
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Study of the intracellular nanoparticle-based radiosensitization mechanisms in F98 glioma cells treated with charged particle therapy through synchrotron-based infrared microspectroscopy
I.
Martínez-Rovira
 *a,
O.
Seksek
bc,
I.
Dokic
de,
S.
Brons
d,
A.
Abdollahi
de and
I.
Yousef
a
*a,
O.
Seksek
bc,
I.
Dokic
de,
S.
Brons
d,
A.
Abdollahi
de and
I.
Yousef
a
aMIRAS beamline BL01, ALBA-CELLS Synchrotron, Carrer de la Llum 2-26, 08290 Cerdanyola del Vallès, Spain. E-mail: immamartinez@gmail.com
bUniversité Paris-Saclay, CNRS/IN2P3, IJCLab, 91405 Orsay, France
cUniversité de Paris, IJCLab, 91405 Orsay, France
dHeidelberg Ion Beam Therapy Center (HIT), Department of Radiation Oncology, Heidelberg University Hospital, 69120 Heidelberg, Germany
eClinical Cooperation Unite Translational Radiation Oncology, German Cancer Consortium (DKTK) Core Center, National Center for Tumor Diseases (NCT), Heidelberg University Hospital (UKHD) and German Cancer Research Center (DKFZ), 69120 Heidelberg, Germany
First published on 29th January 2020
Abstract
The use of nanoparticles (NP) as dose enhancers in radiotherapy (RT) is a growing research field. Recently, the use of NP has been extended to charged particle therapy in order to improve the performance in radioresistant tumors. However, the biological mechanisms underlying the synergistic effects involved in NP-RT approaches are not clearly understood. Here, we used the capabilities of synchrotron-based Fourier Transform Infrared Microspectroscopy (SR-FTIRM) as a bio-analytical tool to elucidate the NP-induced cellular damage at the molecular level and at a single-cell scale. F98 glioma cells doped with AuNP and GdNP were irradiated using several types of medical ion beams (proton, helium, carbon and oxygen). Differences in cell composition were analyzed in the nucleic acids, protein and lipid spectral regions using multivariate methods (Principal Component Analysis, PCA). Several NP-induced cellular modifications were detected, such as conformational changes in secondary protein structures, intensity variations in the lipid CHx stretching bands, as well as complex DNA rearrangements following charged particle therapy irradiations. These spectral features seem to be correlated with the already shown enhancement both in the DNA damage response and in the reactive oxygen species (ROS) production by the NP, which causes cell damage in the form of protein, lipid, and/or DNA oxidations. Vibrational features were NP-dependent due to the NP heterogeneous radiosensitization capability. Our results provided new insights into the molecular changes in response to NP-based RT treatments using ion beams, and highlighted the relevance of SR-FTIRM as a useful and precise technique for assessing cell response to innovative radiotherapy approaches.
1 Introduction
Radiotherapy (RT) is the most important non-surgical modality for cancer treatment since about 50% of cancer patients receive RT during the course of their illness. Latest technological advances in RT brought a substantial gain in the balance between the probability of tumor control and the risk of normal tissue complications. However, despite the use of state-of-the-art RT approaches, the clinical management of some radioresistant tumors (such as gliomas) continues being a challenge since tumor doses are constrained by the surrounding healthy tissue tolerances.The combination of high-atomic number (Z) nanoparticles (NP) as radiosensitizers in RT was born in the last decade with the main goal of enhancing the differential effect between the tumor and the surrounding healthy tissue.1 The size of the NP typically ranges between 1 and 100 nm. Small nanoparticles have a reduced tendency to accumulate in the body after treatment.2 NP leak preferentially into the tumor through active targeting or passively, through enhanced permeability and retention (EPR) effects.3
The therapeutic efficacy of RT in the presence of the NP has been proved in numerous studies using kilovoltage and megavoltage photon beams, both in vitro and in vivo (see ref. 4–6, among others). The initial rationale behind these studies is the enhanced photoelectric cross section of high-Z NP in comparison to water and soft tissue, specially at kilovoltage energies.7
However, despite extensive research on NP radiosensitization using photon beams, few experimental studies have been carried out in charged particle therapy. The pioneering study of Usami and collaborators showed an enhancement in DNA breaks at the molecular scale (plasmid DNA as a probe) using platinum-based complexes combined with helium and iron irradiations.8 The efficacy of the same Pt complexes was later proved in vitro by the same group.9 An enhancement of radiation effects was also demonstrated in proton therapy10–14 and carbon therapy15–18 in several biological studies using different types of high-Z NP. An overview of experimental studies on radiosensitization effects of high-Z compounds in charged particle therapy can be found in the review of Lacombe and collaborators.19
The mechanisms involved in the NP-induced amplification of radiation effects in charged particle therapy is still a matter of controversy since they seem to differ from the ones in standard photon RT. Moreover, the radiosensitization effects involved might be distinct from the well-established pathway of radiation-induced direct DNA damage.20 The amplified radiation damage in the presence of NP was initially associated to the increase in the ionizations and electron emissions induced by the incident ions or the electrons in the secondary tracks on the NP, resulting in a local dose enhancement.15 Recent studies showed a significant increase in the yield of low-energy secondary electrons emitted due to collective electron excitations in the NP (plasmons).21 Other authors proposed that NP could be activated by projectile induced X-rays emission (PIXE), being the origin of intra-atomic cascades of electron emissions.10 At the same time, these electrons enhance cell damage through the increased production of reactive oxygen species (ROS). Indeed, several studies have pointed out the important role of hydroxyl radicals.9,13,14 Furthermore, emerging data supported that radiosensitization effects might be also driven by biological pathways, such as oxidative stress, DNA damage induction, cell cycle effects and potential interference with cell communication.22–25
Both radiation chemistry and biological mechanisms, as well as distinct cell uptake and cell sensitivity as a function of NP type (size, shape, coating, etc.), should be considered since pure physical dose enhancement cannot explain the amplification effect observed in biological studies.26 Therefore, there is an urgent need to understand the mechanisms involved in these novel approaches to progress on the development of NP-based therapeutics.
Within this context, the objective of our study is to shed some light on the NP-based radiosensitization mechanisms at the molecular level in charged particle therapy through synchrotron-based Fourier Transform Infrared Microspectroscopy (SR-FTIRM). Infrared microspectroscopy has become a powerful tool for examining composition and/or conformational changes in biomolecules, including proteins, lipids, carbohydrates and nucleic acids.27 FTIRM methods have been extensively used to study cell functionality, cell cycle, cell death and apoptosis, as well as cell and tissue responses to different treatments (see ref. 27–32, among others). The brilliance of the infrared synchrotron radiation delivered at the MIRAS beamline of ALBA synchrotron allowed an infrared spectroscopic microanalysis at the single-cell level.
Our previous FTIRM works provided new insights into the radiosensitization effects of gold NP (AuNP) and gadolinium NP (GdNP) in photon RT.33–35 In the present study, radiosensitization effects of 1.9 nm AuNP (AuroVist®) and 3 nm GdNP (AGuIX®) were assessed in F98 glioma cells using several charged particle beams (proton, helium, carbon and oxygen). The potential of both NP was demonstrated in previous biological studies.36–39 However, to the best of our knowledge, this is the first study that evaluates radiosensitization of NP in charged particle therapy using SR-FTIRM.
2 Materials and methods
2.1 F98 cell culture
The F98 rat cell line (ATCC® CRL-2397, LGC Standards, Molsheim, France) used in this study is a glioblastoma cell type displaying a tumor growth infiltrative pattern within brain tissue.F98 cells were cultured in high glucose (4.5 g L−1) DMEM growth medium supplemented with 10% Fetal Calf Serum, 1% penicillin–streptomycin, 1% glutamine and 1% sodium pyruvate at 37 °C with 5% CO2 and 95% humidity. Prior to experimentation, cells were plated in order to be kept in the exponential growing phase at around 75–80% confluence. Two days prior to irradiation, 500 μL of a 4 × 105 cells per mL suspension were incubated in each well (48-well microplates) at 37 °C with 5% CO2 in order to reach a 75–80% confluence rate on the irradiation day.
2.2 Gd and Au nanoparticles
Before the irradiation protocol, cells were treated with AuNP or GdNP following our previous protocols.33–35 Gold nanoparticles (1.9 nm AuNP, AuroVist®) were purchased from Nanoprobes (Yaphank, NY). Prior to treatment, 40 mg of AuNP were rapidly dissolved in 0.2 mL PBS and filtered through a 0.2 μm centrifugal filter at 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000g for 8 min. From this stock solution kept at 4 °C, 500 μg mL−1 were applied on the cells during 24 hours prior to irradiation. As for gadolinium nanoparticles (3 nm GdNP, AGuIX®), which were purchased from NH TherAguix (Lyon, France), a 100 mM stock solution was obtained by solubilization with PBS and kept at 4 °C. From this solution, 1 mM NP solution was applied on the cells during 6 hours prior to irradiation. The biochemical effects induced by the NP prior to the irradiations were previously assessed by means of SR-FTIRM and are reported in our previous studies.33–35
000g for 8 min. From this stock solution kept at 4 °C, 500 μg mL−1 were applied on the cells during 24 hours prior to irradiation. As for gadolinium nanoparticles (3 nm GdNP, AGuIX®), which were purchased from NH TherAguix (Lyon, France), a 100 mM stock solution was obtained by solubilization with PBS and kept at 4 °C. From this solution, 1 mM NP solution was applied on the cells during 6 hours prior to irradiation. The biochemical effects induced by the NP prior to the irradiations were previously assessed by means of SR-FTIRM and are reported in our previous studies.33–35
2.3 Cell irradiations
Irradiations were carried out at the fixed horizontal beam experimental station of the Heidelberg Ion Beam Therapy Center (HIT). Several beams were used for the irradiations: proton (p), helium (4He), carbon (12C) and oxygen (16O). The well plates were vertically irradiated and placed at the center of a 10 mm-long spread out Bragg peak (SOPB) region centered at 80 mm-depth. The following physical (absorbed) doses were applied (the energy range, E, and mean dose-averaged LET values are detailed between brackets):• p: 4.5, 9, 18.2 Gy (E = 101.9–108.2 MeV u−1; LET = 5 keV);
• 4He: 3.3, 6.7, 13.3 Gy (E = 101.6–107.9 MeV u−1; LET = 22 keV);
• 12C: 2.5, 5, 10 Gy (E = 194.9–206.9 MeV u−1; LET = 96 keV);
• 16O: 1.7, 3.3, 6.7 Gy (E = 228.2–244.2 MeV u−1; LET = 140 keV);
in order to achieve effective biological doses of approximatively 5 GyRBE, 10 GyRBE and 20 GyRBE for each beam configuration. SOBP beam intensities and energies, (dose-averaged) LET values, as well as physical doses were computed using the experimental planning program TRiP.40–42 The range of doses was selected according to previous radiobiological studies.42,43
2.4 SR-FTIRM sample preparation
24 hours after the irradiations, cells were specifically prepared for SR-FTIRM measurements following our previous protocols.33–35 First, the medium was removed and 100 μL of 0.05% Trypsin-EDTA solution were added to each well and incubated for 5 minutes. Then, 500 μL of supplemented DMEM medium were added and the cell suspension was centrifuged at 1500 rpm for 5 minutes. After discarding the supernatant, the cellular pellet was suspended in PBS and centrifuged again at 1500 rpm for 5 minutes. The final pellet was then re-suspended in 10% formalin neutral buffered solution and incubated for 1 hour at room temperature. Then, samples were rinsed three times in deionized water. Finally, SR-FTIRM was performed on dried cells deposited onto CaF2 coverglasses.2.5 SR-FTIRM data acquisition and data analysis
The Hyperion 3000 microscope coupled to Vertex 70 spectrometer (Bruker Optik GmbH, Germany) installed at the infrared beamline MIRAS of ALBA synchrotron light source was used to perform the SR-FTIRM measurements. The infrared spectra were collected using the transmission operation mode of the FTIR microscope. This microscope is equipped with a 50 μm liquid nitrogen cooled mercury cadmium telluride (MCT) detector optimized for operation in a range covering the mid-infrared spectral region. The FTIRM system is operating with a 36× Schwarzschild magnification objective (NA = 0.65) coupled to a 36× magnification condenser.Between 100 to 120 cells were randomly selected to undergo SR-FTIRM measurements from each sample condition. The SR-FTIRM spectra were acquired using a single masking aperture size of 7 μm × 7 μm. Single point measurements of individual cells were obtained in the mid-infrared range at 4 cm−1 spectral resolution with 128 co-added scans per spectrum. A background spectrum was collected every 5 measurements in order to eliminate the residual contamination (water vapor, CO2) of the room ambient.
Principal Component Analysis (PCA) was performed using the Orange software (Bioinformatics Lab, University of Ljubljana, Slovenia)44 in order to evaluate the variations in the spectral features. Prior to the PCA application, the data was pre-processed by vector normalized second derivative spectra (Savitzky–Golay algorithm; 9 smoothing points; 3rd polynomial order). Vector normalization and PCA were separately performed in the 3025–2800 (‘lipids’) and 1800–950 cm−1 (‘fingerprint’) spectral regions.
A curve-fitting analysis was carried out on the amide I band to evaluate the relative attribution of each component representing a type of secondary structure. The second derivative function was used to assess the minima and, thus, to define the position and number of Gaussian function energy components needed to fit the curve of amide I. The Origin 7.0 software (OriginLab Corporation, Northampton, USA) was used to perform the curve-fitting method. A linear baseline was performed in the amide I range prior to curve-fitting.
In order to evaluate the area under the main spectral bands (see Table 1), the Rubber-band baseline correction method (32 baseline points) was applied to the raw spectra using Orange.44 Then, violin plots were generated to assess the probability density of the data for several spectral ratios that were used as specific markers for indices of the biochemical changes. More details can be found in section 3.
| Main band assignment | Specta region |
|---|---|
| ν sPO2−; deoxyribose/ribose ν C–O (phosphate II; PhII) | 1146–1004 |
| ν asPO2− (phosphate I; PhI) | 1270–1186 |
| δ N–H; ν C–N (proteins: Amide II, AII) | 1590–1483 |
ν C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (proteins: Amide I, AI) O (proteins: Amide I, AI) |
1710–1598 |
|
ν CO ester (lipids) |
1760–1730 |
| ν sCH2 (lipids) | 2862–2842 |
| ν sCH3 (lipids) | 2882–2862 |
| ν asCH2 (lipids) | 2945–2900 |
| ν asCH3 (lipids) | 2980–2945 |
| ν = C–H (lipids) | 3018–3002 |
3 Results and discussion
SR-FTIRM analysis is presented for several beams (p, 4He, 12C, 16O) and doses in the presence (+NP) and absence (−NP) of nanoparticles. SR-FTIRM details on the biochemical effects induced by the GdNP and AuNP are reported separately for the fingerprint (section 3.1.1) and lipids (section 3.1.2) spectral regions.3.1 SR-FTIRM results on the radiosensitization mechanisms of GdNP and AuNP in charged particle therapy
PCA on the Savitzky–Golay second derivative data was employed as an unsupervised method to classify the data and identify the main sources of variation in the SR-FTIRM spectra in the presence of nanoparticles (GdNP or AuNP). Fig. 1–4 depict the PCA scores in the fingerprint (1800–950 cm−1) and in the lipids (3025–2800 cm−1) spectral regions for p, 4He, 12C and 16O irradiations, respectively. The corresponding loading plots are also shown, which represent the influence of the variables (wavenumbers) in each principal component. The first two principal components (PC-1 and PC-2) were used to explain the differences among the samples. The averaged Savitzky–Golay second derivative is also presented in the proteins, DNA and lipids regions in Fig. 5.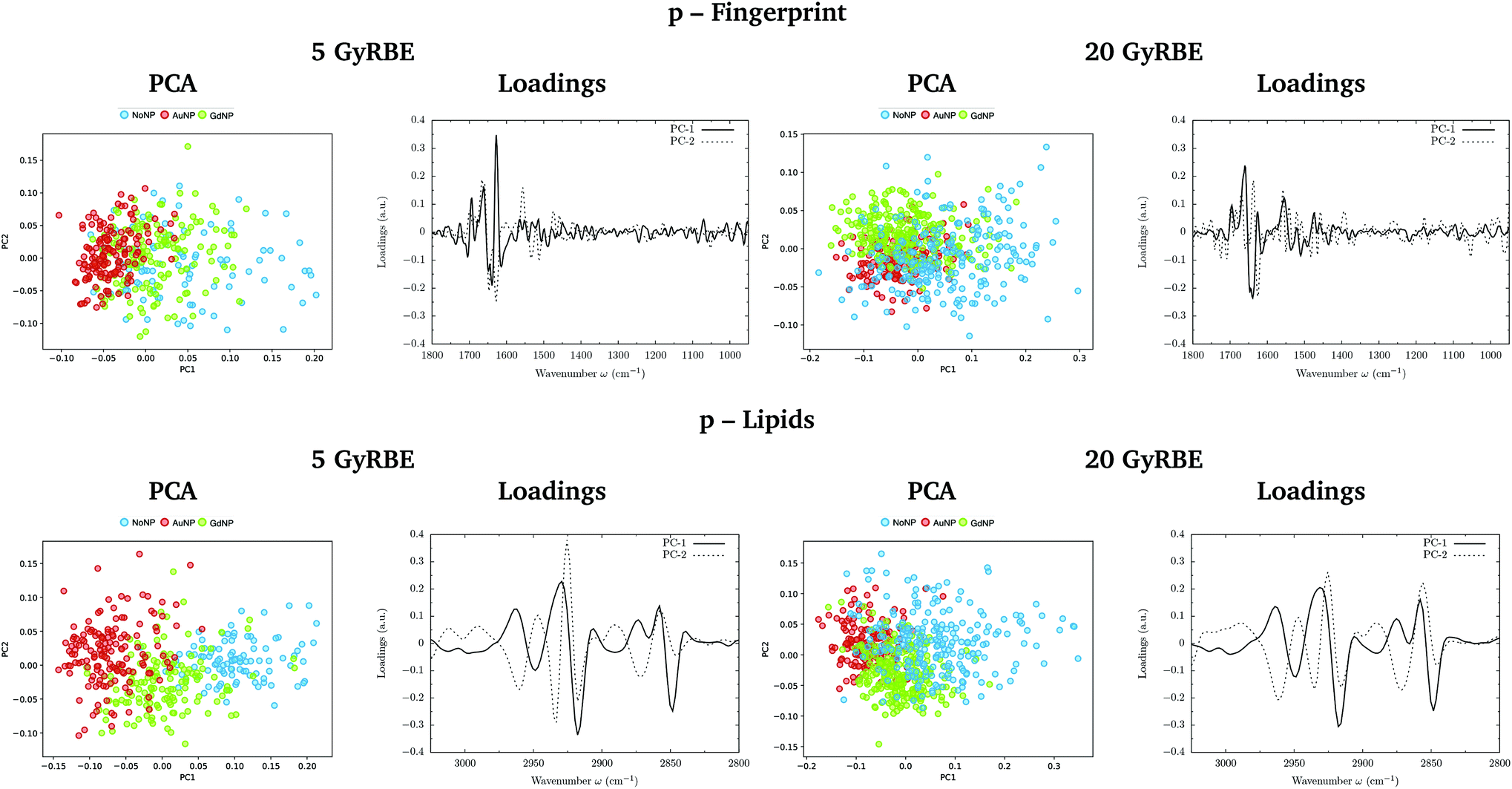 | ||
| Fig. 1 Proton beam irradiations. SR-FTIRM results on the radiosensitization mechanisms of GdNP and AuNP in charged particle therapy. The PCA scores and the loading plots are presented in the fingerprint (1800–950 cm−1, top) and lipids (3025–2800 cm−1, bottom) spectral regions in the absence (NoNP; blue) and in the presence of AuNP (red) and GdNP (green) for 5 GyRBE (left) and 20 GyRBE (right). | ||
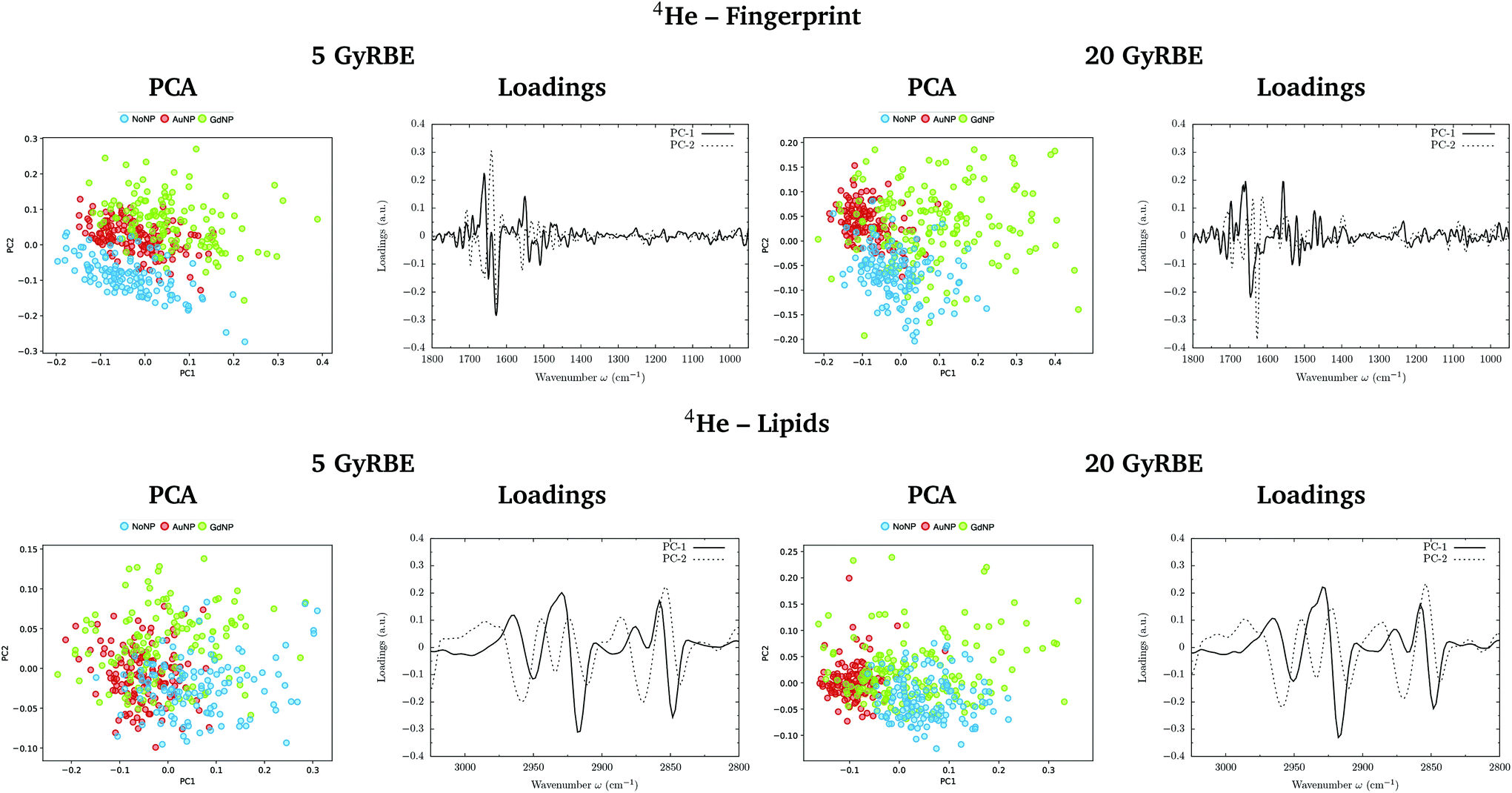 | ||
| Fig. 2 Helium beam irradiations. SR-FTIRM results on the radiosensitization mechanisms of GdNP and AuNP in charged particle therapy. The PCA scores and the loading plots are presented in the fingerprint (1800–950 cm−1, top) and lipids (3025–2800 cm−1, bottom) spectral regions in the absence (NoNP; blue) and in the presence of AuNP (red) and GdNP (green) for 5 GyRBE (left) and 20 GyRBE (right). | ||
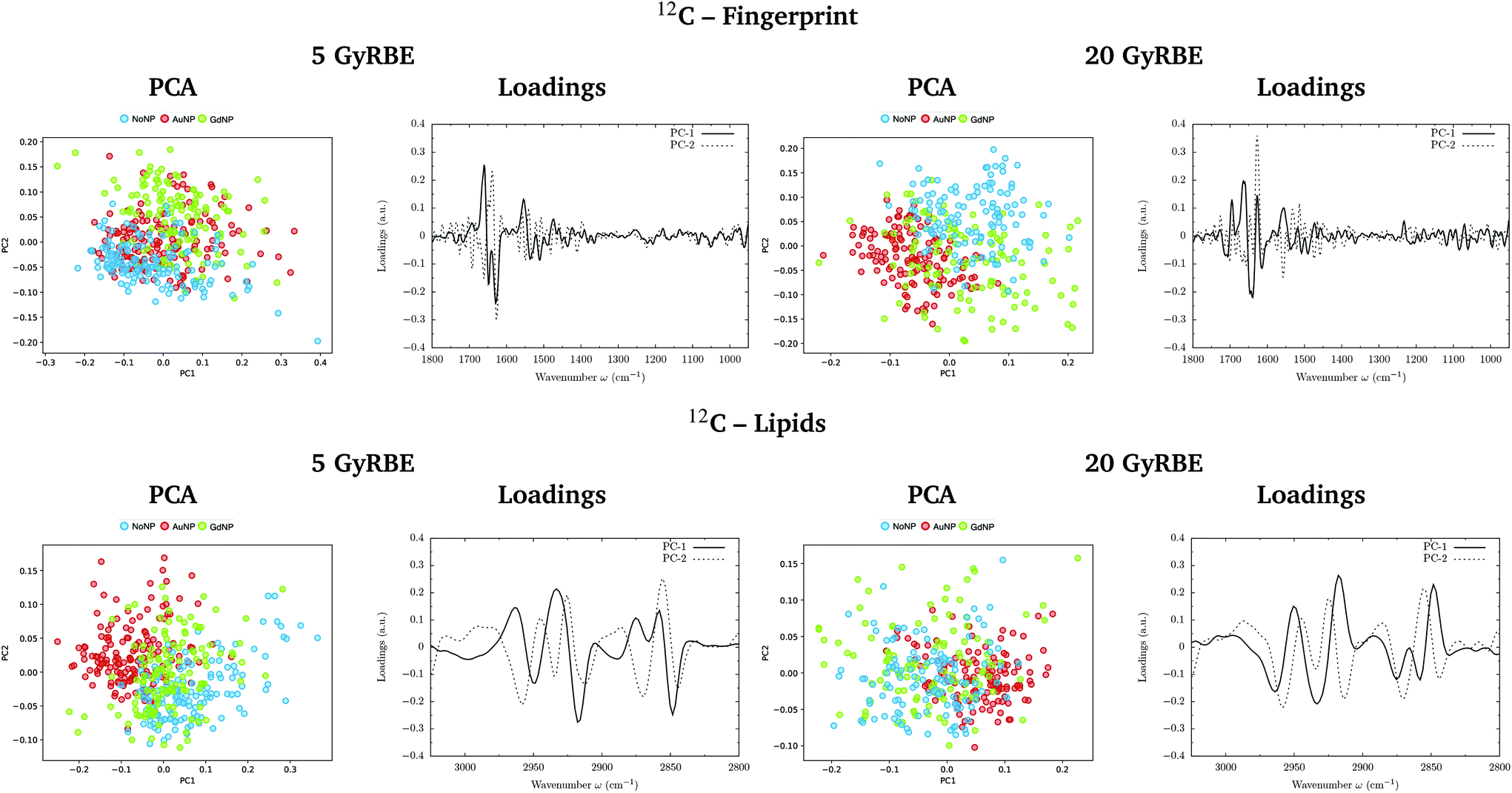 | ||
| Fig. 3 Carbon beam irradiations. SR-FTIRM results on the radiosensitization mechanisms of GdNP and AuNP in charged particle therapy. The PCA scores and the loading plots are presented in the fingerprint (1800–950 cm−1, top) and lipids (3025–2800 cm−1, bottom) spectral regions in the absence (NoNP; blue) and in the presence of AuNP (red) and GdNP (green) for 5 GyRBE (left) and 20 GyRBE (right). | ||
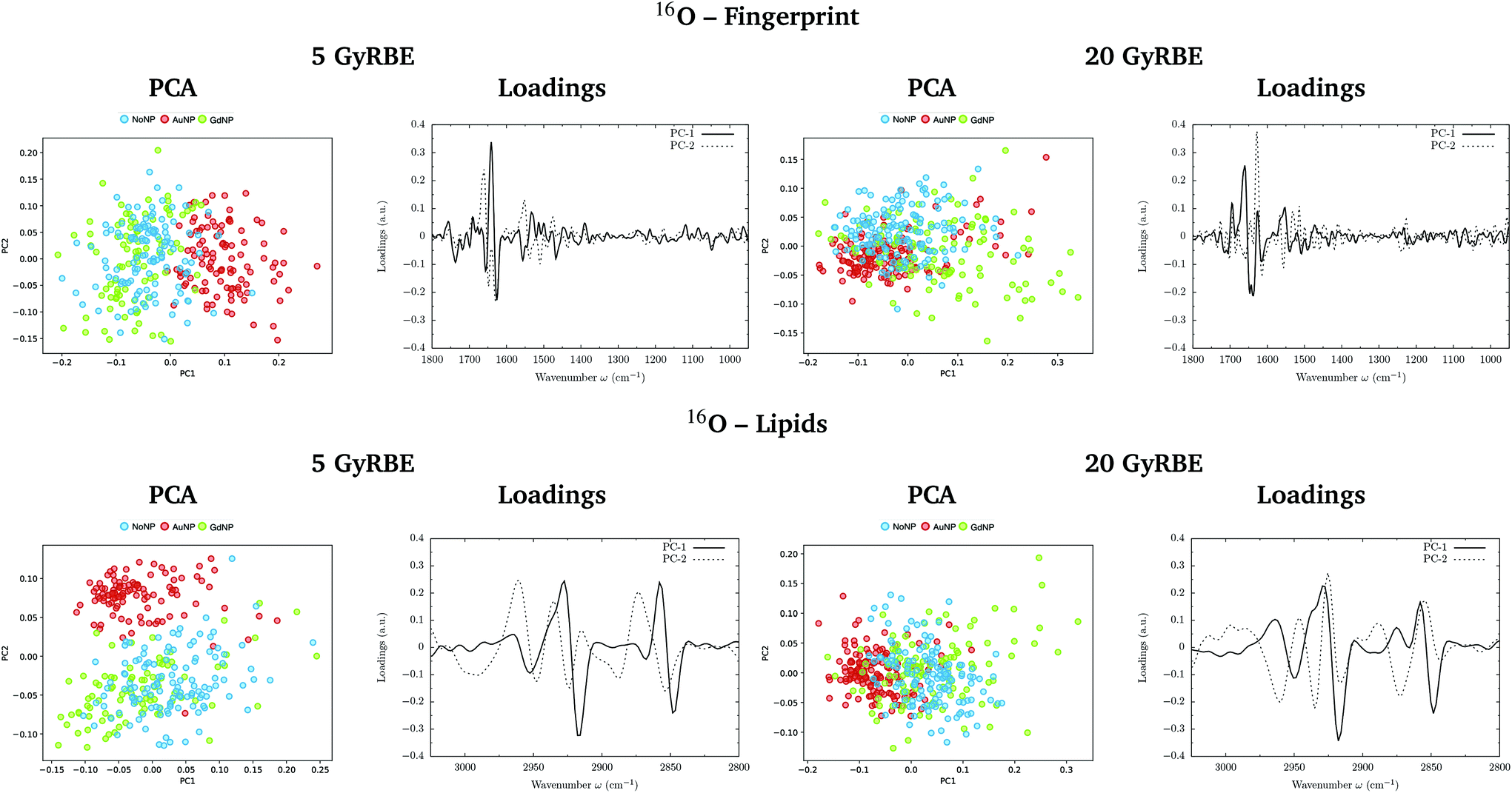 | ||
| Fig. 4 Oxygen beam irradiations. SR-FTIRM results on the radiosensitization mechanisms of GdNP and AuNP in charged particle therapy. The PCA scores and the loading plots are presented in the fingerprint (1800–950 cm−1, top) and lipids (3025–2800 cm−1, bottom) spectral regions in the absence (NoNP; blue) and in the presence of AuNP (red) and GdNP (green) for 5 GyRBE (left) and 20 GyRBE (right). | ||
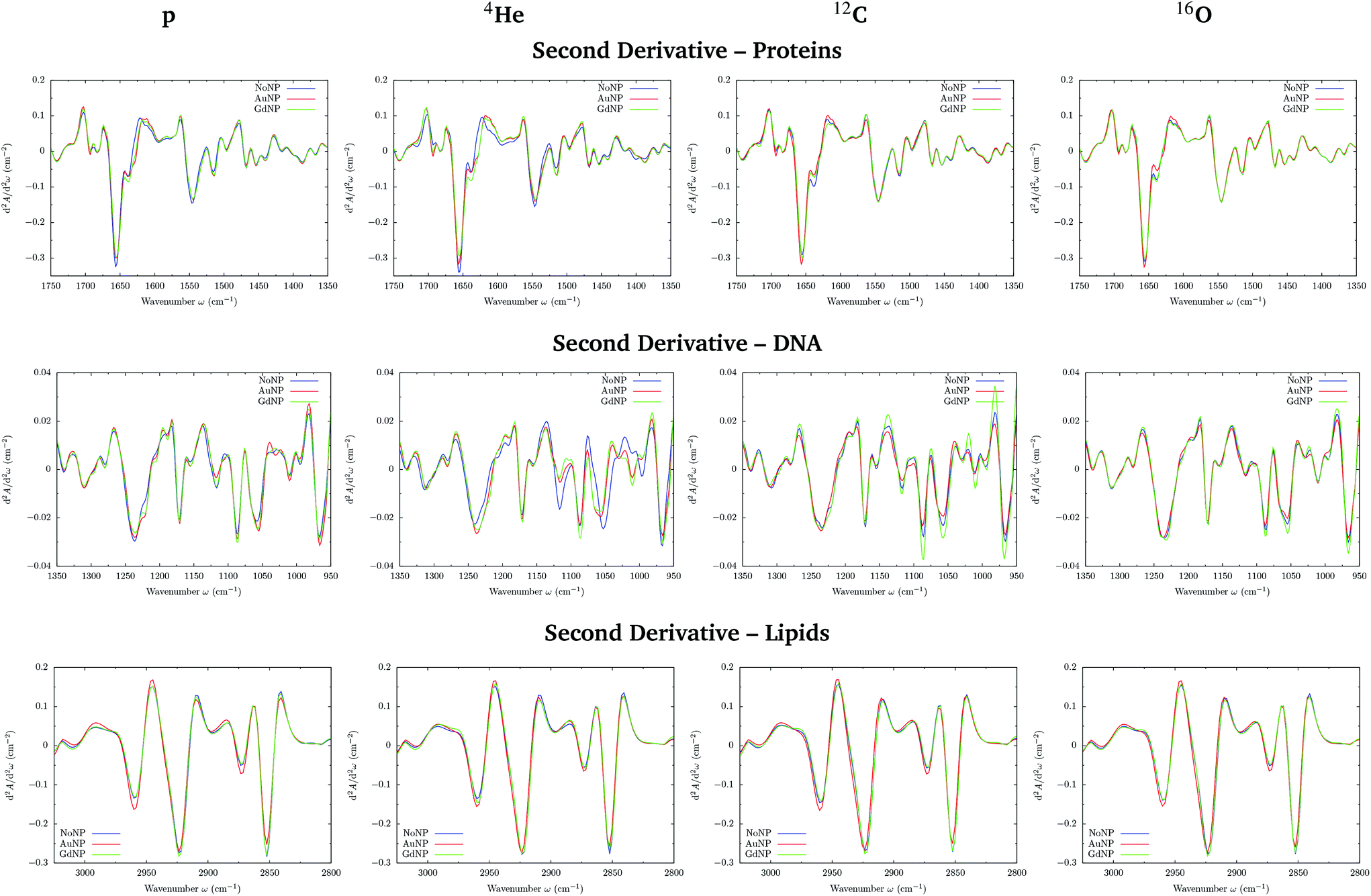 | ||
| Fig. 5 All ions (p, 4He, 12C, 16O; from left to right). SR-FTIRM results on the radiosensitization mechanisms of GdNP and AuNP in charged particle therapy. Savitzky–Golay second derivative of the averaged absorbance spectra in the absence (NoNP; blue) and in the presence of AuNP (red) and GdNP (green) in the proteins (1700–1350 cm−1, top), DNA (1350–950 cm−1; middle) and lipids (3025–2800 cm−1, bottom) spectral regions. | ||
O stretching vibration of amide groups of proteins, weakly coupled with the in-plane NH-bending and the CN-stretching modes.50 The amide II band, located in the 1600–1480 cm−1 region, is mainly due to the combination of NH-bending and CN-stretching vibrations.50 Both the amide I and II bands provide valuable information on the secondary structure of proteins.50 The region between 1350–950 cm−1 consists in a complex bands pattern due to contributions from carbohydrates and phosphates associated with nucleic acids (DNA, RNA). The main bands at 1085 cm−1 and 1238 cm−1 are assigned to the symmetric and asymmetric stretching vibrations of phosphodiester bonds in nucleic acids. Finally, the band in the region 1760–1730 cm−1 is related to the carbonyl ester group stretching in lipids and it will be discussed in the lipids section (3.1.2).
PCA results performed on the Savitzky–Golay second derivative (Fig. 1–4, top) show differences between NP-treated (AuNP and GdNP) groups and NoNP groups for all ion types, with some overlapping in some of the configurations. An analysis of the PCA loading plots reveals that most of the variance accounting for the separation between groups is associated with protein modifications in the region 1700–1600 cm−1 (amide I). The percentage of the total variance described by the first and second principal components is 60–80% and 30–10%, respectively, depending on the configuration.
Fig. 5 (top) shows the vector normalized Savitzky–Golay second derivative of the averaged absorbance spectra in the protein spectral region. The conformation changes in the amide I can be clearly seen in both NP treatments, especially in the case of proton and helium irradiations. Spectra deconvolution of the amide I region was performed by means of curve-fitting to precisely evaluate the differences among the secondary structures of proteins for the several NP treatments, as it can be seen in Fig. 6. The deconvoluted spectra of the amide I band showed a composite of six bands, which are reported in Table 2.51,52 Changes in the band around 1628 cm−1 were observed, which is assigned to the vibrational modes of β-sheet structural components.51,52 The β-sheet intensity increased upon both NP treatments with respect to the absence of NP. Previous works correlated an increase in the β-sheet secondary structure with the hallmark characteristics of cell death,32,53 as well as to lipid-auto-oxidation products that deform the protein secondary structure.54 Therefore, the modifications encountered in the protein spectral region could be correlated to an increase in the ROS production in the presence of NP, which might cause cell damage in the form of protein oxidation (as well as lipids and DNA oxidations), leading to an enhancement in the cell death. We also observed a reduction in the random coil structure content of proteins, also previously detected in dying cells.55
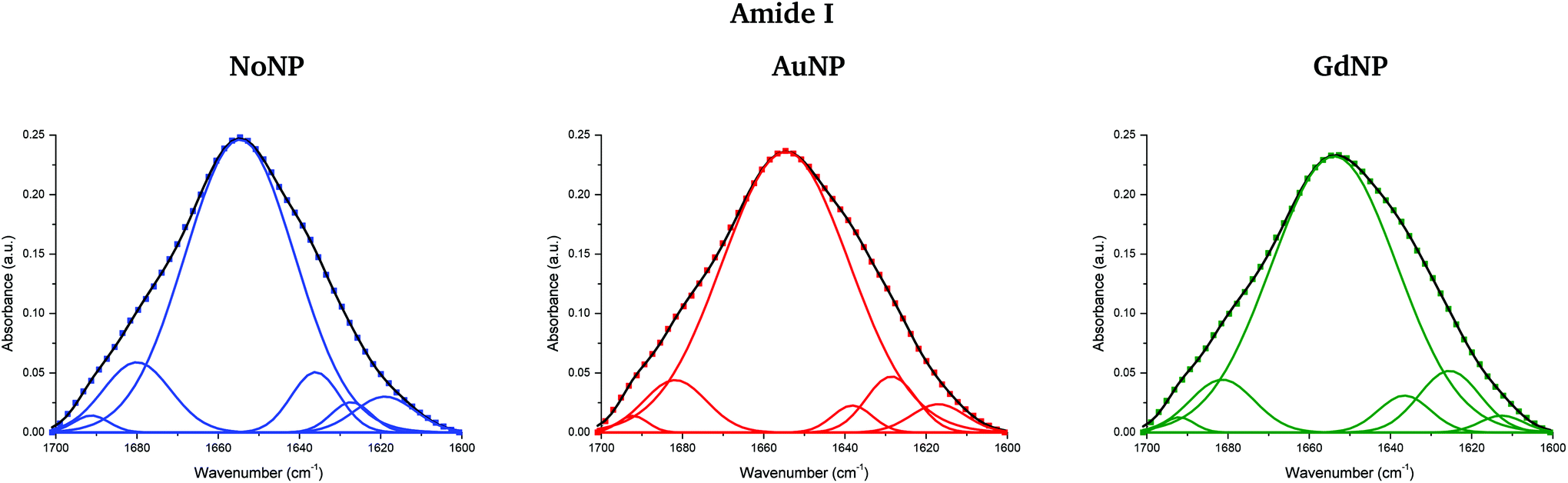 | ||
| Fig. 6 Curve-fitting analysis of the amide I band in the absence (NoNP; blue) and in the presence of AuNP (red) and GdNP (green) (4He). The baseline-corrected spectrum was fitted with six Gaussian band profiles by approximating the number and their positions using the minima of the second derivative function, which fitted the experimental curve (in black). | ||
| Band assignment | Wavenumber |
|---|---|
| Side chain | 1619 cm−1 |
| β-Sheet | 1628 cm−1 |
| Random coil | 1638 cm−1 |
| α-Helix | 1655 cm−1 |
| β-Turn | 1680 cm−1 |
| β anti-parallel sheet | 1691 cm−1 |
The ratio of amide I to amide II was also used to show conformational changes of proteins, since it is directly related to the secondary structure of proteins.56 In the violin plots in Fig. 7, we can clearly observe an increase in the amide I to amide II ratio, especially in the case of AuNP-treated cells. Following previous works,56 these results seem to indicate that cellular protein modifications in the folding and localization have occurred following an enhanced cell death response induced by the presence of the NP. Other works correlated the changes in the amide I and II absorbances with the DNA repairing processes as a result of the radiation damage produced.57
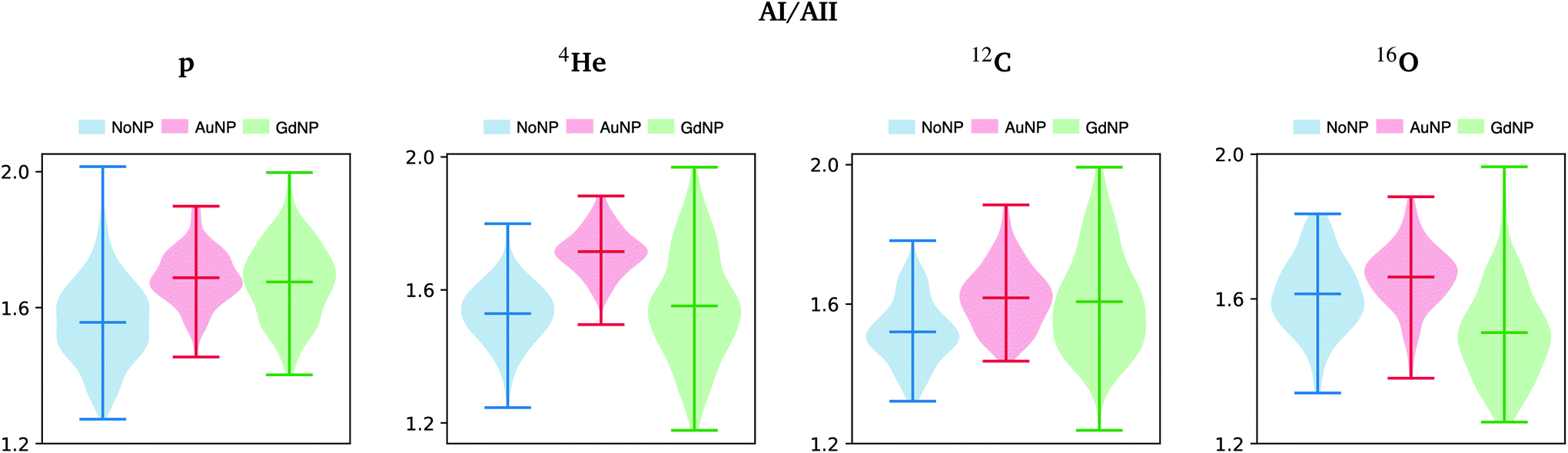 | ||
| Fig. 7 All ions (p, 4He, 12C, 16O; from left to right); 20 GyRBE. SR-FTIRM results on the radiosensitization mechanisms of GdNP and AuNP in charged particle therapy. Distribution of the relative intensities of the amide I (AI) to amide II (AII) spectral band ratio in the absence (NoNP; blue) and in the presence of AuNP (red) and GdNP (green). See Table 1 for more information on the bands assignment. | ||
In the DNA region, several spectral areas appeared to be modified upon NP treatment for all ion beams, as it can be seen in the vector normalized Savitzky–Golay second derivative of the averaged absorbance spectra (see Fig. 5, middle). The multiple changes in the absorbance and position (shift) of various peaks probably indicate a number of different modifications of the DNA conformational changes and/or rearrangements attributed to distortions of the DNA double helix in the presence of NP.
The modifications and the downward shift in the νasPO2− (named phosphate I) band at 1238 cm−1 suggest that the nucleic acid phosphodiester groups suffered conformation modifications in the DNA backbone, such as partial transitions between B-DNA to A-DNA structures due to nucleotide base damage.46,57,58 We also observe changes in the symmetric phosphate stretching band of phosphodiester nucleic acids (1085 cm−1; phosphate II), which was correlated with the degree of intermolecular interactions in the nucleic acids.28
A series of peaks related to ribose and deoxyribose –C–O and –PO2 stretching vibrations are located in the 1170–995 cm−1 range. The second derivative plot shows changes in these infrared bands in NP-treated cells, specially in the peaks around 1060 and 1020 cm−1, suggesting changes in the deoxyribose/ribose structures. They were reported to be correlated with non-repaired damage in the DNA structure.53 In particular, the furanose C–O stretching at 1020 cm−1 is an important marker for base-pairing and base-stacking in RNA.59 This might indicate base cleavage reactions, probably due to ROS damage or DNA repairing processes.60 Previous articles related these changes with modifications in the structure of the RNA that could induce the formation of altered proteins as a result of the incorrect translation processes.59 We observed an increase in such modifications with the presence of both types of NP.
The band near 965 cm−1 is assigned to the ν C–C/C–O stretching vibration involved in the deoxyribose and phosphate moiety of the DNA backbone.47 Modifications in this band were previously related to single-strand breaks (SSB), double-strand breaks (DSB), crosslinks and deoxyribose damage.28,57,60,61 The intensities in the phosphate I and phosphate II bands (with respect to the amide II band) correlate inversely with cell death, as detailed in previous works.46,56 In the violin plots of Fig. 8, AuNP-induced DNA damage is expressed by the decrease in the PhI and PhII bands, which might indicate DNA condensation.28,29,46,53,56,57 A reduction on the absorbance of the phosphate I band was also reported upon oxidative stress.48
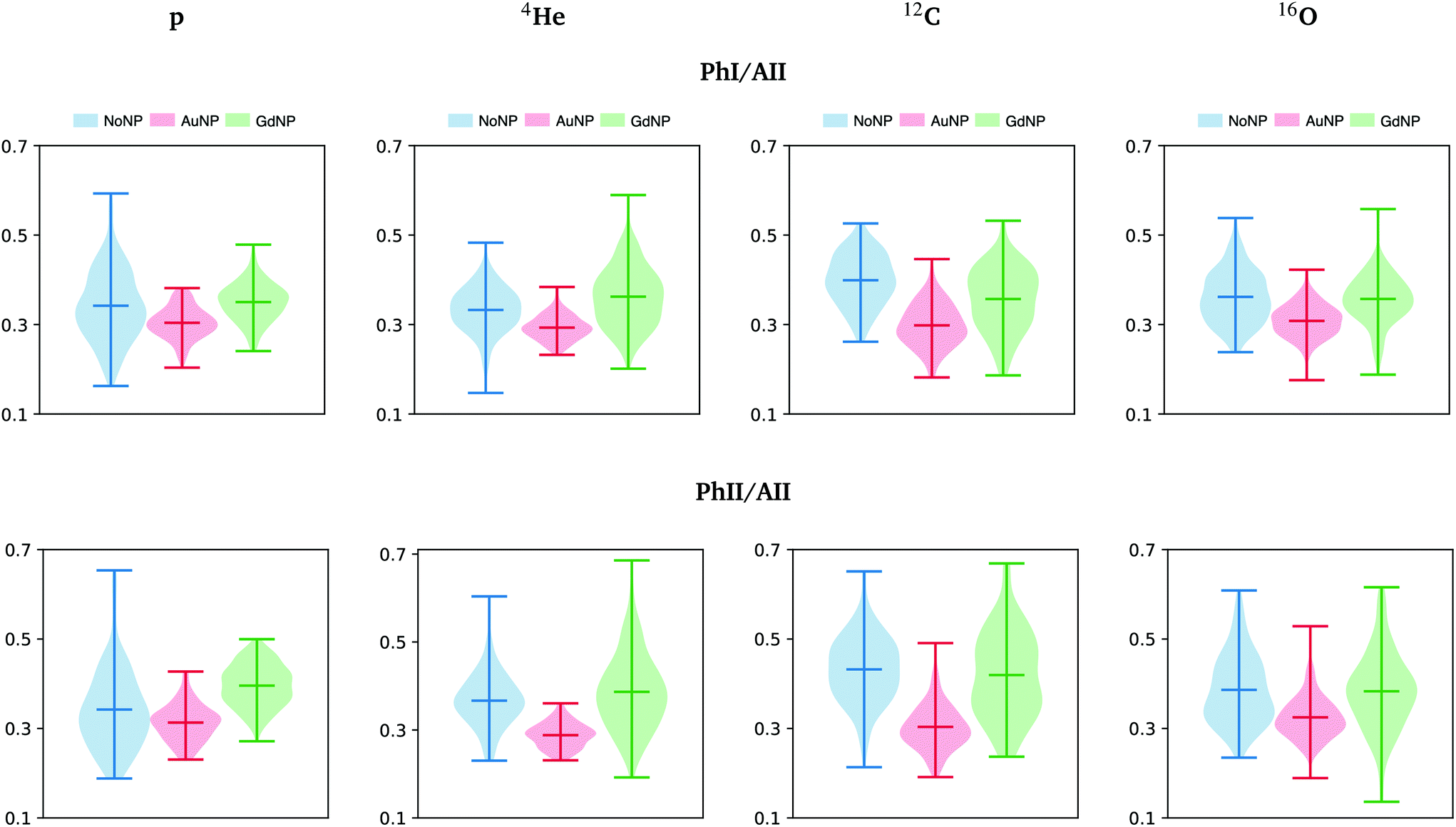 | ||
| Fig. 8 All ions (p, 4He, 12C, 16O; from left to right); 20 GyRBE. SR-FTIRM results on the radiosensitization mechanisms of GdNP and AuNP in charged particle therapy. Distribution of the relative intensities of the phosphate I (PhI) and phosphate II (PhII) bands with respect to the amide II (AII) in the absence (NoNP; blue) and in the presence of AuNP (red) and GdNP (green). See Table 1 for more information on the bands assignment. | ||
The combination of all these spectral changes showed NP-induced DNA damage in different groups of the DNA molecule, which seems to correspond to the course of cell death processes. However, unraveling the specific NP-induced cell processes is a complex task due to the multiple contributions in this region of the spectral range.
Finally the distinct LET of the several beams could be the responsible of the differences observed in the NP-induced biochemical modifications between ions, which might be due to distinct radiation-induced lethal effects. Despite an increase in the ROS production was reported in ion beams, an enhanced DNA-damage complexity and accumulation of non-repairable DSB was observed in the case of high LET particles.42 This, combined with the presence of NP, might induce a difference of lethal effect on the cells as a function of the LET.
O stretching of the phospholipids in the cellular membrane, and it is not overlapped by contributions from proteins and DNA.45
The PC1–PC2 scores scatter plots in the lipids region (Fig. 1–4, bottom) display well-separated clusters between NP-treated groups (GdNP and AuNP) and NoNP groups, mostly along PC-1, for most of the ion beams and doses. PCA suggested a more marked difference for AuNP as the PCA scores showed a clearer separation from NoNP' groups. In all cases, the first two principal components explain around 50% and 10–15% of the total variance, respectively. The corresponding loading plots demonstrated that NP-treated groups, and specially AuNP-treated cells, are mainly correlated with conformational changes in the CHx vibration bands. Despite very subtle shifts can be observed in some lipid bands, the position of the most intense bands is preserved in all configurations (see Fig. 5).
The most important markers for conformational disorders in the cell membrane are the changes in the intensity in the lipids spectral area. In particular, the ratios between the areas of several absorption bands associated to lipids (CH2/CH3, CH2 stretching ratio, CH/CH2 and carbonyl ester to lipids) are presented in Fig. 9 and detailed hereafter. They provide relevant information on the lipids biophysical properties and chemical state modifications within the cell membrane, such as to lipids structure, chain length, oxidative stress, degree of unsaturation and membrane peroxidation.63 These ratios provided indications on the biochemical effects of the NP treatment on the cells.
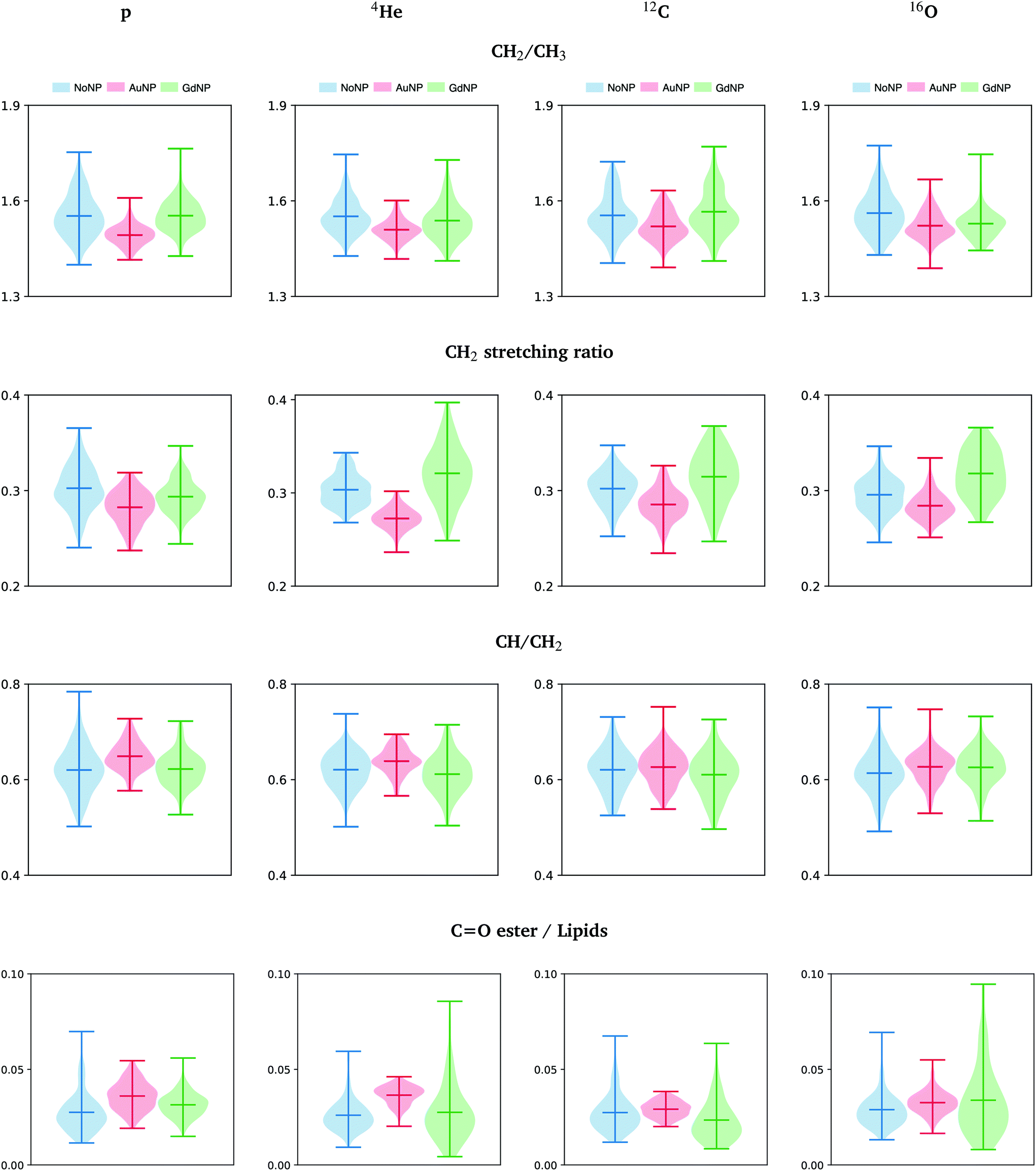 | ||
| Fig. 9 All ions (p, 4He, 12C, 16O; from left to right); 20 GyRBE. SR-FTIRM results on the radiosensitization mechanisms of GdNP and AuNP in charged particle therapy. Distribution of the relative intensities of the CH2/CH3, CH2 stretching ratio, CH/CH2 and CO ester/lipids spectral band ratios in the absence (NoNP; blue) and in the presence of AuNP (red) and GdNP (green). See Table 1 for more information on the bands assignment. | ||
• The asymmetric CH2/CH3 ratio is associated with the hydrocarbon acyl chain length of lipids (increases with chain length). Petibois et al. correlated a reduction of CH2 /CH3 with membrane phospholipid free radical attacks during oxidative stress.48 In our study (see Fig. 9), a slight decrease was observed in AuNP-treated cells with respect to NoNP for all ion beams and doses. In the case of GdNP, no significant changes in the CH2/CH3 ratio were observed.
• The CH2 symmetric to CH2 asymmetric stretching ratio (CH2 stretching ratio) is another way to assess cell membrane morphology alterations.29,64 In particular, a decrease in the CH2 stretching ratio was associated to an increase in the membrane rigidity as a consequence of the presence of membrane stabilizing agents or lipid raft aggregates, which are formed in dying cells.64Fig. 9 depicts the CH2 stretching ratio. In the presence of AuNP, there is a decrease in the CH2 stretching ratio.
• The ratio between unsaturated fatty acids (3018–3002 cm−1) to the symmetric CH2 stretching provides a good index for unsaturation.45 In Fig. 9, we detect a slightly higher concentration of unsaturated bonds in the lipid chains after AuNP treatment with respect to NoNP. The level of unsaturation of the fatty acids in the membrane has a high impact on the characteristics of the membrane. In particular, an increase of unsaturated fatty acids leads to alterations in the membrane fluidity,29 which can highly affect functional properties of the cell.65,66
• The distribution of the carbonyl ester group stretching band (1760–1730 cm−1) found in non-hydrogen bonded and hydrogen-bonded states in phospholipids was reported to be a good indicator of lipid peroxidation.49Fig. 9 shows the distribution of this band with respect to the area under the lipids region in order to normalize to cell thickness differences.49 We detected an increase of the carbonyl ester stretching band in the presence of both types of NP, particularly in the case of AuNP-treated cells. The increase was associated to an increase of oxidized lipids present in the membrane of dying cells or in cells under oxidative stress due to lipid peroxidation products.32,49
Our results showed that RT-NP treatments induced several modifications in the lipids spectral range, which might involve a wide range of biological processes. The modifications observed regarding lipids biophysical properties and chemical state might be related to the oxidative stress produced by the production of ROS from indirect effects of ionizing radiation in the presence of NP, which has been reported to be the main responsible of the amplification of the radiation damage observed in some charged particle therapy studies combined with NP.8,19 Indeed, the induction of AuNP-related oxidative stress was previously reported using the same type of NP via increased production of endogenous reactive oxygen species and depletion of intracellular antioxidants.36,67 The lipid differences in cellular response between AuNP and GdNP might be attributed to the distinct radiosensitization capability of both NP, which might lead to distinct cell damage/death pathways.
4 Conclusions
The objective of our SR-FTIRM study was to shed some light on the (Au and Gd) NP-based radiosensitization mechanisms in F98 cells treated with charged particle therapy (proton, helium, carbon and oxygen). Thanks to the very high signal-to-noise ratio of synchrotron-based FTIRM, modifications in the infrared spectra were assessed at a single-cell level. Observation of PCA results evidenced the separation between the NP-treated cells and the NoNP control cells, with some overlapping in some configurations. PCA in the fingerprint (proteins and DNA) and lipids regions enabled to distinguish the biochemical features induced by the nanoparticles on the main biomolecules present in F98 glioma cells.In the proteins spectral region, a deconvolution study on the amide I revealed an increase in the β-sheet secondary structure in the presence of NP. This, along with examination of the amide I to amide II ratio, indicated cellular protein modifications in the folding and localization induced by the NP. In the DNA region, we revealed a number of structural and conformational changes in the presence of NP, as well as evidences of DNA fragmentation represented by the decrease in the νs PO2− and νas PO2− bands. In the lipids spectral region, several specific markers were used as indices of the changes (CH2/CH3, CH2 stretching ratio, CH/CH2 and CO ester to Lipids), which provided relevant information on the changes induced by the NP in the lipid structure, chain length, oxidative stress, degree of unsaturation and membrane peroxidation. The modulation of the band intensities specific to DNA, proteins and lipids might evidence an enhanced production of ROS in the presence of NP, as well as an enhanced DNA damage response, correlated with cell damage/death. These results are in line with the amplification of radiation effects induced by NP in previous radiobiological studies in charged particle therapy. Finally, vibrational features were NP-dependent since radiosensitization effects differed between different NP (Au/Gd) types.
To conclude, this is the first work that provides new insights into the radiosensitization effects of nanoparticles in charged particle therapy through SR-FTIRM. However, monitoring the overall biochemical changes in the cell upon NP-based RT treatmens is a very complex task since it involves a wide range of biochemical processes. Therefore, the underlying biochemical processes implicated in such novel NP-RT approaches will need to be more thoroughly experimentally investigated.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors acknowledge the granted beamtime at ALBA-CELLS Synchrotron. This project was funded by the European Union Horizon 2020 Research and Innovation Programme under the Marie Sklodowska-Curie grant agreement number 748889. A. Abdollahi acknowledges the support from the German Research Foundation (DFG, SFB 1389, Unite) intramural funds of the National Center for Tumor Diseases (NCT Biodose program).References
- J. F. Hainfeld, D. N. Slatkin and H. M. Smilowitz, Phys. Med. Biol., 2004, 49, 309–315 CrossRef PubMed.
- S. Akhter, M. Z. Ahmad, F. J. Ahmad, G. Storm and R. J. Kok, Expert Opin. Drug Delivery, 2012, 9, 1225–1243 CrossRef CAS PubMed.
- Y. Matsumura and H. Maeda, Cancer Res., 1986, 46, 6387–6392 CAS.
- D. Kwatra, A. Venugopal and S. Anant, Transl. Cancer Res., 2013, 2, 330–342 CAS.
- K. Haume, S. Rosa, S. Grellet, M. A. Smialek, K. T. Butterworth, A. V. Solovyov, K. M. Prise, J. Golding and N. J. Mason, Cancer Nanotechnol., 2016, 7, 1–20 CrossRef PubMed.
- S. Her, D. A. Jaffray and C. Allen, Adv. Drug Delivery Rev., 2017, 109, 84–101 CrossRef CAS PubMed.
- I. Martínez-Rovira and Y. Prezado, Med. Phys., 2011, 38, 4430–4439 CrossRef PubMed.
- N. Usami, Y. Furusawa, K. Kobayashi, H. Frohlich, S. Lacombe and C. Le Sech, Int. J. Radiat. Biol., 2005, 81, 512–522 CrossRef PubMed.
- N. Usami, Y. Furusawa, K. Kobayashi, S. Lacombe, A. Reynaud-Angelin, E. Sage, T.-D. Wu, A. Croisy, J.-L. Guerquin-Kern and C. Le Sech, Int. J. Radiat. Biol., 2008, 84, 603–611 CrossRef CAS PubMed.
- J.-K. Kim, S.-J. Seo, K.-H. Kim, T.-J. Kim, M. H. Chung, K. R. Kim and T. K. Yang, Nanotechnology, 2010, 21, 425102 CrossRef PubMed.
- J.-K. Kim, S.-J. Seo, H.-T. Kim, K.-H. Kim, M.-H. Chung, K.-R. Kim and S.-J. Ye, Phys. Med. Biol., 2012, 57, 8309–8323 CrossRef PubMed.
- J. C. Polf, L. F. Bronk, W. H. P. Driessen, R. Pasqualini and M. Gillin, Appl. Phys. Lett., 2011, 98, 193702 CrossRef PubMed.
- S. Li, S. Penninckx, L. Karmani, A.-C. Heuskin, K. Watillon, R. Marega, J. Zola, V. Corvaglia, G. Genard, B. Gallez, O. Feron, P. Martinive, D. Bonifazi, C. Michiels and S. Lucas, Nanotechnology, 2016, 27, 455101 CrossRef PubMed.
- T. Schlatholter, S. Lacombe, P. Eustache, E. Porcel, D. Salado, L. Stefancikova, O. Tillement, F. Lux, P. Mowat, A. K. Biegun, M.-J. van Goethem, H. Remita and S. Lacombe, Int. J. Nanomed., 2016, 11, 1549–1556 CrossRef PubMed.
- E. Porcel, S. Liehn, H. Remita, N. Usami, K. Kobayashi, Y. Furusawa, C. Le Sech and S. Lacombe, Nanotechnology, 2010, 21, 085103 CrossRef PubMed.
- H. Kaur, G. Pujari, M. Semwal, A. Sarma and D. K. Avasthi, Nucl. Instrum. Methods Phys. Res., Sect. B, 2013, 301, 7–11 CrossRef CAS.
- E. Porcel, O. Tillement, F. Lux, P. Mowat, N. Usami, K. Kobayashi, Y. Furusawa, C. Le Sech, S. Li and S. Lacombe, Nanomedicine, 2014, 10, 1601–1608 CrossRef CAS PubMed.
- Y. Liu, X. Liu, X. Jin, P. He, X. Zheng, F. Ye, W. Chen and Q. Li, J. Nanosci. Nanotechnol., 2016, 16, 2365–2370 CrossRef CAS PubMed.
- S. Lacombe, E. Porcel and E. Scifoni, Cancer Nanotechnol., 2017, 8, 9 CrossRef PubMed.
- M. Sotiropoulos, N. T. Henthorn, J. W. Warmenhoven, R. I. Mackay, K. J. Kirkby and M. J. Merchant, Nanoscale, 2017, 9, 18431 RSC.
- A. V. Verkhovtsev, A. V. Korol and A. V. Solovyov, J. Phys. Chem. C, 2015, 119, 11000–11013 CrossRef CAS.
- K. Butterworth, S. McMahon, F. Currel and K. Prise, Nanoscale, 2012, 4, 4830–4838 RSC.
- S. Rosa, C. Connolly, G. Schettino, K. T. Butterworth and K. M. Prise, Cancer Nanotechnol., 2017, 8, 1–25 CrossRef PubMed.
- D. Choudhury, P. Xavier, K. Chaudhari, R. John, A. Dasgupta, T. Pradeep and G. Chakrabarti, Nanoscale, 2013, 5, 4476–4489 RSC.
- F. Taupin, M. Flaender, R. Delorme, T. Brochard, J.-F. Mayol, J. Arnaud, P. Perriat, L. Sancey, F. Lux, R. Barth, M. Carriere, J.-L. Ravanat and H. Elleaume, Phys. Med. Biol., 2015, 60, 4449–4464 CrossRef PubMed.
- I. Martínez-Rovira and Y. Prezado, Med. Phys., 2015, 42, 6703–6710 CrossRef PubMed.
- M. J. Baker, J. Trevisan, P. Bassan, R. Bhargava, H. J. Butler, K. M. Dorling, P. R. Fielden, S. W. Fogarty, N. J. Fullwood, K. A. Heys, C. Hughes, P. Lasch, P. L. Martin-Hirsch, B. Obinaju, G. D. Sockalingum, J. Sulé-Suso, R. J. Strong, M. J. Walsh, B. R. Wood, P. Gardner and F. L. Martin, Nat. Protoc., 2014, 9, 1771–1791 CrossRef CAS PubMed.
- N. Gault and J. Lefaix, Radiat. Res., 2003, 160, 238–250 CrossRef CAS PubMed.
- W. Mihoubi, E. Sahli, A. Gargouri and C. Amiel, PLoS One, 2017, 12, e0180680 CrossRef PubMed.
- D. Ami, T. Neri, A. Natalello, P. Mereghetti, S. M. Doglia, M. Zanoni, M. Zuccotti, S. Garagna and C. A. Redi, Biochim. Biophys. Acta, Mol. Cell Res., 2008, 1783, 98–106 CrossRef CAS PubMed.
- D. R. Whelan, K. Bambery, L. Puskar, D. McNaughton and B. R. Wood, Analyst, 2013, 138, 3891–3899 RSC.
- H. Holman, M. Martin, E. Blakely, K. Bjornstad and W. McKinney, Biopolymers, 2000, 57, 329–335 CrossRef CAS PubMed.
- I. Yousef, O. Seksek, Y. Prezado, J. Sulé-Suso and I. Martínez-Rovira, Analyst, 2016, 141, 2238–2249 RSC.
- I. Martínez-Rovira, O. Seksek, J. Puxeu, J. Gómez, M. Kreuzer, T. Ducic, M. J. Ferreres, M. Artigues and I. Yousef, Analyst, 2019, 144, 5511–5520 RSC.
- I. Martínez-Rovira, O. Seksek and I. Yousef, Analyst, 2019, 144, 6352–6364 RSC.
- J. A. Coulter, S. Jain, K. T. Butterworth, L. E. Taggart, G. R. Kickson, S. J. McMahon, W. B. Hyland, M. F. Muir, C. Trainor, A. R. Hounsell, J. M. O'Sullivan, G. Schettino, F. J. Currell, D. G. Hirst and K. M. Prise, Int. J. Nanomed., 2012, 7, 2673–2685 CrossRef CAS PubMed.
- K. T. Butterworth, J. A. Coulter, S. Jain, J. Forker, S. J. McMahon, G. Schettino, K. M. Prise, F. J. Currell and D. G. Hirst, Nanotechnology, 2010, 21, 2673–2685 CrossRef PubMed.
- L. Sancey, F. Lux, S. Kotb, S. Roux, S. Dufort, A. Bianchi, Y. Cremillieux, P. Fries, J. Coll, C. Rodriguez-Lafrasse, M. Janier, M. Dutreix, M. Barberi-Heyob, F. Boschetti, F. Denat, C. Louis, E. Porcel, S. Lacombe, G. Le Duc, E. Deutsch, J. Perfettini, A. Detappe, C. Verry, R. Berbeco, K. Butterworth, S. McMahon, K. Prise, P. Perriat and O. Tillement, Br. J. Radiol., 2014, 87, 20140134 CrossRef CAS PubMed.
- L. Sancey, S. Kotb, C. Truillet, F. Appaix, A. Marais, E. Thomas, B. van der Sanden, J. Klein, B. Laurent, M. Cottier, R. Antoine, P. Dugourd, G. Panczer, F. Lux, P. Perriat, V. Motto-Ros and O. Tillement, ACS Nano, 2015, 9, 2477–2488 CrossRef CAS PubMed.
- M. Krämer, O. Jäkel, T. Haberer, G. Kraft, D. Achardt and U. Weber, Phys. Med. Biol., 2000, 45, 3299–3317 CrossRef PubMed.
- M. Krämer and M. Scholz, Phys. Med. Biol., 2000, 45, 3319–3330 CrossRef PubMed.
- I. Dokic, A. Mairani, M. Niklas, F. Zimmermann, N. Chaudhi, D. Krunic, T. Tessonier, A. Ferrari, K. Parodi, O. Jakel, J. Debus, T. T. Haberer and A. Abdollahi, Oncotarget, 2016, 7, 56676–56689 CrossRef PubMed.
- S. Combs, J. Bohl, T. Elsasser, K.-J. Weber, D. Schulz-Ertner, J. Debus and W. Weyrather, Int. J. Radiat. Biol., 2009, 85, 126–137 CrossRef CAS PubMed.
- J. Demšar, T. Curk, A. Erjavec, V. Gorup, T. Hočevar, M. Milutinovič, M. Možina, M. Polajnar, M. Toplak, A. Starič, M. Štajdohar, L. Umek, L. Žagar, J. Žbontar, M. Žitnik and B. Zupan, J. Mach. Learn. Res., 2013, 14, 2349–2353 Search PubMed.
- R. Gasper, J. Dewelle, R. Kiss, T. Mijatovic and E. Goormaghtigh, Biochim. Biophys. Acta, 2009, 1788, 1263–1270 CrossRef CAS PubMed.
- G. Birarda, D. E. Bedolla, E. Mitri, S. Pacor, G. Grenci and L. Vaccari, Analyst, 2014, 139, 3097 RSC.
- C. Sandt, C. Nadaradjane, R. Richards, P. Dumas and V. See, Analyst, 2016, 141, 870 RSC.
- C. Petibois and G. Deleris, Analyst, 2004, 129, 912–916 RSC.
- B. Vileno, S. Jeney, A. Sienkiewicz, P. R. Marcoux, L. M. Miller and L. Forro, Biophys. Chem., 2010, 152, 164–169 CrossRef CAS PubMed.
- A. Barth, Biochim. Biophys. Acta, 2007, 1767, 1073–1101 CrossRef CAS PubMed.
- H. Ghimire, M. Venkataramani, Z. Bian, Y. Liu and A. G. U. Perera, Sci. Rep., 2017, 7, 16993 CrossRef PubMed.
- C. Chadefaux, A. Le Ho, L. Bellot-Gurlet and I. Reiche, e-Preserv. Sci., 2009, 6, 129–137 CAS.
- N. Gault, O. Rigaud, J. Poncy and J. Lefaix, Int. J. Radiat. Biol., 2005, 81, 767–779 CrossRef CAS PubMed.
- A. Lamberti, C. Sanges and P. Arcari, Spectroscopy, 2010, 24, 535–546 CrossRef CAS.
- U. Zelig, J. Kapelushnik, R. Moreh, S. Mordechai and I. Nathan, Biophys. J., 2009, 97, 2107–2114 CrossRef CAS PubMed.
- F. Gasparri and M. Muzio, Biochem. J., 2003, 369, 239–248 CrossRef CAS PubMed.
- E. Lipiec, K. Bambery, P. Heraud, C. Hirschmugl, J. Lekki, W. Kwiatek, M. Tobin, C. Vogel, D. Whelan and B. Wood, J. Mol. Struct., 2014, 1073, 134–141 CrossRef CAS.
- D. Whelan, K. Bambery, P. Heraud, M. Tobin, M. Diem, D. McNaughton and B. Wood, Nucleic Acids Res., 2011, 39, 5439–5448 CrossRef CAS PubMed.
- E. Lipiec, G. Birarda, J. Kowalska, J. Lekki, L. Vaccari, A. Wiechec, B. Wood and W. Kwiatek, Radiat. Phys. Chem., 2013, 93, 135–141 CrossRef CAS.
- A. Meade, H. Byrne and F. Lyng, Mutat. Res., 2010, 704, 108–114 CAS.
- N. Gault, O. Rigaud, J. Poncy and J. Lefaix, Radiat. Res., 2007, 167, 551–562 CrossRef PubMed.
- Z. Movasaghi, S. Rehman and I. Rehman, Appl. Spectrosc. Rev., 2008, 43, 134–179 CrossRef CAS.
- F. S. Ruggeri, C. Marcott, S. Dinarelli, G. Longo, M. Girasole, G. Dietler and T. Knowles, Int. J. Mol. Sci., 2008, 19, 2582 CrossRef PubMed.
- S. van der Driesche, F. Iuliano, C. Haiden, D. Pucciarelli, H. Breiteneder, S. Pastorekova, C. Hafner and M. J. Vellekoop, Sens. Actuators, B, 2013, 179, 150–156 CrossRef.
- S. Baritaki, P. Kanellou, S. Apostolakis, D. A. Spandidos and B. Bonavida, Proc. Amer. Assoc. Cancer Res., 2006, 47, 1262 Search PubMed.
- M. K. Mishra, D. Ghosh, R. Duseja and A. Basu, Neurochem. Int., 2009, 54, 464–470 CrossRef CAS PubMed.
- Y. Pan, A. Leifert, D. Ruau, S. Neuss, J. Bornemann, G. Schmid, W. Brandau, U. Simon and W. Jahnen-Dechent, Small, 2009, 5, 2067–2076 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2020 |