the absence of any non-functionalized GST-CpOGAD298N and unpolymerized AI-OGA. Subsequently, we demonstrated that AI-OGA undergoing the above chemical modification still retained its affinity for O-GlcNAc proteins by using gel-shift assays, in which AI-OGA formed a stable protein complex with a model protein gTAB1, but not with TAB1 (Fig. 2b and S3†). The acrylamide-modified sites of AI-OGA were then mapped with mass spectrometry and shown in Fig. 2c and detailed in ESI (Fig. S4 and Excel file).†
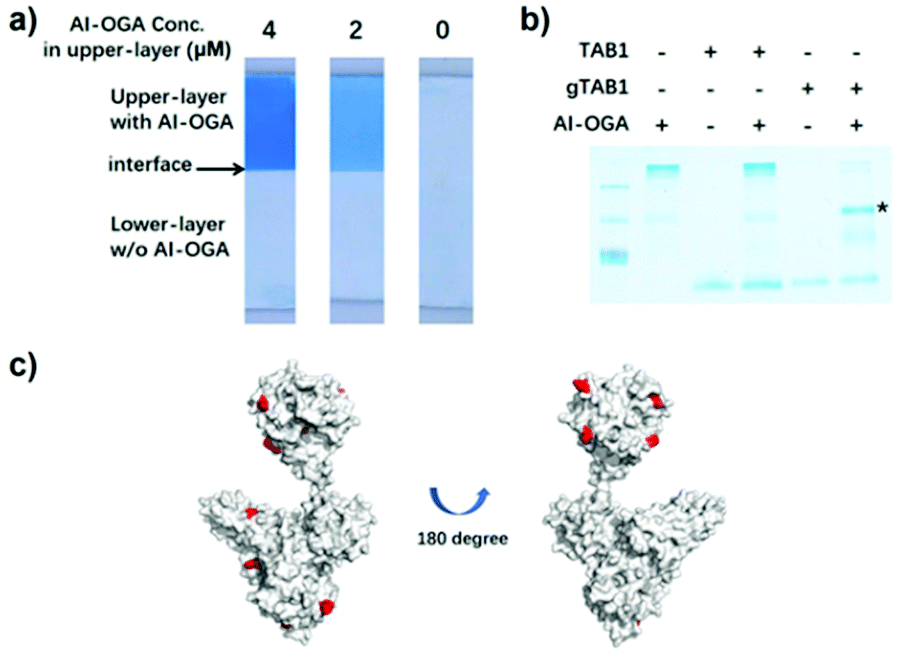 | ||
| Fig. 2 Characterization of AI-OGA. (a) Two-layer gels with AI-OGA added to the upper-layer at indicated concentration. The clear interface between two layers was indicated with arrow. (b) AI-OGA/gTAB1 complex revealed by gel-shift assay. Briefly, 10 pmol AI-OGA was mixed with 15 pmol of TAB1 or gTAB1. The mixture was subjected to native-PAGE and the stable complex was indicated with asterisk. (c) Acrylamide modified lysine residues on AI-OGA. The AI-OGA structure was modelled with PHYRE2 software using PDB 2CBJ and 4AKG as templates. 11 modified lysine with a Delta score > 40 were labelled in red. 6 of these sites located in the GST region. | ||
To assess the feasibility of SOPAGE analysis, AI-OGA was added at a concentration of 2 μM to the conventional gel mixture prior to initiation of polymerization with TEMED. As such, AI-OGA was covalently and uniformly embedded in a gel. Three model protein samples (TAB1, gTAB1 and gTAB1 treated with wild type CpOGA) were used for validation. TAB1, which acts as a signalling intermediate between TGFβ receptors and TAK1, is O-GlcNAcylated on Ser395, providing an ideal model for studying proteins with single O-GlcNAc moieties.23 gTAB1 was prepared by an in vitro reaction with OGT, followed by size-exclusion chromatography to remove OGT. Complete conversion of TAB1 to gTAB1 was achieved by adding an excess of OGT over prolonged reaction times (∼3 days). After that, proteins were subjected to SOPAGE, and then electroblotted onto nitrocellulose membranes. The addition of 0.04% (w/v) SDS to the transfer buffer was necessary for disrupting the interaction between the immobilized AI-OGA and O-GlcNAc proteins, facilitating electrotransfer. The membrane was then visualized with a mixture of anti-TAB1 and anti-gTAB1 antibodies. As shown in Fig. 3a, gTAB1 exhibited slower migration than TAB1, resulting in a markedly up-shifted band. Treatment of gTAB1 with CpOGA eliminated this migratory retardation. In addition, the level of separation of TAB1 and gTAB1 bands correlated with the AI-OGA concentration used in the gel (Fig. 3b). We next evaluated SOPAGE for multi-O-GlcNAcylated protein. To this end, the first PRO repeat of host cell factor 1 (HCF1-rep1) which bears >20 O-GlcNAc sites was chosen as a model.32 As a result, HCF1-rep1 was also spatial separated from its O-GlcNAcylated counterpart (Fig. S5†). Taken together, these data show that immobilized AI-OGA reduces the electrophoretic mobility of O-GlcNAc proteins, paving the way for native detection of O-GlcNAc levels.
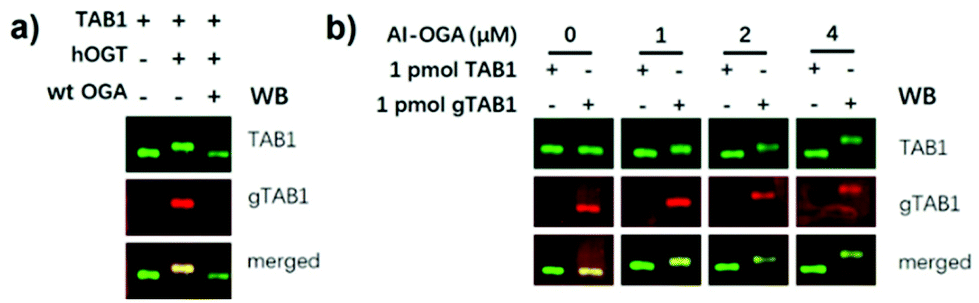 | ||
| Fig. 3 (a) Spatial separation of gTAB1 and TAB1 by SOPAGE. TAB1 was O-GlcNAcylated by hOGT to yield gTAB1. OGA treatment of gTAB1 rescued such retardation. (b) The migration distance between TAB1 and gTAB1 bands separated by SOPAGE was associated with the AI-OGA concentration (0–4 μM). | ||
SOPAGE quantification of O-GlcNAc stoichiometry allows determination of OGT kinetics
We next attempted to use SOPAGE to measure the kinetics of OGT on intact protein substrates. To this end, we first used SOPAGE to monitor the in vitro conversion of TAB1 over time by OGT. As shown in Fig. 4, approximate 70% of TAB1 was converted to gTAB1 within 24 h, which was consistent with recent results obtained by native protein MS.33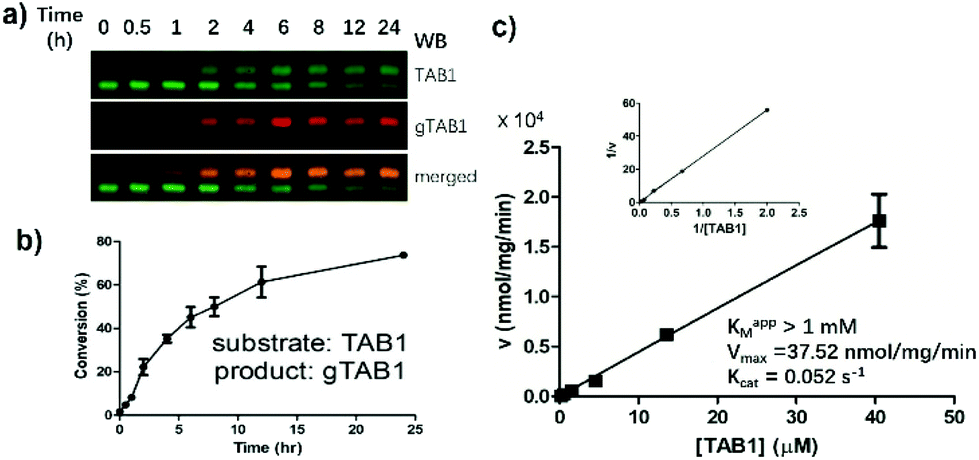 | ||
| Fig. 4 Measuring OGT kinetics by SOPAGE. (a) Monitoring hOGT reaction dynamics by SOPAGE. The signal was visualized with a mixture of TAB1 and gTAB1 Abs. (b) The conversion of TAB1 to gTAB1 by hOGT over time. (c) Determination of OGT steady-state kinetics for TAB1. The reactions were carried out with 0.17–41 μM TAB1, 100 μM of UDP-GlcNAc and 0.45 μM of hOGT at 37 °C for 1 h. After SOPAGE and blotting with TAB1 Abs, TAB1 and gTAB1 were quantified with Image Studio software. Kinetic parameters were calculated by double-reciprocal plotting. The turnover was <10% in all reactions. Assays in a-c were in triplicate. | ||
Next, a series of OGT reactions were set up with varying concentrations of TAB1, while the concentrations of hOGT and UDP-GlcNAc were kept constant. After SOPAGE and western blotting, the spatially separated TAB1 and gTAB1 bands in each lane were developed using anti-TAB1 as the sole primary antibody to allow signals to be compared. The band intensities of TAB1 (Ifast) and gTAB1 (Islow) were quantified, and the fraction of gTAB1 (gTAB1%) in each individual reaction was calculated by the following formula:
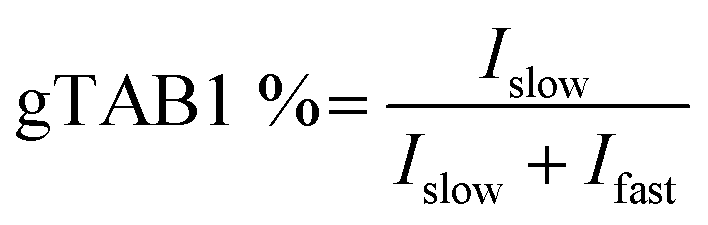
Plotting of initial velocities against TAB1 concentration allowed us to establish steady-state kinetics (Fig. 4c). Unlike when short peptides were used as substrates, this curve did not reach a plateau. Accordingly, we used a previously reported double-reciprocal plotting to calculate the kinetic parameters.18 The maximum velocity (Vmax), the apparent Michaelis constant (Km,app) and the catalytic rate constant (kcat) of hOGT for TAB1 were 37.52 nmol mg−1 min−1, >1 mM and 0.052 s−1, respectively. Taken together, SOPAGE quantification of O-GlcNAc stoichiometry allows determination of OGT kinetics using intact TAB1 as a substrate.
SOPAGE applies to a wide dynamic range of O-GlcNAc levels
In the above kinetics experiment, only low levels of O-GlcNAc modification (<10% turnover) were used to maintain steady-state conditions. We next challenged SOPAGE with samples of near-stoichiometric O-GlcNAc modification. In such cases, if the affinity of the ligand (AI-OGA in this study) was too weak, or the ligand co-migrated with O-GlcNAc proteins due to un-immobilization, the theoretical plate number of SOPAGE gel will decline,34 resulting in the failure to retard O-GlcNAc species. Besides, the applicability of SOPAGE to other types of proteins also needed to be evaluated. To this end, we chose substrate-hOGT fusions as models in which peptides derived from the widely studied O-GlcNAc proteins (TAB1, CK2, and CRMP2) were fused to the OGT N-terminus, yielding TAB1-hOGT, CK2-hOGT and CRMP2-hOGT, respectively. As we previously reported, these fusions were auto-glycosylated using endogenous UDP-GlcNAc as the donor during expression in E. coli, and showed high O-GlcNAc stoichiometry.35 We used these proteins together with an HCF1-hOGT fusion lacking an glycosylation site as a negative control, together with CpOGA treatment, in a SOPAGE experiment.As a result, the migration patterns of TAB1-hOGT, CK2-hOGT and CRMP2-hOGT were changed after CpOGA treatment, but not that of the negative control HCF1-hOGT (Fig. 5). Besides, the band intensities corresponding to the non-glycosylated forms were increased. The overall O-GlcNAc levels of these proteins purified from E. coli was determined to be 90 ± 5%, 58 ± 4%, and 58 ± 5%, based on triplicates. Taken together, these data show determination of O-GlcNAc levels by SOPAGE covers a wide dynamic range.
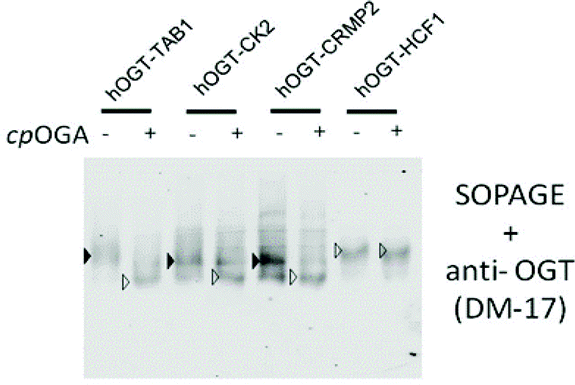 | ||
| Fig. 5 Determining O-GlcNAc stoichiometry of selected proteins. Peptide-hOGT fusions were analysed by SOPAGE and blotted with anti-OGT antibody. O-GlcNAcylated and non-glycosylated bands were identified by comparing the pattern before and after CpOGA treatment, and are indicated by solid and empty triangles, respectively. | ||
Conclusion
Compared to the limited methods developed for determining native protein O-GlcNAc stoichiometry, SOPAGE offers a number of advantages. First, SOPAGE only requires the addition of the AI-OGA monomer, obtainable to large quantities by recombinant expression (>40 mg L−1), to a standard PAGE gel. Second, SOPAGE does not require sophisticated sample processing by chemical and/or enzymatic methods, or any advanced equipment (e.g., mass spectrometer or radioactive detector) beyond a standard protein electrophoresis set up. This makes SOPAGE more convenient and economical that is accessible to any laboratory. Third, the inherent high affinity of CpOGAD298N, and its higher specificity than other affinity-ligands, such as WGA or succinylated WGA (sWGA), makes this approach specifically suitable for O-GlcNAc analysis.30 In most cases, a thin layer of SOPAGE gel (∼5 mm in length) between the stacking and separation gel was sufficient to separate O-GlcNAc proteins into a sharp band. In general, the detection limitation of SOPAGE is similar to that of conventional western blotting and is limited by the antibodies and instruments used (e.g. Li-Cor, chemiluminescence scanner or film) Last but not least, SOPAGE was compatible with many existing technologies because of the electrophoretic basis. For instance, in the future, SOPAGE could be combined with the Phospho-tag gel36 to differentiate phosphorylated and O-GlcNAcylated protein on the same gel, or integrated into “on-chip” electrophoresis and “μWestern” technology37 to enable miniaturization and automation of analysis.In summary, a facile, versatile, and economical gel-based method was developed for measuring OGT kinetics on protein substrates and determining native protein O-GlcNAc levels. This high-performance approach involved the retardation of electrophoretic mobility of O-GlcNAc proteins by immobilized O-GlcNAcase inactive mutant AI-OGA copolymerized with acrylamide/bis-acrylamide. In the future, the synthesized AI-OGA monomer could be exploited to develop a next-generation matrix for weak affinity chromatography. We envision that SOPAGE will be a useful tool to help to advance the understanding of the physiological functions of O-GlcNAcylation.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by a Wellcome Trust Senior Research Fellowship (110061) to D.M.F.v.A. We also thank Dr. A. Gorelik, Dr. M. Gundogdu, and Dr. A.T. Ferenbach for their helpful discussion and proofreading of the manuscript.Notes and references
- G. W. Hart, M. P. Housley and C. Slawson, Nature, 2007, 446, 1017 CrossRef.
- J. A. Hanover, M. W. Krause and D. C. Love, Biochim. Biophys. Acta, 2010, 1800, 80 CrossRef.
- P. Wang, B. D. Lazarus, M. E. Forsythe, D. C. Love, M. W. Krause and J. A. Hanover, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 17669 CrossRef.
- H. J. Tarbet, C. A. Toleman and M. Boyce, Biochemistry, 2018, 57, 13 CrossRef.
- N. Khidekel, S. B. Ficarro, E. C. Peters and L. C. Hsieh-Wilson, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 13132 CrossRef CAS.
- N. Selvan, R. Williamson, D. Mariappa, D. G. Campbell, R. Gourlay, A. T. Ferenbach, T. Aristotelous, I. Hopkins-Navratilova, M. Trost and D. M. F. Van Aalten, Nat. Chem. Biol., 2017, 13, 882 CrossRef CAS.
- C. M. Woo, P. J. Lund, A. C. Huang, M. M. Davis, C. R. Bertozzi and S. J. Pitteri, Mol. Cell. Proteomics, 2018, 17, 764 CrossRef CAS.
- N. E. Zachara and G. W. Hart, Biochim. Biophys. Acta, 2004, 1673, 13 CrossRef CAS.
- L. M. Andres, I. W. Blong, A. C. Evans, N. G. Rumachik, T. Yamaguchi, N. D. Pham, P. Thompson, J. J. Kohler and C. R. Bertozzi, ACS Chem. Biol., 2017, 12, 2030 CrossRef CAS.
- K. Vosseller, L. Wells, M. D. Lane and G. W. Hart, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 5313 CrossRef CAS.
- C. Slawson and G. W. Hart, Nat. Rev. Cancer, 2011, 11, 678 CrossRef CAS.
- M. Morris, G. M. Knudsen, S. Maeda, J. C. Trinidad, A. Ioanoviciu, A. L. Burlingame and L. Mucke, Nat. Neurosci., 2015, 18, 1183 CrossRef CAS.
- A. P. Willems, M. Gundogdu, M. J. E. Kempers, J. C. Giltay, R. Pfundt, M. Elferink, B. F. Loza, J. Fuijkschot, A. T. Ferenbach, K. L. I. van Gassen, D. M. F. van Aalten and D. J. Lefeber, J. Biol. Chem., 2017, 292, 12621 CrossRef CAS.
- N. Selvan, S. George, F. J. Serajee, M. Shaw, L. Hobson, V. Kalscheuer, N. Prasad, S. E. Levy, J. Taylor, S. Aftimos, C. E. Schwartz, A. M. Huq, J. Gecz and L. Wells, J. Biol. Chem., 2018, 293, 10810 CrossRef CAS.
- V. M. Pravata, V. Muha, M. Gundogdu, A. T. Ferenbach, P. S. Kakade, V. Vandadi, A. C. Wilmes, V. S. Borodkin, S. Joss, M. P. Stavridis and D. M. F. van Aalten, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 14961 CrossRef CAS.
- J. E. Rexach, P. M. Clark and L. C. Hsieh-Wilson, Nat. Chem. Biol., 2008, 4, 97 CrossRef CAS.
- J. Ma and G. W. Hart, Clin. Proteomics, 2014, 11, 8 CrossRef.
- W. A. Lubas and J. A. Hanover, J. Biochem., 2000, 275, 10983 CAS.
- J. W. Thompson, M. E. Griffin and L. C. Hsieh-Wilson, Methods Enzymol., 2018, 598, 101 CAS.
- K. Vosseller, J. C. Trinidad, R. J. Chalkley, C. G. Specht, A. Thalhammer, A. J. Lynn, J. O. Snedecor, S. Guan, K. F. Medzihradszky, D. A. Maltby, R. Schoepfer and A. L. Burlingame, Mol. Cell. Proteomics, 2006, 5, 923 CrossRef CAS.
- A. K. Nagel, M. Schilling, S. Comte-Walters, M. N. Berkaw and L. E. Ball, Mol. Cell. Proteomics, 2013, 12, 945 CrossRef CAS.
- C. F. Teo, S. Ingale, M. A. Wolfert, G. A. Elsayed, L. G. Nöt, J. C. Chatham, L. Wells and G.-J. Boons, Nat. Chem. Biol., 2010, 6, 228 CrossRef.
- S. Pathak, V. S. Borodkin, O. Albarbarawi, D. G. Campbell, A. Ibrahim and D. M. F. Van Aalten, EMBO J., 2012, 31, 1394 CrossRef CAS.
- R. J. Chalkley and A. L. Burlingame, J. Am. Soc. Mass Spectrom., 2001, 12, 1106 CrossRef CAS.
- J. J. P. Maury, D. Ng, X. Bi, M. Bardor and A. B. H. Choo, Anal. Chem., 2014, 86, 395 CrossRef CAS.
- J. E. Rexach, C. J. Rogers, S. H. Yu, J. Tao, Y. E. Sun and L. C. Hsieh-Wilson, Nat. Chem. Biol., 2010, 6, 645 CrossRef CAS.
- Y. Kubota, K. Fujioka and M. Takekawa, PLoS One, 2017, 12, e0180714 CrossRef.
- L. Leickt, M. Bergström, D. Zopf and S. Ohlson, Anal. Biochem., 1997, 253, 135 CrossRef CAS.
- F. V. Rao, H. C. Dorfmueller, F. Villa, M. Allwood, I. M. Eggleston and D. M. F. Van Aalten, EMBO J., 2006, 25, 1569 CrossRef CAS.
- M. Schimpl, V. S. Borodkin, L. J. Gray and D. M. F. Van Aalten, Chem. Biol., 2012, 19, 173 CrossRef CAS.
- D. Mariappa, N. Selvan, V. Borodkin, J. Alonso, A. T. Ferenbach, C. Shepherd, I. H. Navratilova and D. M. F. VanAalten, Biochem. J., 2015, 470, 255 CrossRef CAS.
- F. Capotosti, S. Guernier, F. Lammers, P. Waridel, Y. Cai, J. Jin, J. W. Conaway, R. C. Conaway and W. Herr, Cell, 2011, 144, 376 CrossRef CAS.
- A. C. Leney, K. Rafie, D. M. F. van Aalten and A. J. R. Heck, ACS Chem. Biol., 2017, 12, 2078 CrossRef CAS.
- Z. Li, J. Kim and F. E. Regnier, Anal. Chem., 2018, 90, 1668 CrossRef CAS.
- K. Rafie, O. Raimi, A. T. Ferenbach, V. S. Borodkin, V. Kapuria and D. M. F. van Aalten, Open Biol., 2017, 7(6), 170078 CrossRef.
- P. M. Clark, J. F. Dweck, D. E. Mason, C. R. Hart, S. B. Buck, E. C. Peters, B. J. Agnew and L. C. Hsieh-Wilson, J. Am. Chem. Soc., 2008, 130, 11576 CrossRef CAS.
- R. E. Gerver and A. E. Herr, Anal. Chem., 2014, 86, 10625 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9an02506e |
| This journal is © The Royal Society of Chemistry 2020 |