
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Miniaturisation of a peptide-based electrochemical protease activity sensor using platinum microelectrodes
Ahmet
Ucar
 a,
Eva
González-Fernández
b,
Matteo
Staderini
b,
Nicolaos
Avlonitis
b,
Alan F.
Murray
a,
Mark
Bradley
*b and
Andrew R.
Mount
*b
a,
Eva
González-Fernández
b,
Matteo
Staderini
b,
Nicolaos
Avlonitis
b,
Alan F.
Murray
a,
Mark
Bradley
*b and
Andrew R.
Mount
*b
aSchool of Engineering, Institute for Bioengineering, The University of Edinburgh, The King's Buildings, Mayfield Road, Edinburgh EH9 3JL, UK
bEaStCHEM, School of Chemistry, University of Edinburgh, Joseph Black Building, West Mains Road, Edinburgh, EH9 3FJ, UK. E-mail: a.mount@ed.ac.uk; mark.bradley@ed.ac.uk; Tel: +44 (0)131-650-4747
First published on 5th December 2019
Abstract
Proteases are ideal target biomarkers as they have been implicated in many disease states, including steps associated with cancer progression. Electrochemical peptide-based biosensors have attracted much interest in recent years. However, the significantly large size of the electrodes typically used in most of these platforms has led to performance limitations. These could be addressed by the enhancements offered by microelectrodes, such as rapid response times, improved mass transport, higher signal-to-noise and sensitivity, as well as more localised and less invasive measurements. We present the production and characterisation of a miniaturised electrochemical biosensor for the detection of trypsin, based on 25 μm diameter Pt microelectrodes (rather than the ubiquitous Au electrodes), benchmarked by establishing the equivalent Pt macroelectrode response in terms of quantitative response to the protease, the kinetics of cleavage and the effects of non-specific protein binding and temperature. Interestingly, although there was little difference between Au and Pt macroelectrode response, significant differences were observed between the responses of the Pt macroelectrode and microelectrode systems indicative of increased reproducibility in the microelectrode SAM structure and sensor performance between the electrodes, increased storage stability and a decrease in the cleavage rate at functionalised microelectrodes, which is mitigated by measurement at normal body temperature. Together, these results demonstrate the robustness and sensitivity of the miniaturised sensing platform and its ability to operate within the clinically-relevant concentration ranges of proteases in normal and disease states. These are critical features for its translation into implantable devices.
Introduction
Proteolytic enzymes such as proteases and their dysregulation are widely recognised for playing significant roles in many disease states.1 As such, they are attracting widespread attention as key biomarkers of disorders including cardiovascular diseases, HIV, Alzheimer's disease, thrombosis, diabetes and cancer.2–9 Therefore, the development of both sensitive and selective assays and sensors that monitor protease activity has generated considerable interest. According to Ong et al., these can be classified into two main groups: homogeneous assays (that include those based on colorimetry, mass spectrometry and fluorescence resonance energy transfer) and heterogeneous systems (that include electrochemical assays, surface-enhanced Raman scattering and surface plasmon resonance).2 In heterogeneous systems, the probe is typically immobilised on a solid surface (which often provides ready interrogation, signal generation and a detection interface) with the target analyte present in the neighbouring aqueous medium, whereas in homogeneous systems, both probe and analyte are present in the aqueous medium, which can present contamination and detection challenges. The principle of detection for both systems is largely based on protease recognition and catalytic cleavage of a specific peptide sequence attached to a reporter e.g. a fluorophore, quantum dot or redox tag. The reporter exerts analyte detection through the generation of an output signal following cleavage by the target protease.10–13Amongst the various methods used for protease sensing, there has been considerable growth in the popularity of electrochemical peptide-based biosensors, particularly those where the peptide-sequence with an attached redox tag is immobilised onto an electrode surface. These sensors offer high sensitivity, rapid response times, ready applicability to computer control and multiplexing, the use of cheap instrumentation, and ease of miniaturisation for point-of-care (PoC) applications.14 Such electrochemical peptide-based biosensors typically use a specific substrate peptide as the selective recognition moiety and the protease as the analyte. There is extensive literature reported on protease detection using this approach.13–22 For example, Liu et al. described a method that allowed detection of matrix metalloproteinases (MMPs), whose proteolytic activities have close association with cancer progression, at very low concentrations (with a limit of detection of 3.4 pM).15 An alternative approach was developed by Lee et al. who reported on the reference electrode-free sensing of MMP-9 using the self-gating effect on a concentric electrode system which consisted of an island and enclosing electrode.22
We have previously proposed, produced, characterised and optimised a macroelectrode-based sensor system for the detection of protease activity using a self-assembled monolayer (SAM) on an electrode surface, which was successfully translated to the detection of other proteases such as human neutrophil elastase, allowing clinically relevant measurements of its activity in human blood.20,23 However, the electrodes used in most of these systems are still macroscopic which leads to limitations with respect to sensor performance as well as their applicability to implantation. Smaller, less invasive footprint, microelectrodes offer potential advantages over macroelectrodes that include more rapid diffusion (which can lead to shorter and more controlled response times through enhanced and more reproducible mass transport) and higher signal-to-noise which leads to higher sensitivity; together this makes using microelectrodes an attractive route for electrochemical biosensors.24,25 In addition, miniaturised technologies offer advantages for biosensor design such as integrated high fidelity manufacturing with lower manufacturing costs per sensor, the ability to work with small quantities of materials and samples and ease of multiplexed measurement options.26,27 Despite these strong drivers for the development of miniaturised SAM-based electrochemical systems for biosensors for the detection of proteases, although some examples of in vitro and/or in vivo microelectrode use in biosensing are found in the literature,28–31 there has been little focus on the comparison of macro- and microelectrodes, especially those that are SAM-based.
The purpose of this study was to investigate the miniaturisation of our sensing platform onto novel Pt-based microelectrodes as a step towards their development and application as implantable sensors. Microelectrode sensors were produced, the resulting properties were optimised and finally evaluated in terms of sensor performance, including determining the kinetics and thermodynamics of enzyme–substrate cleavage and the effects of non-specific protein binding. This performance was compared to the analogous Au and (for the first time) equivalent Pt-based macroelectrode systems.
Experimental
Instrumentation
All electrochemical measurements were carried out using a conventional three-electrode electrochemical cell which was driven by a computer-controlled AutoLab PGstat-30 potentiostat by running the NOVA 1.11 software (Metrohm Autolab B.V., The Netherlands). An in-house built platinum-coated silicon dioxide chip was used as an auxiliary electrode, with 2 mm and 25 μm diameter platinum disc electrodes (IJ Cambria, UK) used as working electrodes for the macro- and microelectrode measurements, respectively. All the working electrode potentials, E, were applied with respect to (and are reported relative to) a Ag|AgCl|KCl (3 M) reference electrode (Bioanalytical Systems, Inc., USA). A Lauda Eco Silver thermostatic bath (VWR International Ltd, UK) with an external pumping system and a water-jacketed glass cell was used to control the temperature of all the experiments conducted at 25 °C or 37 °C.Reagents and materials
Trypsin (MW 23.4 kDa), bovine serum albumin (BSA), casein, ethanol, 6-mercaptohexanol (MCH), 2,2′-(ethylenedioxy) diethanethiol (DT) and 10× PBS were purchased from Sigma Aldrich (UK) and used as received. All reagents were of analytical grade and all solutions were prepared using protease-free deionised water.Synthetic methods
Two methylene blue-labelled peptides were synthesised in-house following the synthetic experimental procedures described previously.20,21 Briefly, the redox labelled peptides (probe substrate and the control) (MB-Phe–Arg–Arg-PEG-6–Cys) were synthesised using Fmoc solid-phase chemistry on a polystyrene resin with a Rink-amide linker. First, cysteine was coupled to the resin, followed by a polyethylene glycol unit containing 6 ethylene glycol units (PEG-6) as a spacer, then a cleavable short peptide sequence (specific to trypsin) (Phe–Arg–Arg (FRR)) and finally the methylene blue redox label (MB). Finally, the probe was cleaved from the resin with a TFA cleavage mixture and purified by RP-HPLC.Cleaning, pre-treatment and preparation of electrodes
Platinum working electrodes were first immersed in concentrated H2SO4 (95%) for 10 min to remove any organic residue on the surface and then successively polished to a progressively finer surface finish using a polishing cloth and a sequence of aqueous slurries containing first 1, then 0.3 and finally 0.05 μm alumina particles (Buehler, Germany). After that, each electrode was subjected to an electrochemical cleaning step by carrying out cyclic voltammetry (CV), performing cycles of E between −0.3 and +1.6 V in 0.1 M H2SO4 at a potential scan rate of 100 mV s−1 until the characteristic voltammogram of clean platinum was obtained.32 Extending the established macroelectrode protocol,20 the surface substrate monolayer was then formed as a mixed SAM (labelled peptide, DT and MCH) on this platinum electrode surface by immersing the electrode overnight at 4 °C in a 40 μM solution of the methylene blue-labelled peptide (either the substrate containing cleavable L-amino acids or the control containing uncleavable D-amino acids) and freshly prepared DT (600 μM for macroelectrodes, 150 μM for microelectrodes) in ethanol. After washing with ethanol, the resulting SAM-modified electrode was immersed in 1 mM MCH in ethanol (1 h for the macroelectrodes and 10 min for the microelectrodes). It should be noted that these reductions in concentration and/or time in the optimised SAM microelectrode deposition protocol are consistent with the expected enhancement in sensitivity and mass transport kinetics of reactions at microelectrodes. Finally, washing was carried out, firstly with ethanol and then with phosphate-buffered saline (PBS) and the modified electrodes were stored in PBS at 4 °C until use.Electrochemical sensor measurements
The SAM-modified working electrodes were immersed in 1× PBS (which has an optimum pH of 7.4 for trypsin activity) and subjected to electrochemical measurements using square wave voltammetry (SWV, applying E at a frequency of 60 Hz, with an amplitude of 25 mV and a step potential of 5 mV) until a stable background signal was obtained. After the addition of the target enzyme (trypsin), or the proteins, casein and BSA for characterisation of non-specific binding, the SWV signal was continuously monitored with time. Following the established analysis method for Au macroelectrodes,20,21 this resulting signal is expressed as the percentage relative change in the SWV peak current with respect to the initial peak current (henceforth called the % signal change).Results and discussion
Successful SAM modification of both macroelectrode and microelectrode surfaces was first demonstrated by recording cyclic voltammograms (CVs) in ferri/ferrocyanide ([Fe(CN)6]3−/[Fe(CN)6]4−), 1× PBS solution (Fig. 1). Notwithstanding the expected difference of peaks (macroelectrodes) and waves (microelectrodes) in the resulting clean electrode redox responses arising from linear and hemispherical diffusional control, respectively, the CVs recorded before and after the overnight incubation of the electrodes with the probe solution show the expected significant inhibition of the redox reaction of the external redox agent characteristic of blocking SAM layers.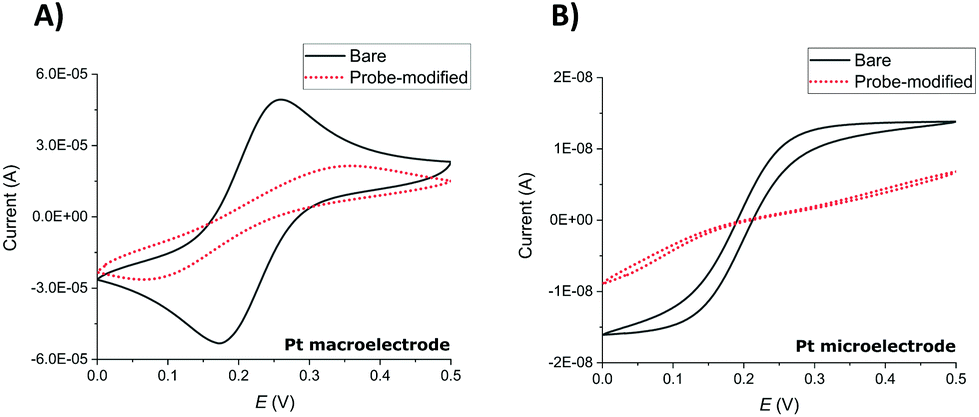 | ||
| Fig. 1 CVs of (A) a Pt macroelectrode and (B) a Pt microelectrode recorded before (black) and after (red) SAM probe immobilisation in 5 mM potassium ferri/ferrocyanide in 1× PBS. CVs were recorded between potentials of 0 and 0.5 V at a scan rate of 100 mV s−1. | ||
SWV redox peak MB currents of 3.1 ± 1.0 μA (macroelectrodes) and 1.3 ± 0.4 nA (microelectrodes) were obtained in 1× PBS solution (n ≥ 9 functionalised electrodes). This is consistent with the ratio of macroelectrode to microelectrode area of 6400 and demonstrates that these SWV parameters, previously established and optimised for Au macroelectrodes,20,23 again provided a clear MB peak and a stable and low background signal required for both Pt macroelectrode and microelectrode SAM analysis in this study.
The electrochemical detection principle is based on the specific proteolytic cleavage of the redox-tagged peptide probes anchored onto the platinum electrode surface by the target protease, trypsin, which should lead to the release of the soluble redox-tagged peptide fragment and a corresponding decrease in the redox peak as measured by the square wave voltammetry (SWV) technique (Fig. 2A). Based on previous macroelectrode optimisation work,20,21 the sensing layer consisted of a mixed SAM; the probe with the target peptide was tagged with MB and attached to a thiol-terminated PEG-6 spacer, backfilling MCH to minimise pinholes in the SAM layer and co-adsorbent PEG-based DT molecules to support the orientation, specificity and accessibility of the probe on the electrode surface. Two different MB-labelled peptides were used, the L-amino acid sequence for a trypsin-cleavable substrate or the D-amino acid sequence analogue as a trypsin-uncleavable control. The general sequence structure is depicted in Fig. 2B.
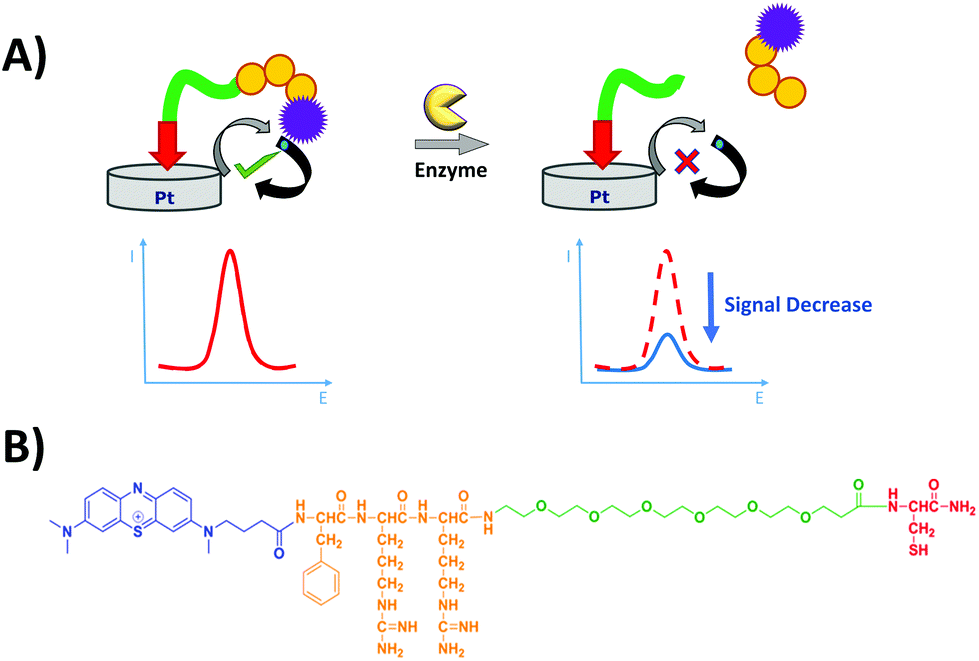 | ||
| Fig. 2 (A) Principle of detection of the peptide-based electrochemical platform. The protease (trypsin) catalyses the cleavage of the immobilised redox-labelled peptide releasing the redox-containing fragment into solution and leading to a decrease of the electrochemical signal which is measured by SWV. (B) Chemical structure of the probe, containing methylene blue (blue) as the redox tag, phenylalanine–arginine–arginine (orange) as the peptide and PEG-6 (green) as the spacer and cysteine (red) as the anchor. | ||
Analytical performance comparison of the macro- and microelectrodes as an electrochemical platform for trypsin analysis
In order to compare the analytical characteristics offered by the macro and micro platinum electrodes in protease sensing, both macro- and microelectrode surfaces were modified with the mixed SAM as described previously. They were then immersed in buffer solutions containing varying concentrations of trypsin (1–100 nM) and the electrochemical signal was interrogated by SWV with time. The addition of trypsin caused the expected decrease in signal for the macroelectrode (Fig. 3A), with the initial rate of % signal change observed to be proportional to the trypsin concentration (Fig. 3B). Given that this system has been shown to follow Langmuir kinetics,20 this is to be expected at these relatively low concentrations of trypsin compared to the Michaelis constant, where the proportion of surface covered by the trypsin–protein complex is expected to be low and proportional to the concentration of trypsin and the resulting rate of proteolytic cleavage is determined by this. As predicted by the method developed in previous work,23 the natural logarithm of A(t), the percentage of signal left to change at time t, calculated as A(t) = [((% signal change at t) − (% signal change as t → ∞))/((% signal change at t = 0) − (% signal change as t → ∞ and for the highest trypsin concentration, as [E] → ∞))] × 100%, is proportional to the trypsin concentration, [E] (Fig. 3B inset), which confirms that the response is first order with respect to [E]. | ||
Fig. 3 (A) Typical background-subtracted SWV curves for a SAM-functionalised probe substrate macroelectrode recorded for 100 nM trypsin at different incubation times between 0 and 60 min (with intervals of 5 min) in 1× PBS. (B) Plot of % signal decrease vs. time for SAM-functionalised probe macroelectrodes immersed in varying trypsin concentrations in PBS (0, 1, 5, 10, 25, 50, 100 nM for the substrate probe and 100 nM for the negative control probe, containing D-amino acids). All data represent the average (and standard deviations) from (typically) 3 functionalised electrodes. Inset: Natural logarithm of the adjusted signal, A(%), after 92 min, plotted against the concentration of trypsin. The straight line corresponds to the best linear regression fit (ln![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) A = −0.075 [trypsin]/nM + 4.498; r2 = 0.96). The point of [trypsin] = 50 nM was not included in the fit due to having large replicate errors compared to the other concentrations. A = −0.075 [trypsin]/nM + 4.498; r2 = 0.96). The point of [trypsin] = 50 nM was not included in the fit due to having large replicate errors compared to the other concentrations. | ||
An equivalent proteolytic trypsin cleavage behaviour was also recorded when using microelectrodes (Fig. 4A and B and inset), also indicating a first order cleavage process, but it is interesting that the corresponding cleavage rates were significantly slower at the same trypsin concentration when using microelectrodes compared to macroelectrodes.
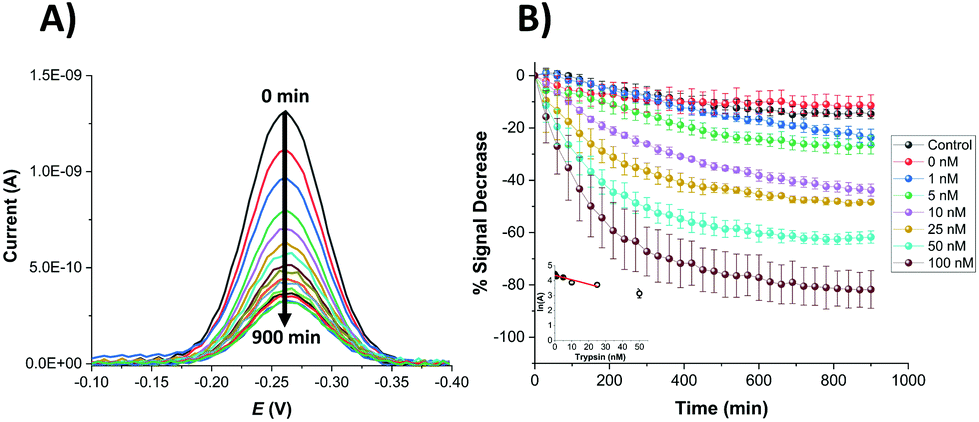 | ||
| Fig. 4 (A) Typical background-subtracted SWV curves recorded for a SAM-functionalised microelectrode and 100 nM trypsin at different incubation times between 0 and 900 min (with intervals of 60 min) in 1× PBS. (B) Plot of % signal decrease vs. time for SAM-functionalised microelectrodes immersed in varying trypsin concentrations in 1× PBS (0, 1, 5, 10, 25, 50, 100 nM for the probe substrate and 100 nM for the negative control probe, containing D-amino acids). All data represent the average (and standard deviations) from (typically) 3 functionalised electrodes. Inset: Natural logarithm of A(%), after 900 min, plotted against the concentration of trypsin. The straight line corresponds to the best linear regression fit (lnA = −0.030 [trypsin/nM] + 4.354; r2 = 0.84). The point of [trypsin] = 50 nM was not included again in the fit due to having large replicate errors compared to the other concentrations. | ||
Microelectrodes have been reported many times to show enhanced diffusional kinetics due to hemispherical diffusion compared to the linear diffusion observed for macroelectrodes.33 Therefore, it is clear from these observed cleavage rates that this difference in the rate of proteolytic cleavage is not as a result of the rate of trypsin diffusion. Given that the rate must therefore be determined by surface reactions, it is likely that this difference results from the macro- and microelectrode surfaces having a different SAM structure and/or probe or target surface disposition. It was previously noted that macro- and microelectrodes might differ from each other regarding the deposition and disposition of SAMs on their surfaces, which could affect the resulting properties such as electron transfer.34 Although the trypsin cleavage reaction is not under diffusional control, such distinctions could arise from the differences in diffusional rates during SAM film formation and/or the differences in the uniformity of diffusion to macro and microelectrode surfaces when under diffusional control. A difference in the observed initial SWV peak potentials for macro and microelectrodes of around 70 mV supports a dissimilarity in the average redox environment and overall film structure. It is interesting that higher error bars were obtained for the data sets recorded for the macroelectrodes compared to the microelectrodes, reflected by the errors in the linear fit data (Fig. 3 and 4 insets). This suggests a more reproducible microelectrode film structure and resulting cleavage rate.
Using the observed and expected linear dependency of lnA(t) to concentration (Fig. 3 and 4 insets), the limit of detection (LOD) for [E] was estimated for each electrode type to be equivalent at 2.5 nM for the macroelectrode and 2.9 nM for the microelectrode with a linear dependency for sensing up to 25 nM. The calculated linear response range and LOD successfully completely cover the clinically relevant range for trypsin levels in, for example, normal patients (11 ± 4 nM), and partially cover that for chronic renal failure (47 ± 25 nM) and chronic pancreatitis conditions (60 ± 27 nM).35,36
Kinetic analysis of proteolytic cleavage
Although single time point analysis and linear calibration is a simple method, this does not apply across the entire clinically relevant range for all conditions and time-dependent analysis of multiple data points is likely to be more robust and sensitive. When combined with Michaelis–Menten kinetic analysis, which models the non-linearity of trypsin binding and the resulting response with [E], analysis should be possible over a wider range of trypsin concentration. The measured % signal changes for varying trypsin concentrations were therefore analysed as a function of time using the previously established Michaelis–Menten kinetic model.20 According to this model, the % signal change, A, is expressed as a variation in the fraction (θ(t) = 1 − A(t)) of the cleavable peptide which has been cleaved at any time, t. Data for each trypsin concentration are then fitted to the equation:| θ = 1 − e−kefft | (1) |
Although good fits were obtained to this equation for the Pt macroelectrode system, consistent with our previous observations on gold macroelectrodes, this was not the case for the Pt microelectrode system. Inspection of Fig. 4 indicates that this is likely due to an increase in the fraction of the cleavable probe with increasing [E], as shown by the increasing maximum % signal change as t → ∞. All data were therefore fitted to eqn (2), which includes the additional concentration-dependent variable, a, which is the fraction of the cleavable peptide that can be cleaved at each trypsin concentration, [E], (with a → 1 and eqn (2) → eqn (1) as [E] → ∞ and at all [E] for macroelectrodes):
| θ = a[1 − e−kefft] | (2) |
This simple equation was shown to fit well to all data and generally enabled the extraction of both a and keff (effective rate constant) values for each trypsin concentration (both Fig. 5, for which a = 1 and eqn (2) collapses to eqn (1), and Fig. 6). The only exception was for the lowest [E] in Fig. 6, where over this measurement time range the product kefft was sufficiently small for eqn (2) to become effectively linear and only a combined constant akeff could be determined.
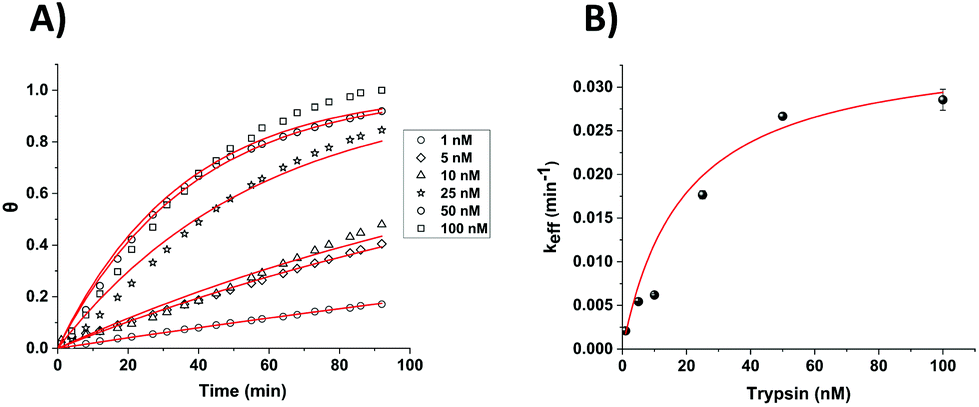 | ||
| Fig. 5 (A) Calculated fractional cleavage, θ, vs. time plots for data for macroelectrode cleavage data from Fig. 3. The data points (from bottom to top) correspond to immersion in varying trypsin concentrations in 1× PBS (namely 1, 5, 10, 25, 50 and 100 nM), whilst each line shows the best fit to eqn (1). (B) Effective rate constant, keff, as a function of the bulk trypsin solution concentration. Data represented by dots correspond to the experimental data obtained from the fits to the data in (a) and the solid red line shows the fitting processed according to eqn (3). | ||
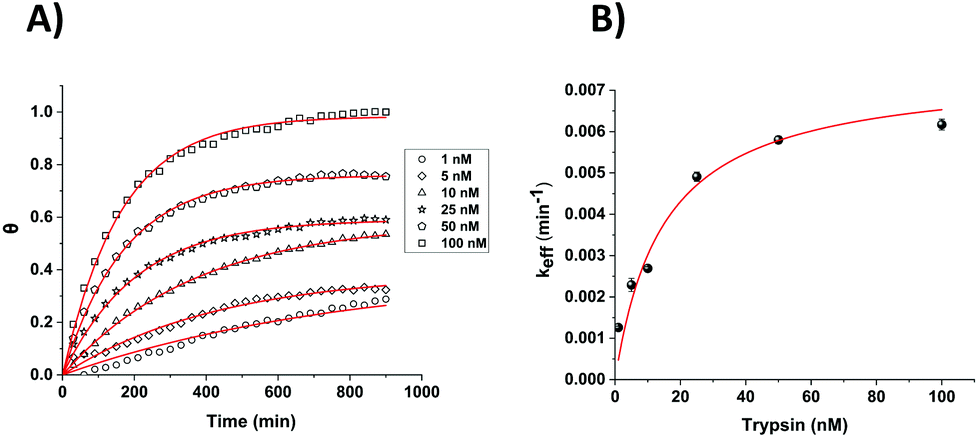 | ||
| Fig. 6 (A) Calculated fractional cleavage, θ, vs. time plots for data for microelectrode cleavage data from Fig. 4. The data points (from bottom to top) correspond to immersion in varying trypsin concentrations in 1× PBS (namely 1, 5, 10, 25, 50 and 100 nM), whilst each line shows the best fit to eqn (2) from which values of a = 0.39, 0.39, 0.58, 0.59, 0.76 and 0.98 respectively have been obtained. (B) Effective rate constant, keff, as a function of the bulk trypsin solution concentration. Data represented by dots correspond to the experimental data obtained from the fits to the data in (a) and the solid red line shows the best iterative fit to eqn (3). | ||
For the estimation of kcat (enzyme turn-over number) and KM (Michaelis–Menten binding constant) values, these extracted keff values were plotted as a function of trypsin concentration (Fig. 5B and 6B) and fitted to the Michaelis–Menten enzyme cleavage model using the equation:
| keff = kcat/(1 + KM/[E]) | (3) |
This non-linear fitting enabled us to estimate the values of kcat and KM as 0.035 min−1 and 19 ± 3 nM for macroelectrodes and, 0.0075 min−1 and 15 ± 3 nM for microelectrodes, respectively.
These calculated KM values are comparable to each other and also to previously reported values by Anne et al. (∼17 nM) and also to our previous work on gold macroelectrodes (28 ± 3 nM).13,20 These comparable KM values suggest that there is little difference between the thermodynamics of the trypsin binding to the probe substrate to form the enzyme–substrate complex in all cases. However, the markedly lower kcat value indicates that it is the significantly lower kinetics of the reaction of this enzyme–substrate complex on the SAM-functionalised microelectrodes which gives rise to the markedly lower cleavage rates. This indicates that the catalytic efficiency (kcat/KM) of the enzyme is lower for this surface, and that although absolute and relative enzyme–substrate association and dissociation rates appear similar (which given the size and multiple interactions of the enzyme with the surface is likely to be reflective of more general enzyme-SAM surface association and dissociation) the trypsin–substrate complex cleavage rates are different (which are likely to reflect variations in probe accessibility). Therefore, the rationale for slower proteolytic cleavage observed is lower probe accessibility to the enzyme active site on the SAM microelectrode surface, which would result in lower probe reactivity. It is possible that this (and the fact that a < 1 in eqn (2) for microelectrodes) is due to the relative inaccessibility of the enhanced proportion of the probe nearer to the electrode edge, due to the dramatic increase in the amount and importance of edge per unit area on decreasing from the macro to the microelectrode dimension. This is in addition to the uncleavable subset of the immobilised peptides at all [E] for both electrode types, previously attributed to electrode roughness and probe orientation variation as the sources of local site inaccessibility.21,23
It is clear that this analysis method has now extended the trypsin measurement range to 100 nM, covering the clinically relevant range even for chronic pancreatitis conditions (60 ± 27 nM).
Characterisation of selectivity for the microelectrodes
In order to assess the selectivity of the miniaturised trypsin sensor, the modified microelectrodes were exposed to solutions containing BSA and casein, as models for evaluating the potential for interference in real-world samples arising from non-specific binding of proteins. Fig. 7 shows the % signal decrease of both the substrate (orange) and the control (cyan) SAM structures upon the addition either of the non-specific proteins or the target enzyme trypsin. Although there was a significant amount of signal decrease recorded for both casein and BSA at these very long measurement times, no statistically significant difference between the substrate and control probe responses was observed, which confirms that the signal decrease in both cases was due to non-specific binding occurring presumably due to the consequent reduction in probe flexibility that hinders the redox tag–electrode interaction and therefore, the redox activity. This is in contrast to the maximum signal decrease of ∼80% recorded for substrate-modified surfaces upon the addition of trypsin, which was markedly and statistically different to the relatively small response of the control-modified probe surface. This clear difference highlights that the proposed microelectrode-based sensing system is highly selective towards trypsin, and that a combination of substrate and control measurement offers potential for direct measurements in real-world samples containing proteins.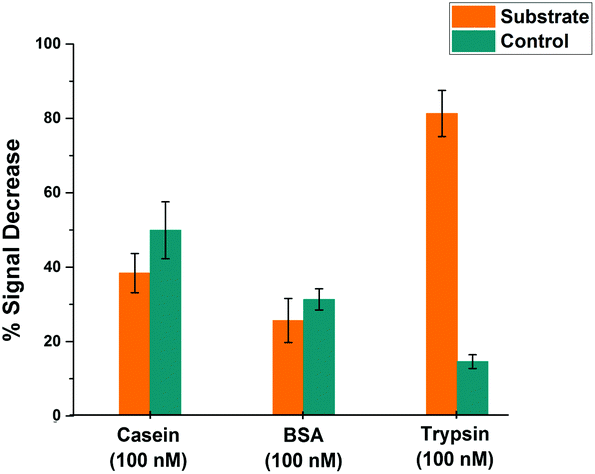 | ||
| Fig. 7 Comparative % signal decrease recorded for SAM-functionalised microelectrodes after 900 minutes upon the addition of 100 nM trypsin or non-specific binding proteins; casein and BSA for both substrate- (orange) and control-modified (cyan) sensing layers. Average data and error bars are typically from 3 individual SAM sensing layers. | ||
Effect of temperature on microelectrode-based proteolytic cleavage
It is known that temperature has an important role in the functional activity characteristics of most enzymes.37 Therefore, the kinetics of proteolytic trypsin cleavage was determined on the microelectrode-based sensor not only at room temperature (25 °C) but also at the clinically relevant normal body temperature (37 °C). Fig. 8A shows the comparative signal decrease recorded for both control- and substrate-modified microelectrodes at these temperatures with time. For both temperature cases, it is clearly seen that the signal decrease for the trypsin-cleavable substrate-modified electrodes is higher than the control-modified electrodes, which again demonstrates the trypsin selectivity of the sensor. A higher rate of signal decrease (∼30% after 200 min) was observed for the control-modified surfaces at 37 °C, compared to that (∼10% after 200 min) recorded at 25 °C, which is indicative of an enhanced detachment and loss of the probe from the surface at these elevated temperatures. The fact that this is an additional and parallel process also present in the substrate-modified surfaces which, like trypsin cleavage, results in probe signal loss is shown by subtracting the time dependent control-modified electrode response from that of the substrate-modified electrode. This was then converted to fractional cleavage, θ and A(t) following the same process as detailed before, but in this case this was normalised to the signal for these data as t → ∞ at this value of [E] (Fig. 8B). This shows the expected characteristic change in signal with time due only to probe loss arising from trypsin cleavage, as shown by the good fit to eqn (2) (which is equivalent to eqn (1), as this normalisation by definition fixes a as 1 in eqn (2)) (Fig. 8B). This fit gives keff = 0.082 ± 0.004 min−1 at 37 °C, which is around four-times larger than the 0.021 ± 0.002 min−1 obtained at 25 °C. This enhancement in kinetics is consistent with the previously reported work,38 which suggests an activation energy of around 90 kJ mol−1 and indicates the potential for markedly faster trypsin measurement in vivo on implanted microelectrodes. It is also worth noting that the undesirable need for control data subtraction arising from the significant parallel probe loss at normal body temperature could be addressed by use of a stronger tri-branched thiol anchor as previously demonstrated on a similarly functionalised electrode surface.39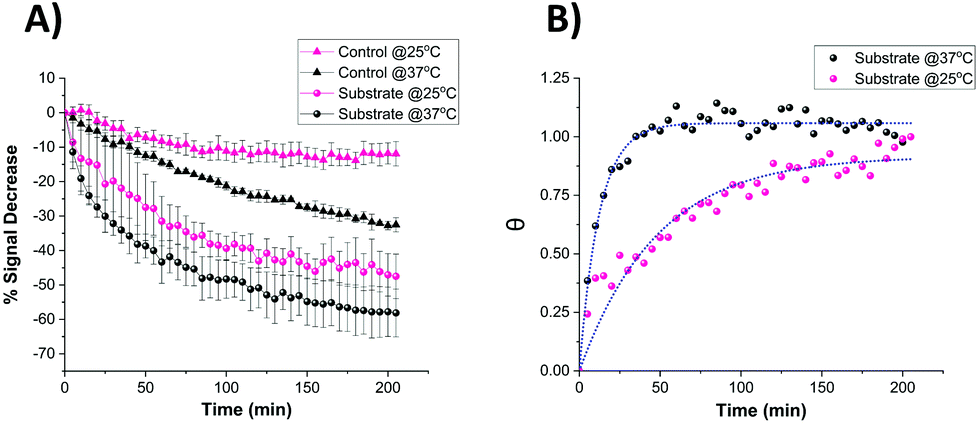 | ||
| Fig. 8 (A) Comparative signal decrease vs. time curves recorded for the miniaturised sensor upon the addition of 100 nM trypsin in 1× PBS at 25 °C (pink) and 37 °C (black) for both control- (triangle) and substrate-modified (circle) microelectrodes. (B) Calculated fractional cleavage, θ, vs. time plots for control-subtracted substrate data from (A). The data points correspond to temperatures of 25 °C (pink) and 37 °C (black) whilst the green dotted lines show the best iterative fits to eqn (1) with keff = 0.021 ± 0.002 min−1 and 0.082 ± 0.004 min−1 respectively. | ||
Characterisation of sensor storage stability
It is widely accepted that the storage stability is quite significant for SAM-based biosensors and much effort is being devoted in the literature to enhancing this storage lifetime.40 Therefore, Pt macro- and microelectrodes were modified with the probe as detailed above and their storage stability at 4 °C was investigated by monitoring their SWV signal over one week period. Fig. 9 shows that the macroelectrodes showed a higher rate of decrease in signal (∼40%) than the microelectrodes (∼20%) over this period. This amount of decrease observed in the Pt macroelectrodes was also consistent with the previous work,39 where the storage stability of Au macroelectrodes was assessed for 30 days, confirming that Au or Pt macroelectrode surfaces do not differ significantly in this respect. Additionally, there is some evidence that the drop in the signal on microelectrodes occurs within one day, with little evidence of a statistically significant decrease after this. Again, there is also more variation between individual electrodes in the macroelectrode set, compared to the microelectrode set. This supports the suggestion that more reproducible as well as durable SAM probes are formed on these miniaturised electrodes.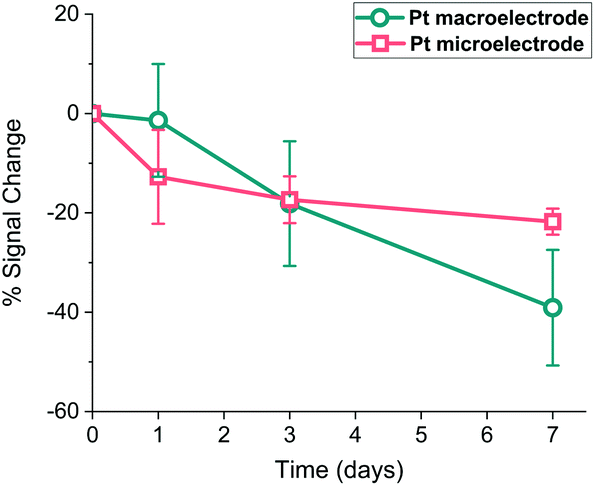 | ||
| Fig. 9 Percentage signal change recorded for probe-modified Pt macroelectrodes (circles, cyan) and microelectrodes (squares, pink) when stored in 1× PBS at 4 °C for 7 days. Mean signal losses and associated errors recorded after 7 days are 2.14 ± 1.72 μA (macroelectrodes) and 0.24 ± 0.06 nA (microelectrodes). | ||
Conclusions
This paper presents results which demonstrate the successful development of a peptide-based electrochemical microelectrode biosensor system for protease detection. Using a previously developed detection mechanism based on the signal change due to the proteolytic cleavage by trypsin of a SAM-immobilised peptide sequence resulting in the loss of the redox tag, the performance of this miniaturised electrode in terms of trypsin detection was assessed and compared to the results of previously reported (gold) and new (platinum) macroelectrodes. These comparisons demonstrated the feasibility of using microelectrodes, with comparable analytical performance being observed in terms of target binding and specificity, and with enhanced reproducibility of the response between electrodes, which we attribute to the enhanced reproducibility of SAM film formation and the resulting structure. We have also shown insensitivity to the non-specific adsorption of proteins through comparison of the responses of control and substrate systems, showing its potential for measurement in natural biological media. This system was also shown to give a quantitative response across a measured concentration range which encompassed the clinically relevant concentration range for trypsin detection for normal and diseased states, and fits well to a Michaelis–Menten surface cleavage model, which enabled the estimation of kcat and KM values and the ready extraction of trypsin concentration from measured signals. Some interesting differences were also observed between the microelectrode and macroelectrode systems, attributed to the differences in the SAM film structure. First the reproducibility of response was enhanced on microelectrodes, indicating less variability in SAM formation and structure. It is to be recognised that analysis of additional microelectrode data collected across this concentration range should further increase the accuracy of modelling and concentration determination. Secondly the overall cleavage rate was seen to be markedly slower at room temperature on microelectrodes, as a result of a slower trypsin-substrate cleavage rate. Although this was significantly increased at normal body temperature, reducing required measurement times, future work will focus on minimising response times and translating these findings to electrode-on-silicon chip technologies.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by funding from the UK Engineering and Physical Sciences Research Council (EPSRC), through the Implantable Microsystems for Personalised Anti-Cancer Therapy (IMPACT) programme grant (EP/K034510/1). AU acknowledges the doctoral scholarship from the Republic of Turkey, Ministry of National Education through the YLSY programme.Data used within this publication can be accessed at: https://doi.org/10.7488/ds/2720.
Notes and references
- H. Neurath and K. A. Walsh, Proc. Natl. Acad. Sci. U. S. A., 1976, 73, 3825–3832 CrossRef CAS PubMed.
- I. L. H. Ong and K.-L. Yang, Analyst, 2017, 142, 1867–1881 RSC.
- R. D. A. Wilkinson, R. Williams, C. J. Scott and R. E. Burden, Biol. Chem., 2015, 396, 867–882 CAS.
- Y. Hua and S. Nair, Biochim. Biophys. Acta, Mol. Basis Dis., 2015, 1852, 195–208 CrossRef CAS PubMed.
- J. Yuan and B. A. Yankner, Nature, 2000, 407, 802–809 CrossRef CAS PubMed.
- H. Haim, I. Salas and J. Sodroski, J. Virol., 2013, 87, 1884–1889 CrossRef CAS PubMed.
- A. Doucet and C. M. Overall, Mol. Aspects Med., 2009, 29, 339–358 CrossRef PubMed.
- S. H. Havale and M. Pal, Bioorg. Med. Chem., 2009, 17, 1783–1802 CrossRef CAS PubMed.
- J. E. Koblinski, M. Ahram and B. F. Sloane, Clin. Chim. Acta, 2000, 291, 113–135 CrossRef CAS.
- E. Pazos, O. Vázquez, J. L. Mascareñas and M. E. Vázquez, Chem. Soc. Rev., 2009, 38, 3348 RSC.
- T. Zeng, T. Zhang, W. Wei, Z. Li, D. Wu, L. Wang, J. Guo, X. He and N. Ma, ACS Appl. Mater. Interfaces, 2015, 7, 11849–11856 CrossRef CAS PubMed.
- S. B. Lowe, J. A. G. Dick, B. E. Cohen and M. M. Stevens, ACS Nano, 2012, 6, 851–857 CrossRef CAS PubMed.
- A. Anne, A. Chovin and C. Demaille, Langmuir, 2012, 28, 8804–8813 CrossRef CAS.
- M. Puiu and C. Bala, Bioelectrochemistry, 2018, 120, 66–75 CrossRef CAS.
- G. Liu, J. Wang, D. S. Wunschel and Y. Lin, J. Am. Chem. Soc., 2006, 128, 12382–12383 CrossRef CAS.
- H. Xiao, L. Liu, F. Meng, J. Huang and G. Li, Anal. Chem., 2008, 80, 5272–5275 CrossRef CAS PubMed.
- J. Adjémian, A. Anne, G. Cauet and C. Demaille, Langmuir, 2010, 26, 10347–10356 CrossRef PubMed.
- N. Zhao, Y. He, X. Mao, Y. Sun, X. Zhang, C. Z. Li, Y. Lin and G. Liu, Electrochem. Commun., 2010, 12, 471–474 CrossRef CAS.
- D.-S. Shin, Y. Liu, Y. Gao, T. Kwa, Z. Matharu and A. Revzin, Anal. Chem., 2012, 85, 220–227 CrossRef PubMed.
- E. González-Fernández, N. Avlonitis, A. F. Murray, A. R. Mount and M. Bradley, Biosens. Bioelectron., 2016, 84, 82–88 CrossRef PubMed.
- E. González-Fernández, M. Staderini, N. Avlonitis, A. F. Murray, A. R. Mount and M. Bradley, Sens. Actuators, B, 2018, 255, 3040–3046 CrossRef.
- J. W. Lee, J. Y. Yun, W. C. Lee, S. Choi, J. H. Lim, H. Jeong, D. S. Shin and Y. J. Park, Sens. Actuators, B, 2017, 240, 735–741 CrossRef CAS.
- E. González-Fernández, M. Staderini, A. Yussof, E. Scholefield, A. F. Murray, A. R. Mount and M. Bradley, Biosens. Bioelectron., 2018, 119, 209–214 CrossRef PubMed.
- J. Heinze, Angew. Chem., Int. Ed. Engl., 1993, 32, 1268–1288 CrossRef.
- R. J. Forster, Chem. Soc. Rev., 1994, 23, 289–297 RSC.
- B. Derkus, Biosens. Bioelectron., 2016, 79, 901–913 CrossRef CAS PubMed.
- L. Soleymani and F. Li, ACS Sens., 2017, 2, 458–467 CrossRef CAS PubMed.
- E. Llaudet, S. Hatz, M. Droniou and N. Dale, Anal. Chem., 2005, 77, 3267–3273 CrossRef CAS PubMed.
- K. Wassum, V. Tolosa, J. Wang, E. Walker, H. Monbouquette and N. Maidment, Sensors, 2008, 8, 5023–5036 CrossRef CAS.
- R. E. Ionescu, C. Fillit, N. Jaffrezic-Renault and S. Cosnier, Biosens. Bioelectron., 2008, 24, 489–492 CrossRef CAS.
- L. Shi, X. Rong, Y. Wang, S. Ding and W. Tang, Biosens. Bioelectron., 2018, 102, 41–48 CrossRef CAS.
- L. Jacobse, S. J. Raaijman and M. T. M. Koper, Phys. Chem. Chem. Phys., 2016, 18, 28451–28457 RSC.
- K. Stulík, C. Amatore, K. Holub, V. Marecek and W. Kutner, Pure Appl. Chem., 2000, 72, 1483–1492 Search PubMed.
- M. Sheffer, V. Vivier and D. Mandler, Electrochem. Commun., 2007, 9, 2827–2832 CrossRef CAS.
- J. M. Artigas, M. E. Garcia, M. R. Faure and A. M. Gimeno, Postgrad. Med. J., 1981, 57, 219–222 CrossRef CAS.
- W. S. Ruddell, C. J. Mitchell, I. Hamilton, J. P. Leek and J. Kelleher, Br. Med. J., 1981, 283, 1429–1432 CrossRef CAS.
- G. N. Somero, Annu. Rev. Ecol. Syst., 1978, 9, 1–29 CrossRef CAS.
- E. D. Stevens and J. M. McLeese, Am. J. Physiol.: Regul., Integr. Comp. Physiol., 1984, 246, 487–494 CrossRef.
- M. Staderini, E. González-Fernández, A. F. Murray, A. R. Mount and M. Bradley, Sens. Actuators, B, 2018, 274, 662–667 CrossRef CAS.
- F. Kuralay, S. Campuzano and J. Wang, Talanta, 2012, 99, 155–160 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2020 |