
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Paper-based microchip electrophoresis for point-of-care hemoglobin testing†
Muhammad Noman
Hasan
 a,
Arwa
Fraiwan
a,
Ran
An
a,
Yunus
Alapan
ab,
Ryan
Ung
a,
Asya
Akkus
a,
Julia Z.
Xu
c,
Amy J.
Rezac
d,
Nicholas J.
Kocmich
d,
Melissa S.
Creary
e,
Tolulope
Oginni
f,
Grace Mfon
Olanipekun
g,
Fatimah
Hassan-Hanga
h,
Binta W.
Jibir
i,
Safiya
Gambo
j,
Anil K.
Verma
k,
Praveen K.
Bharti
k,
Suchada
Riolueang
l,
Takdanai
Ngimhung
l,
Thidarat
Suksangpleng
l,
Priyaleela
Thota
m,
Greg
Werner
n,
Rajasubramaniam
Shanmugam
k,
Aparup
Das
k,
Vip
Viprakasit
lo,
Connie M.
Piccone
pq,
Jane A.
Little
rst,
Stephen K.
Obaro
dg and
Umut A.
Gurkan
*au
a,
Arwa
Fraiwan
a,
Ran
An
a,
Yunus
Alapan
ab,
Ryan
Ung
a,
Asya
Akkus
a,
Julia Z.
Xu
c,
Amy J.
Rezac
d,
Nicholas J.
Kocmich
d,
Melissa S.
Creary
e,
Tolulope
Oginni
f,
Grace Mfon
Olanipekun
g,
Fatimah
Hassan-Hanga
h,
Binta W.
Jibir
i,
Safiya
Gambo
j,
Anil K.
Verma
k,
Praveen K.
Bharti
k,
Suchada
Riolueang
l,
Takdanai
Ngimhung
l,
Thidarat
Suksangpleng
l,
Priyaleela
Thota
m,
Greg
Werner
n,
Rajasubramaniam
Shanmugam
k,
Aparup
Das
k,
Vip
Viprakasit
lo,
Connie M.
Piccone
pq,
Jane A.
Little
rst,
Stephen K.
Obaro
dg and
Umut A.
Gurkan
*au
aCase Biomanufacturing and Microfabrication Laboratory, Department of Mechanical and Aerospace Engineering, Case Western Reserve University, Cleveland, Ohio 44106, USA. E-mail: umut@case.edu; Tel: +1 216 368 6447
bPhysical Intelligence Department, Max Planck Institute for Intelligent Systems, Stuttgart 70569, Germany
cDepartment of Medicine, Duke University Medical Center, Durham, North Carolina 27707, USA
dDivision of Pediatric Infectious Diseases, College of Medicine, University of Nebraska Medical Center, Omaha, Nebraska 68198, USA
eDepartment of Health Management and Policy, University of Michigan School of Public Health, Ann Arbor, Michigan 48109, USA
feHealth Africa, Kano, Nigeria
gInternational Foundation against Infectious Disease in Nigeria, Abuja, Nigeria
hBayero University, Aminu Kano Teaching Hospital, Kano, Nigeria
iHasiya Bayero Pediatric Hospital, Kano, Nigeria
jMurtala Muhammad Specialist Hospital, Kano, Nigeria
kICMR-National Institute of Research in Tribal Health, Nagpur Road, Jabalpur, 482003, Madhya Pradesh, India
lSiriraj Thalassemia Center, Faculty of Medicine Siriraj Hospital, Mahidol University, Bangkok 10700, Thailand
mHemex Health, Inc., Portland, Oregon 97239, USA
nThogus Products, Avon Lake, Ohio 44012, USA
oDivision of Haematology/Oncology, Department of Pediatrics, Faculty of Medicine Siriraj Hospital, Mahidol University, Bangkok, 10700, Thailand
pUniversity Hospitals Rainbow Babies & Children Hospital, Cleveland, Ohio 44106, USA
qDepartment of Pediatrics, School of Medicine, Case Western Reserve University, Cleveland, Ohio 44106, USA
rUniversity Hospitals Cleveland Medical Center, Cleveland, Ohio 44106, USA
sDepartment of Hematology and Oncology, School of Medicine, Case Western Reserve University, Cleveland, Ohio 44106, USA
tDivision of Hematology and Oncology, School of Medicine, University of North Carolina, Chapel Hill, North Carolina 27516, USA
uDepartment of Biomedical Engineering, Case Western Reserve University, Cleveland, Ohio 44106, USA
First published on 2nd March 2020
Abstract
Nearly 7% of the world's population live with a hemoglobin variant. Hemoglobins S, C, and E are the most common and significant hemoglobin variants worldwide. Sickle cell disease, caused by hemoglobin S, is highly prevalent in sub-Saharan Africa and in tribal populations of Central India. Hemoglobin C is common in West Africa, and hemoglobin E is common in Southeast Asia. Screening for significant hemoglobin disorders is not currently feasible in many low-income countries with the high disease burden. Lack of early diagnosis leads to preventable high morbidity and mortality in children born with hemoglobin variants in low-resource settings. Here, we describe HemeChip, the first miniaturized, paper-based, microchip electrophoresis platform for identifying the most common hemoglobin variants easily and affordably at the point-of-care in low-resource settings. HemeChip test works with a drop of blood. HemeChip system guides the user step-by-step through the test procedure with animated on-screen instructions. Hemoglobin identification and quantification is automatically performed, and hemoglobin types and percentages are displayed in an easily understandable, objective way. We show the feasibility and high accuracy of HemeChip via testing 768 subjects by clinical sites in the United States, Central India, sub-Saharan Africa, and Southeast Asia. Validation studies include hemoglobin E testing in Bangkok, Thailand, and hemoglobin S testing in Chhattisgarh, India, and in Kano, Nigeria, where the sickle cell disease burden is the highest in the world. Tests were performed by local users, including healthcare workers and clinical laboratory personnel. Study design, methods, and results are presented according to the Standards for Reporting Diagnostic Accuracy (STARD). HemeChip correctly identified all subjects with hemoglobin S, C, and E variants with 100% sensitivity, and displayed an overall diagnostic accuracy of 98.4% in comparison to reference standard methods. HemeChip is a versatile, mass-producible microchip electrophoresis platform that addresses a major unmet need of decentralized hemoglobin analysis in resource-limited settings.
Introduction
Hemoglobin disorders are among the world's most common monogenic diseases. Nearly 7% of the world's population live with hemoglobin gene variants, with the most prevalent hemoglobinopathies or structural hemoglobin variants being the recessive β-globin gene mutations, βS or S, βC or C, and βE or E.1–3 Hemoglobin S is highly prevalent in sub-Saharan Africa4 and in tribal populations of Central India.5 Hemoglobin C is common in West Africa,6 and hemoglobin E is common in Southeast Asia.7 Hemoglobin S results from a single point mutation in the 6th codon on the β-globin gene replacing the normal amino acid glutamine with the hydrophobic amino acid valine.8–10 Sickle cell disease (SCD) causes the highest morbidity and mortality among hemoglobin disorders.11 SCD arises when these mutations are inherited homozygously (Hb SS or SCD-SS) or paired with another β-globin gene mutation, such as hemoglobin C (Hb SC or SCD-SC) or β-thalassemia (compound heterozygous, Hb Sβthal+/0). In SCD, abnormal polymerization of deoxygenated hemoglobin S makes red blood cells (RBCs) stiff, changes membrane properties, alters shape, and triggers deleterious activation of inflammatory and endothelial cells.12–14 Sickled RBCs are non-deformable and adhesive in the microcirculation,15–20 particularly in parts of the body where the oxygen tension is relatively low, such as the kidney or spleen.13,17,21,22 Abnormally shaped RBCs result in microvascular occlusion and a vicious cycle of enhanced sickling, hemoglobin desaturation, and further vascular occlusion.23–25 In childhood, recurrent splenic infarction from sickled RBCs increases the risk for life-threatening infections.10,26–29 In addition, young children with SCD are at risk of life-threatening cerebral vasculopathy.29 Afflicted patients who survive to adulthood can suffer both acute and chronic painful crises as well as cumulative organ damage and early mortality.29,30 Infections, stroke, and numerous other SCD-related complications can be mitigated by newborn/neonatal screening and comprehensive medical care.29,31,32 Individuals who inherit one copy of hemoglobin S and one copy of the normal hemoglobin A have sickle cell trait (Hb AS or SCD Trait). These people are healthy carriers, but have a 25% chance of transmitting SCD to their offspring. Individuals who carry one copy of hemoglobin C or hemoglobin βthal+/0 and one copy of hemoglobin A may likewise transmit hemoglobin C disease or thalassemia.33 Approximately 70% of individuals with SCD have homozygous Hb SS and over 25% have compound heterozygous Hb SC or Hb Sβthal+/0.8Hemoglobin E is another common structural hemoglobin variant, but it is unique in that it also decreases expression of the β-globin gene.7,34 Individuals with hemoglobin E trait (Hb AE or Hb E Trait) are asymptomatic, and homozygous hemoglobin E (Hb EE or Hb E Disease) causes a mild microcytic anemia. However, hemoglobin E in combination with βthal (Hb Eβthal+/0) causes thalassemia of varying severity.35 Hemoglobin SE disease is similar to Hb Sβthal in terms of clinical manifestations and treatment, though uncommon due to the geographical separation of the S and E variants.35,36 With increasing migration and inter-racial marriages, combinations of these hemoglobin diseases are being encountered all around the world.35,36 Regardless of their geographic origin, all of these common hemoglobin variants need to be screened for, so that individuals with disease can be diagnosed early and managed timely. Newborn/neonatal screening, currently available in resource-rich countries, is needed for optimal management of hemoglobinopathies worldwide.37–39 For example, in high-income countries, which have less than 1% of the global disease burden, over 90% of babies born with SCD survive into adulthood due to established national screening programs and comprehensive care.29 In contrast, in low-income countries, due to lack of nationwide screening and comprehensive care programs, up to 80% of babies born with SCD are undiagnosed and less than half of them survive beyond 5 years of age.29 More than 300![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 babies are born each year with SCD, with 2–3 per 100 births in some high-burden countries with expected under-5 mortality of 50–90%. It is projected that by 2050, about 400000 babies will be born with SCD annually worldwide.29,40 Screening for hemoglobin disorders is not currently feasible in many low-income countries with the high disease burden.29 For example, in Nigeria, 150000 babies are born with the disease every year.41 An estimated 50–90% of these babies die before age 5, in part because they are not diagnosed and hence not treated.15,42–45
000 babies are born each year with SCD, with 2–3 per 100 births in some high-burden countries with expected under-5 mortality of 50–90%. It is projected that by 2050, about 400000 babies will be born with SCD annually worldwide.29,40 Screening for hemoglobin disorders is not currently feasible in many low-income countries with the high disease burden.29 For example, in Nigeria, 150000 babies are born with the disease every year.41 An estimated 50–90% of these babies die before age 5, in part because they are not diagnosed and hence not treated.15,42–45
Hemoglobinopathy screening after birth is mandated by all 50 states in the United States and in the District of Columbia, as well as in many other high-resource states, including The United Kingdom and France.45–49 Newborn screening programs in the United States and other high-resource countries typically involve collection and shipping of blood samples to centralized laboratories.46,50,51 While decentralized blood sample collection and centralized testing works in high-resourced settings, this approach is not practical in resource-limited regions due to the logistical and infrastructural limitations. Hemoglobinopathy screening studies conducted in low-resource settings, using centralized laboratories, have reported that up to 50% of test results never reach to the patients due to infrastructural challenges.52–54
In resource-rich countries, standard clinical laboratory tests (high-performance liquid chromatography (HPLC) or hemoglobin electrophoresis are typically used in the diagnosis of hemoglobin disorders.33 Additionally, genetic testing, usually polymerase chain reaction-based, can be used to precisely identify globin gene mutations.55 However, these advanced laboratory techniques require trained personnel and state-of-the-art facilities, which are lacking or in short supply in countries where the prevalence of hemoglobin disorders is the highest.29 Furthermore, these tests are costly in terms of time, labor, and resources.15,56 For example, it may take days to weeks to receive the test results from centralized clinical laboratories,56 and when screening is conducted in remote areas, locating those who test positive may be difficult or impossible.15,44,56 Therefore, there is a need for affordable, portable, easy-to-use, accurate point-of-care tests to facilitate decentralized hemoglobin testing in resource-constrained countries.20,29 The realities of resource-limited environments demand a fundamentally different approach to diagnosis: one that is affordable, portable, and easily administered by entry-level healthcare workers in local health service settings or in rural areas at the point-of-need.15,44,56 Importantly, test results must be available while the patient is still present so that the test result can be given to the patient or legal guardian(s) immediately, and the treatment and education can begin without losing the patient to follow-up. The World Health Organization (WHO) estimates that more than 70% of SCD related deaths are preventable with early diagnosis and widely available cost-efficient interventions (e.g., pneumococcal vaccinations, daily penicillin, hydroxyurea).27–29,45,57–62 For example, hydroxyurea use has been shown to reduce the vaso-occlusive crises, stroke, infections, malaria, transfusions, and death in children with SCD, warranting the need for wider access to this treatment.29,59–62 Hydroxyurea treatment has been shown to be feasible, safe, and effective in children with SCD living in India63 and in sub-Saharan Africa.29,59,64,65
In this manuscript, we describe the design, development, manufacturing, and clinical testing of a novel point-of-care hemoglobin test, HemeChip. HemeChip is a paper-based, microchip electrophoresis technology that helps with the diagnosis of hemoglobin disorders in resource-limited settings. HemeChip system is composed of a single-use, disposable cartridge and a portable, affordable reader. HemeChip cartridge can be mass-produced at low cost. HemeChip reader houses the cartridge and it is used to separate, image, and track hemoglobin variants in real-time during electrophoresis. The fundamental principle behind the HemeChip technology is hemoglobin electrophoresis, in which different variants can be separated based on charge differences, when subjected to an electric field in the presence of a carrier substrate.66,67 The HemeChip test works with a standard finger-prick or a heel-prick blood sample, or with a venous blood sample. HemeChip test is completed in less than ten minutes, and can be run at the point-of-care, hence, the results would be available during a patient's visit. Furthermore, the compact design of the HemeChip system allows portability for decentralized testing and use at the point-of-need, which eliminates blood sample transfer to central laboratories.
Materials and methods
Fully integrated microchip electrophoresis cartridge allows high-volume manufacturing
The HemeChip cellulose acetate paper-based microchip electrophoresis system (Fig. 1C & D) facilitates, for the first time, real-time tracking and quantitative analysis of hemoglobin electrophoresis process (Fig. 2A & B, Video S1†). The HemeChip cartridge is composed of two injection molded plastic parts made of Optix® CA-41 Polymethyl Methacrylate Acrylic. This single-use, cartridge-based design was transformed from a proof-of-concept laboratory prototype to a version that supports low-cost mass-production via injecting molding (Fig. S1†) as described in ESI.† The top and bottom parts (Fig. S2†) were manufactured with a 1 + 1 injection mold. Cartridge design embodies specific geometrical features and a precisely designed energy director (Fig. S2†) for rapid ultrasonic welding after assembly. Cellulose acetate paper was chosen because of its stability over environmental conditions.68 Cartridge layout, plastic material selection, and injection molding process were engineered to achieve structural integrity, uniform optical clarity, and high light transmission (up to 80%) in the visible spectrum (Fig. S3†). The injection molded HemeChip cartridge embodies a pair of round corrosion-resistant,69 biomedical grade stainless steel 316 electrodes.70 HemeChip electrodes provide oxidative resistance, stability against electrochemical reactions during operation, and reliability of electrical connection with the power source (Fig. 1A). The combination of high stability cellulose acetate paper,68 injection molded Polymethyl Methacrylate Acrylic plastic, and corrosion-resistant biomedical grade stainless-steel electrodes69,70 results in a shelf life of at least two years. The cartridge also houses a pair of buffer pools that are in direct contact with the electrode top surface and the cellulose acetate paper strip (Fig. 1B & C). One corner of the HemeChip cartridge is chamfered to facilitate correct orientation during use (Fig. 1A & B).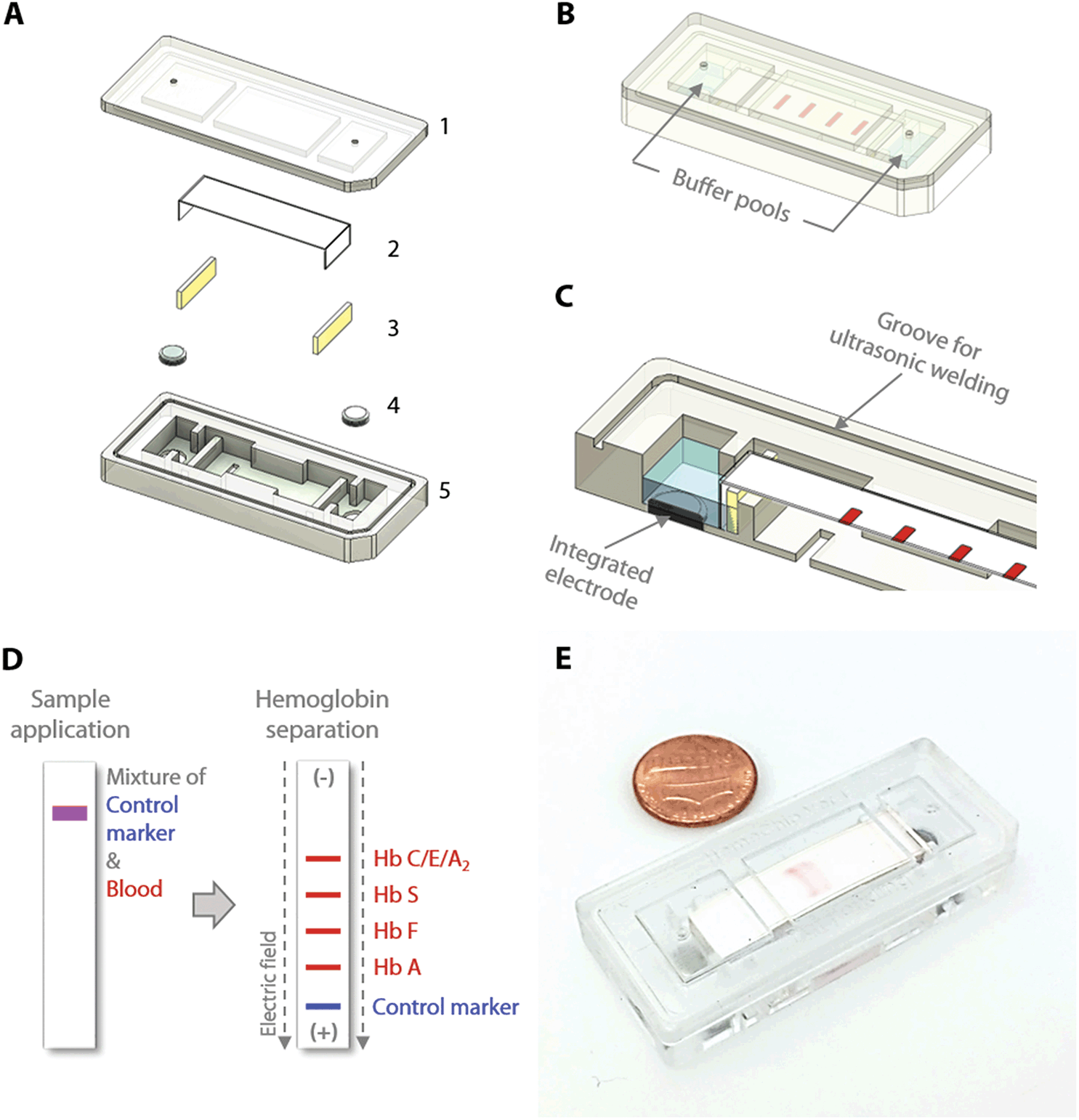 | ||
| Fig. 1 HemeChip: paper-based microchip electrophoresis for hemoglobin testing. HemeChip is a miniaturized, fully integrated cartridge-based microchip electrophoresis system that can be mass-produced. (A) Top (1) and bottom (5) plastic parts are manufactured via injection molding. Cartridge contains a single strip of cellulose acetate paper (2), a pair of blotting pads (3), and integrated stainless-steel electrodes (4). (B) HemeChip cartridge design is compact, fully integrated, and self-contained, including liquid compartments (buffer pools). One corner of the HemeChip cartridge is chamfered to restrict orientation and facilitate correct placement during use. (C) A partial section view of the internal components of HemeChip, showing the cross-section of an electrode and the cellulose acetate paper. (D) A schematic representation of the separation of hemoglobin variants in HemeChip: normal hemoglobin (Hb A), fetal hemoglobin (Hb F), sickle hemoglobin (Hb S), and hemoglobins C/E/A2 that co-migrate. A blue control marker (xylene cyanol) is pre-mixed with blood before sample application into the cartridge. (E) A fully assembled injection molded HemeChip is shown after a completed test with hemoglobin S and C bands that are visible. | ||
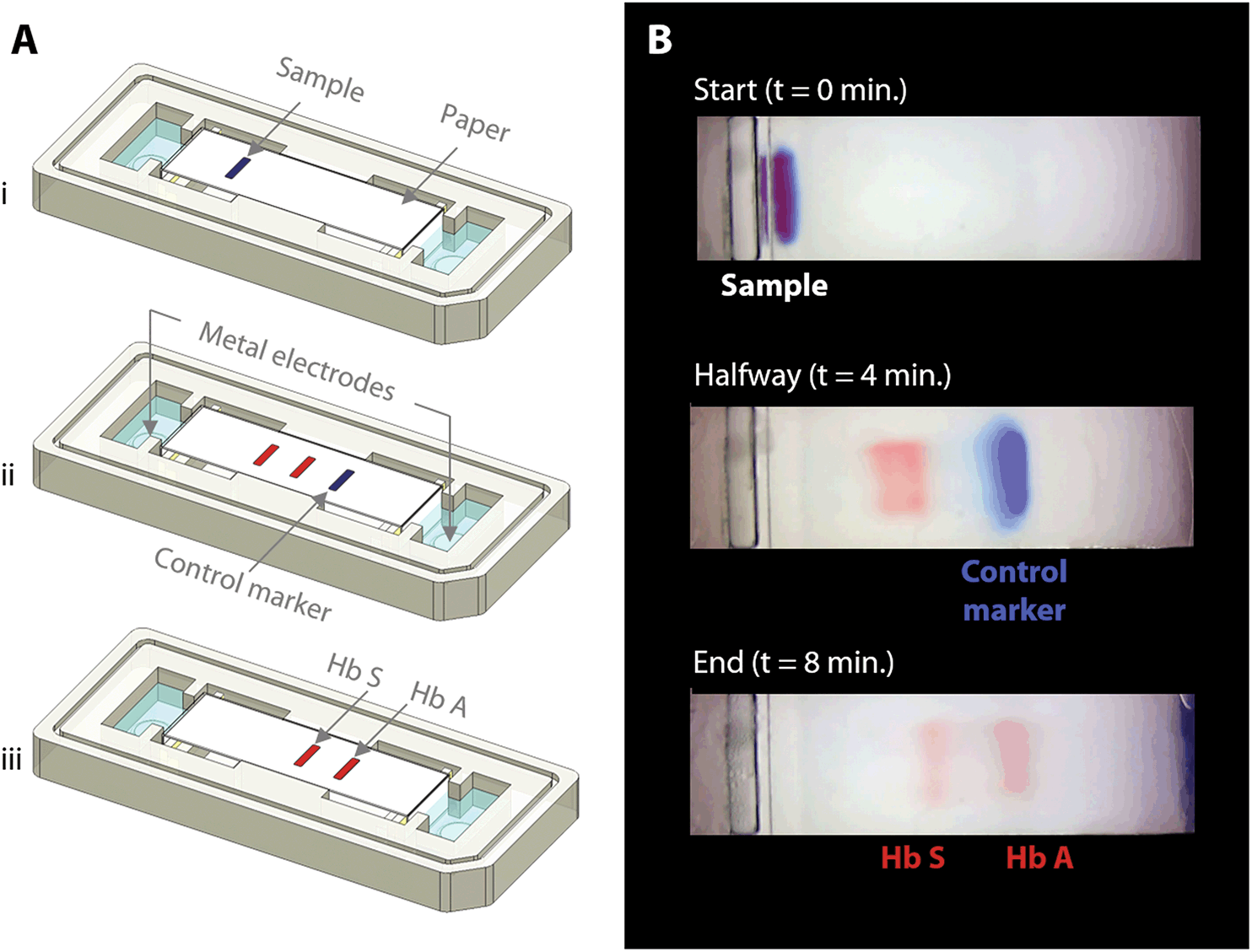 | ||
| Fig. 2 Overview of HemeChip operation and hemoglobin variant separation with a control marker. (A) Schematic illustration of hemoglobin separation and the blue control marker (xylene cyanol) migration in HemeChip. (i) Beginning of the test is shown with blood sample and marker mixture applied. (ii) Hemoglobin bands and the control marker start migrating. Migration of the blue control marker is used in image processing algorithm to confirm that the test is running as expected. (iii) Fully separated hemoglobin bands appear at the end of the test. Control marker migrates all the way to the end and leaves the field of view, at which time, hemoglobin bands are imaged and analyzed in their final positions. (B) Time lapse images captured during a HemeChip test demonstrate the separation of hemoglobin A and S bands, and the migration of the control marker. | ||
Design transformation of HemeChip from laboratory prototype to a mass-producible cartridge
The HemeChip cartridge was previously fabricated by a lamination approach composed of five distinct plastic layers and did not incorporate integrated electrodes (Fig. S1A & B†). In the injected molded design, the number of plastic parts was reduced to two, a top part, and a bottom part, which can be ultrasonically welded to encompass the internal components and integrated stainless-steel electrodes (Fig. S1C & D†). This reduction in the number of injection-molded parts reduced the final assembly time and effort. The current design employs integrated stainless-steel electrodes (Fig. S1C & D†). The choice for electrode material was constrained by the cost and the corrosion resistance of the electrode material. The use of noble metals and/or their alloys, which are the preferred materials for specialized applications,71–74 would have increased the cost of the cartridges significantly. In addition, noble metals can corrode when subjected to electrochemical processes.74–77 The 316 stainless steel, which we employed, is known for its high corrosion resistance.70 It is inexpensive and is available in a variety of stock shapes that are easily machined or processed into the required dimensions. Although the design is sophisticated in view of all the delicate interior features, this adaptation provided more control over the design of the complex interior features, which were unattainable with the lamination-based fabrication approach (Fig. S1A†). These interior features include custom designed buffer pools, buffer basin ribs, metal electrode enclosures, alignment and positioning features for the cellulose acetate paper strips, sample and buffer ports, fiducial markers, and product artwork (Fig. 1A & B). Another improvement to the HemeChip design was the inclusion of the in-chip blotting mechanism (Fig. S1C†). The blotting mechanism is designed to handle any excess buffer that may be applied to the cellulose acetate paper. A notable addition to the transformed design is the energy director and ultrasonic welding groove for the purpose of ultrasonic welding to fully seal the HemeChip cartridge top and bottom pieces (Fig. S2A & B†).HemeChip portable reader design
One advantage of point-of-care technologies is that they can be used in remote locations where use of existing technologies is not feasible.78–80 HemeChip has been designed as a battery-powered, portable test platform to enable hemoglobin testing in remote locations (Fig. 3). HemeChip reader consists of a rechargeable battery power supply, a data acquisition system, and an imaging and image analysis unit (Fig. S4†). The portable HemeChip Reader, once fully charged, allows a minimum of ten hours of testing, which corresponds to at least 48 tests per charge. The reader is equipped with a rechargeable 12 V lithium ion battery with a capacity of 11000 mA h, yielding 132-Watt hours. Each test consumes about 2-Watt hours. The reader enables automated interpretation of test results, local and remote test data storage, and includes geolocation (Global Positioning System). The user is guided through the test process, and the test result is shown on the reader's display and the reader stores the results. The reader can link via Bluetooth or Wi-Fi to a cell phone or PC to transfer the information for review or retention. The reader is equipped to send a PDF version of the results page to a wireless network printer. The data acquisition system is a national instruments data acquisition board (USB-6001) controlled by a custom-built LabView program that records the voltage and current values for the duration of the test. The imaging system consists of an ELP video camera (ELP-USB500W02M) that captures real-time video and the run-time images (Fig. S4†). The imaging system includes 85° wide angle 5 mega pixel HD USB camera and the camera optics are modified with a 3.6 mm, F1.3 mega pixel, CCTV Board Lens. The imaging system and the developed software are calibrated to correct for the fish-eye effect (due to the wide-angle lens) and to normalize the nonlinear pixels-to-distance relation. The power supply, imaging system and data acquisition system are assembled inside a rugged Pelican 1400 case (Fig. 3A). An embedded tablet computer runs the custom-built software to control the reader as well as to acquire, process, and analyze the run-time data. The chamber door houses the backlight used in the imaging system and a magnetic door sensor to ensure that the high voltage supply turns on only when the door is closed (Fig. 3B). An array of white light LEDs is used as the light source for the imaging system. The HemeChip cartridges are imaged using a transmissive light mode.
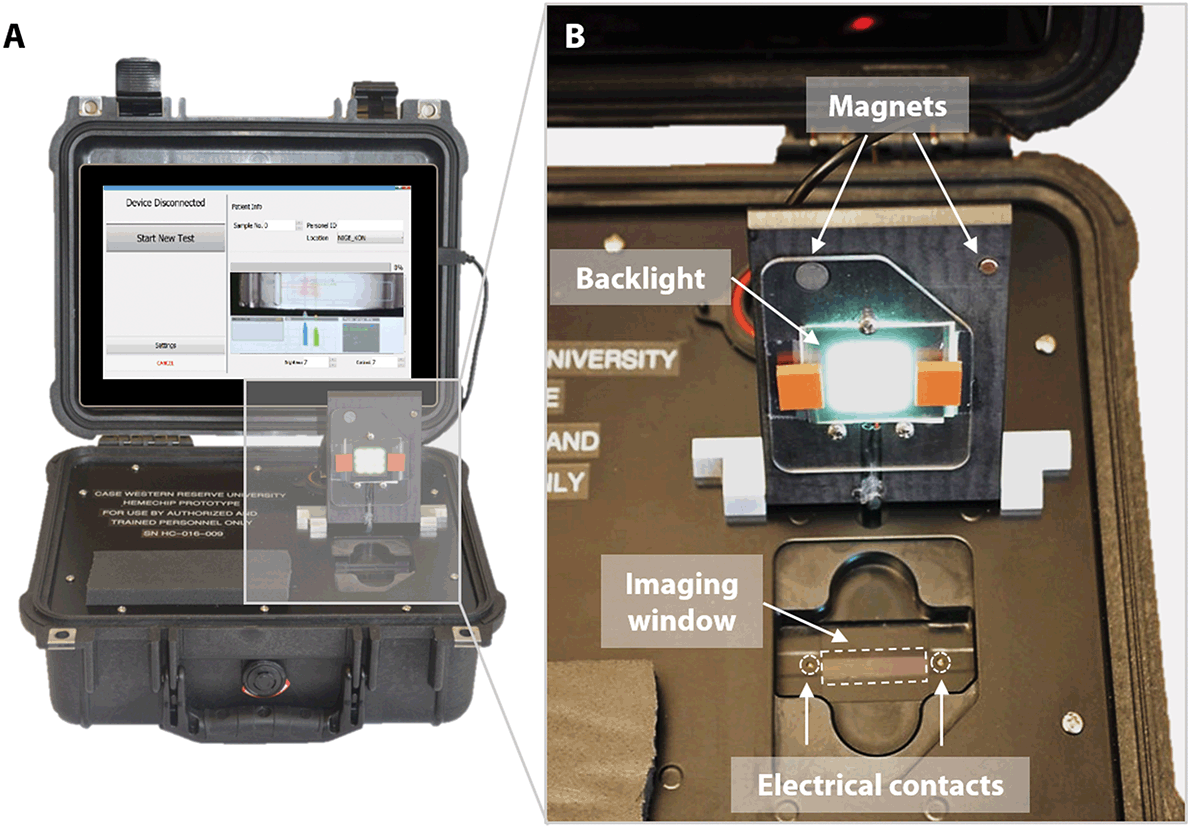 | ||
| Fig. 3 Portable reader for HemeChip. (A) The portable HemeChip prototype readers were built for research use only in a robust Pelican Protector Case for clinical studies worldwide. The reader includes a rechargeable battery, a touch-screen tablet computer, and an integrated imaging system. The reader guides the user through the test procedure via on-screen instructions, allows real-time imaging, automated data analysis, result storage, and wireless transmission of test results. (B) The cartridge chamber in the reader houses electrical contacts, an imaging window, and a backlight for imaging in transmission mode. The chamber door is equipped with magnetic contacts as a safety feature for sensing the door status as open or closed. | ||
User interface and automated image analysis
We developed a user-friendly user interface (UI) that guides the user through the test, performs quality checks, and analyzes the results. This UI has the following functionalities (Fig. S5†): (i) perform hemoglobin separation in HemeChip cartridge by controlling the reader electronic circuit, (ii) collect runtime data for quality checks, (iii) guide the user through the test procedure using step-by-step instructions (Fig. S6 and Video S2†), and (iii) analyze post-run data and images. The UI performs the test control and run-time data collection simultaneously and automatically without any interference or assistance from the user. The UI is designed to generate the data files with specific file name and format based on the information provided before the test, which contains the patient, sample and test identifier. HemeChip tests are monitored real-time with a custom developed application. The real-time tracking of hemoglobin bands (Fig. 4 and Video S1†) and automated image analysis provide necessary information for the identification and quantification of the test results. The application is configured to automatically detect the sample application point and the separated hemoglobin bands. Relative pixel intensities along the paper are used to identify the peaks corresponding to each type of hemoglobin band. The area under each peak is calculated to obtain the relative hemoglobin percentages (Fig. 5A–F). The area under each peak is outlined using the valley-to-valley method commonly used in gas chromatography.81 The HemeChip image analysis algorithm was developed to find the position and intensity of the single hemoglobin band (for homogeneous hemoglobin types) or multiple hemoglobin bands (for heterogeneous hemoglobin types) that appear on the cellulose acetate paper strip inside the HemeChip after a test has been performed. The captured RGB image is analyzed to determine how far the hemoglobin band(s) propagated and the relative percentage of each band based on pixel intensity data.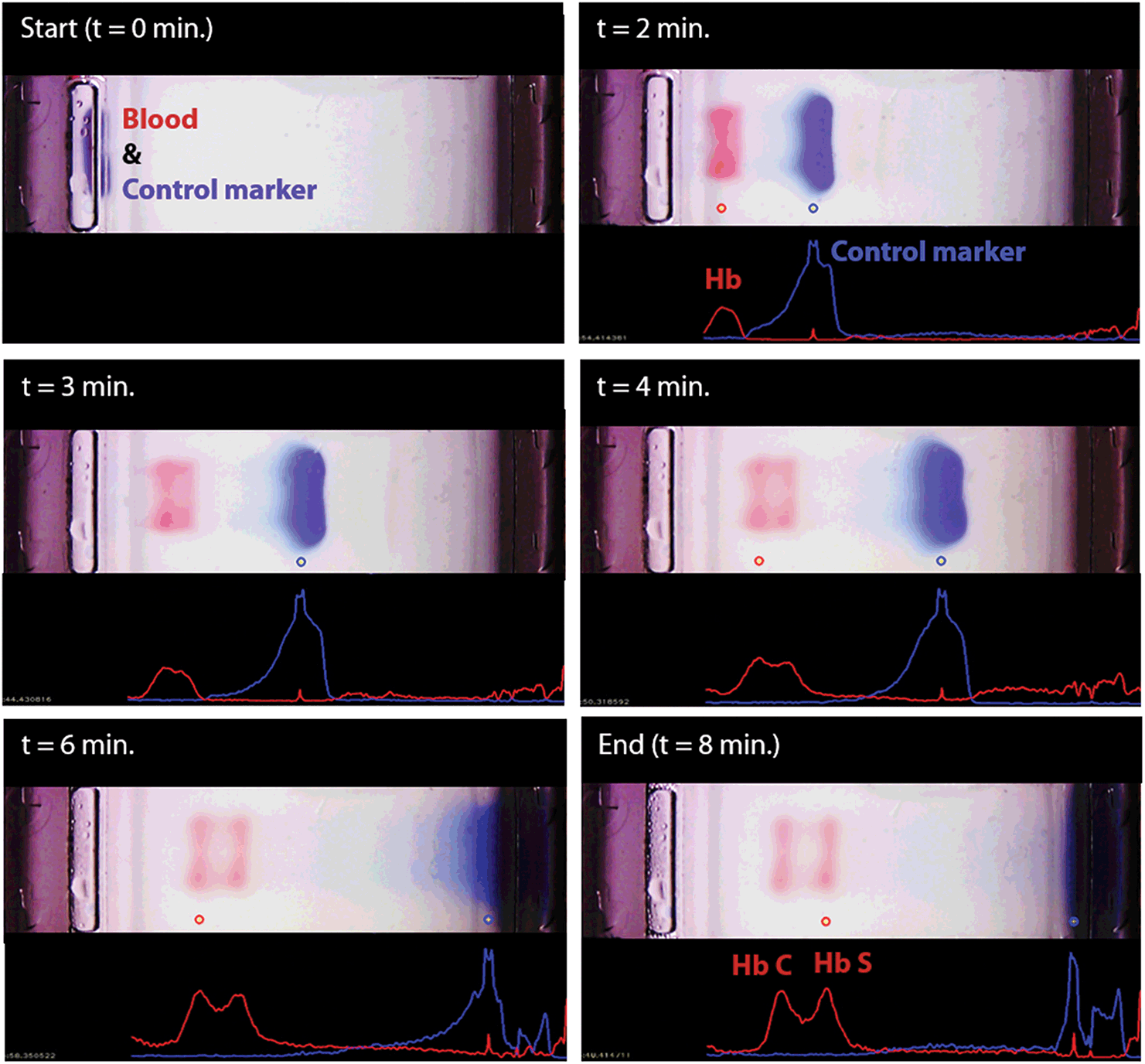 | ||
| Fig. 4 Real-time imaging, image analysis, and tracking of control marker and hemoglobin bands in HemeChip. The time-lapse images show the real-time tracking of the blue control marker and hemoglobin bands. Hemoglobin bands, and the control marker are imaged, automatically recognized, and identified. Movement of the control marker is tracked and used to confirm that the test is running as expected. In this example, hemoglobin types S and C were identified by the algorithm. | ||
Clinical study design and participants
Clinical study design, study participants, sample size calculation, and details on test methods are described according to the Standards for Reporting Diagnostic Accuracy (STARD) guidelines.82 Institutional Review Board (IRB) approved study protocols included the following common objectives: to validate HemeChip technology as a point-of-care platform for hemoglobin testing, to compare the screening results obtained from HemeChip with that obtained from laboratory electrophoresis and/or HPLC as the standard reference methods, to determine the diagnostic accuracy of the test including sensitivity and specificity, and to determine the feasibility of using HemeChip as a point-of-care testing platform in low and middle income countries. We obtained approvals from Institutional Review Boards at University Hospitals Cleveland Medical Center (UHCMC IRB# 04-17-15), University of Nebraska Medical Center (UNMC IRB# 754-17-CB), Amino Kano Teaching Hospital in Kano, Nigeria (AKTH/MAC/SUB/12A/P-3/VI/2102), the Kano State Ministry of Health in Nigeria (MOH/Off/797/T.I/377). The study was registered in United States Library of Medicine's ClinicalTrials.gov (Identifier: NCT03948516). The study protocol in India was approved by the institutional ethical committee of ICMR (Indian Council of Medical Research) National Institute of Research in Tribal Health, Jabalpur, Madhya Pradesh, India (Letter vide no. NIRTH/IEC/1153/2017). The study protocol in Thailand was approved by the Siriraj Institutional Review Board (SiRB Protocol no. 906/2561(EC1)). Informed consents were obtained from all participants of these clinical studies.Clinical sample size estimation was based on published methods by incorporating disease prevalence into sample size calculation83 with 95% confidence interval and with estimated sensitivity and specificity of 98% (Table S3†). We estimated the SCD-SS (homozygous S) disease prevalence in the study population in Nigeria at 3%,84 which resulted in a minimum sample size of 251. SCD-SS prevalence was estimated at 2.7% in Central India,85 which resulted in a minimum sample size of 279 in India. In Bangkok, Thailand, since homozygous Hb E disease prevalence is low (1.8%), we utilized a combined homozygous and heterozygous Hb E (with or without α-thalassemia) prevalence estimated at 8.7%,86 which resulted in a minimum sample size of 87.
315 children (6 weeks to 5 years of age) were tested in Kano, Nigeria. Study participants were enrolled as part of the existing Community-Acquired Pneumonia and Invasive Bacteremia Disease study at three government hospitals, Amino Kano Teaching Hospital, Murtala Muhammad Specialist Hospital, and Hasiya Bayero Pediatric Hospital. Inclusion criteria for the Nigeria study included: 6 weeks to 5 years of age, fever or hypothermia and one of the following conditions: prostration, excessive crying, poor feeding, altered consciousness, convulsion, difficulty breathing, profuse vomiting, diarrhea, and a provision of signed and dated informed consent by the parent or guardian. Exclusion criteria for the Nigeria study included parent or child choosing to opt out of the study after initial consent, and blood transfusion within 3 months of the study enrollment. In Cleveland, Ohio, 31 adult participants were recruited to the study as part of an ongoing clinical trial (ClinicalTrials.gov Identifier: NCT02824471). The main purpose of the clinical study in Cleveland was to test SCD-SC blood samples, which are rare in Nigeria and India.
124 subjects (7 weeks to 63 years old) were tested in Bangkok, Thailand by local laboratory personnel. Surplus blood samples were obtained from patients undergoing clinical laboratory testing at Siriraj Hospital in Bangkok. Hb AA, Hb AE, and Hb EE (with or without alpha-thalassemia) samples were identified by laboratory personnel using a combination of complete blood count, low-pressure liquid chromatography (LPLC) or HPLC, and clinical history of hemoglobin disorders. Inclusion criteria for the Thailand study included: any age or gender, residual blood sample available with a known date of collection within 1 month, and predicted hemoglobin types of Hb AA, Hb AE, or Hb EE. Exclusion criteria for the Thailand study included: unknown date of sample collection, blood sample older than 1 month at the time of use, any findings on complete blood count, LPLC or HPLC, or clinical history to suggest the presence of a beta-globin variant other than Hb E, such as a high quantitative Hb A2 in the range suggestive of beta-thalassemia.
298 subjects (8 months to 65 years old) were tested at a referral testing facility of ICMR-National Institute of Research in Tribal Health located at Late Baliram Kashayap Memorial Medical College, Jagdalpur, Chhattisgarh, India. Inclusion criteria included: subjects of 6 weeks or older, with or without the signs and symptoms of pallor, jaundice, abdominal pain, joint pain and provision of a signed informed consent (either by the subject or by the parent as appropriate if the subject is a minor). Subjects who withdrew their consent after enrolling were excluded from the study.
Blood sample acquisition and testing
In all clinical studies, blood samples were collected as part of the standard clinical care and only surplus de-identified blood samples were utilized for testing. The whole blood samples were tested with both HemeChip and the reference standard HPLC (VARIANT™ II, Bio-Rad Laboratories, Inc., Hercules, California) in Cleveland, Ohio, Nigeria, and Thailand. In India, blood samples were tested with both HemeChip and the standard laboratory-based reference standard cellulose acetate electrophoresis, followed by HPLC testing for discordant results. HPLC and cellulose acetate electrophoresis procedures were performed according to manufacturer's guidelines. HPLC was considered to be the reference standard for our comparisons. In any disagreement between HemeChip, cellulose acetate electrophoresis and HPLC, HPLC result was considered to be correct. HPLC %Hb results were normalized to a total of 100% for the cases in which the summation of reported Hb% values were less than 100%. In India, hemoglobin percentage values were reported for only Hb S, A2, F by the standard reference laboratory, hence the balance percentage was assumed to be Hb A. The HemeChip reader guides the user step-by-step through the test procedure (Fig. S6†) with animated on-screen instructions to minimize user errors. Local users were trained to use a custom-designed micro-applicator (Fig. S7†), which is included in a kit (Fig. S8†) with visual instructions for use (Fig. S9†). Hemoglobin identification and quantification is automatically performed with a custom software on the reader and results are reported to the user in a clear and objective way (Fig. S10–S15†).Clinical information and reference test results were not available to the performers of the test or the study team at the time of testing. Similarly, HemeChip test results were not available to the performers of the standard reference tests. In Nigeria, HemeChip tests were performed on eHealth Africa campus in Kano, Nigeria, by local healthcare workers using blood samples collected at the nearby Hasiya Bayero Pediatric Hospital, Murtala Muhammad Specialist Hospital, and Aminu Kano Teaching Hospital. Clinical standard (HPLC) testing was done independently by the International Foundation Against Infectious Disease in Nigeria (IFAIN, Abuja, Nigeria) for the blood samples obtained in Kano. In Cleveland, Ohio, HemeChip tests were performed at Case Biomanufacturing and Microfabrication laboratory by the research team members. HPLC testing was done independently by the University Hospitals Cleveland Medical Center Clinical Laboratories (Cleveland, Ohio) for the blood samples obtained in Cleveland. In Thailand, HemeChip tests were performed at Siriraj Thalassemia Center by local laboratory personnel. Reference standard testing (HPLC) was independently performed at ATGenes Co. Ltd (Bangkok, Thailand). In India, HemeChip tests and standard reference cellulose acetate electrophoresis tests were performed by local laboratory personnel at the testing facility of ICMR-National Institute of Research in Tribal Health located at Late Baliram Kashyap Memorial Medical College located in Jagdalpur, Chhattisgarh, and HPLC testing was performed at the central laboratory of ICMR-National Institute of Research in Tribal Health, Jabalpur, Madhya Pradesh, India.
Statistical methods
Hemoglobin band separation data was analyzed statistically using one-way Analysis of Variance (ANOVA) test with p-value set at 0.001 for statistically significant difference (Fig. 6A–F). Tukey post-hoc test was used for multiple comparisons, where applicable. Hemoglobin percentage quantification agreement between HemeChip and the reference standard method (HPLC) was assessed by Bland-Altman analysis for heterozygous samples tested (Fig. 6G–I). For homozygous samples tested, Bland-Altman analysis was not performed, since for such samples, both HemeChip and the reference standard method display only one Hb%, which is greater than 90%. The Bland-Altman method was used to determine the repeatability of HemeChip quantification using residual analysis by comparison to the standard method. The coefficient of repeatability was set as 1.96 times the standard deviations of the differences between the two measurements. Diagnostic accuracy of HemeChip test in comparison to HPLC was determined as the percent ratio of correct tests results, and summation of correct tests results and incorrect test results for a hemoglobin variant category (Table 2). Sensitivity was determined as the ratio of true positive results divided by the summation of true positive results and false negative results. Specificity was determined as the ratio of true negative results divided by the summation of true negative results and false positive results (Table 3). Sensitivity, specificity, positive predictive value (PPV), and negative predictive value (NPV) were analyzed and reported for binary pairs of SCD-SS vs. others, SCD-SC vs. others, SCD Trait vs. others, Hb E Disease vs. others, Hb E Trait vs. others (Table 3).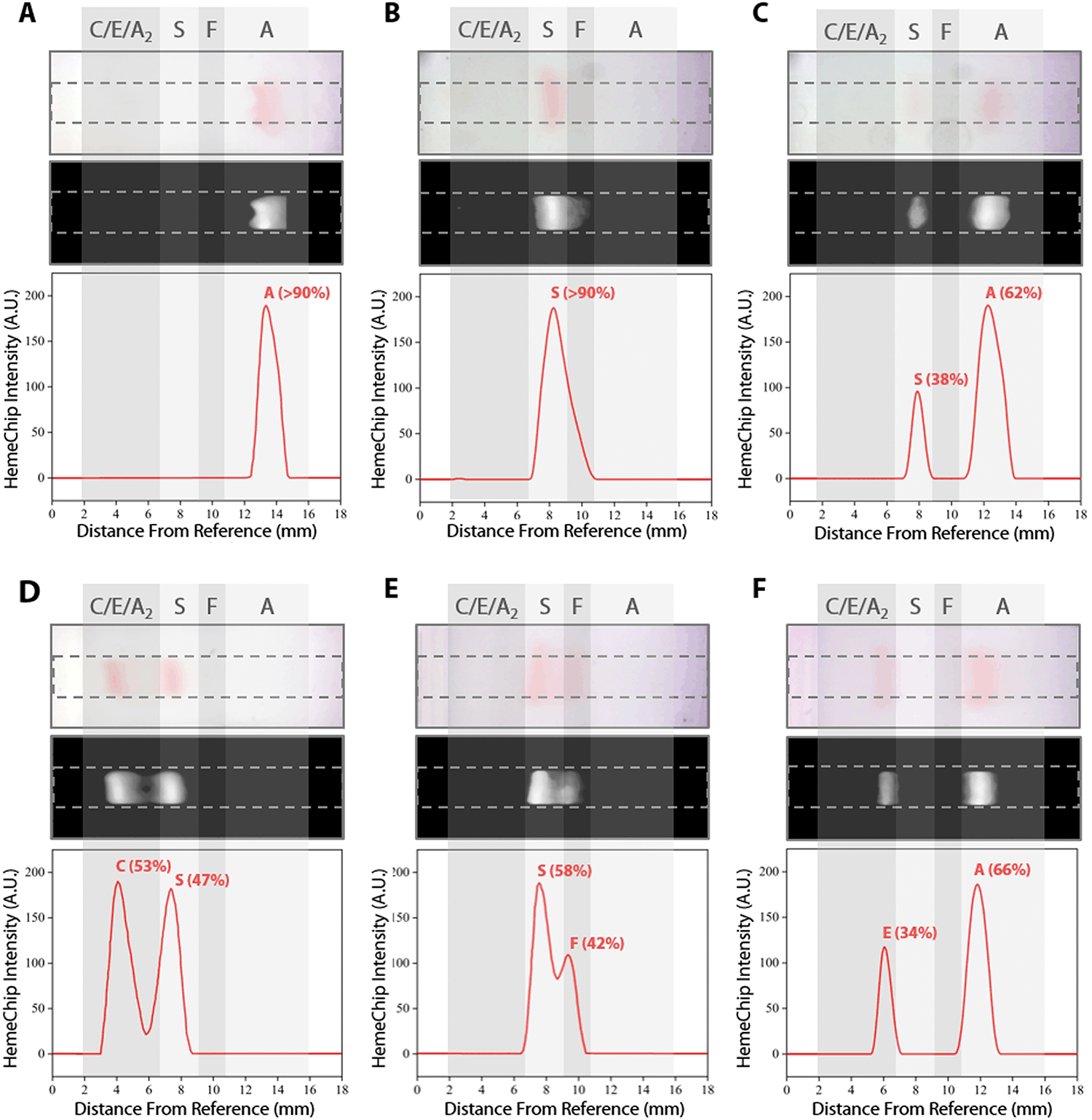 | ||
| Fig. 5 Identification of hemoglobin types and quantification of hemoglobin percentages by HemeChip. Typical HemeChip results are presented for the main hemoglobin variants tested. For each subset, the top image represents the raw image captured by the HemeChip reader at the end of the test. The middle black and white image is generated by the image analysis algorithm to produce the intensity curves (bottom image) for peak identification and Hb% quantification. Hb% values displayed were determined by HemeChip, which showed agreement with the reference standard method (HPLC). Gray regions indicate the approximate zones in which A, F, S, and C/E/A2 bands appear at the end of the test. (A) Normal (no abnormal Hb) with Hb AA (>90%). (B) SCD-SS with Hb SS (>90%). (C) SCD Trait with Hb AS (A: 62%, S: 38%). (D) SCD-SC with Hb SC (S: 47%, C: 53%). (E) SCD-SS with Hb SF (S: 58%, F: 42%). (F) Hb E Trait with Hb AE (A: 66%, E: 34%). | ||
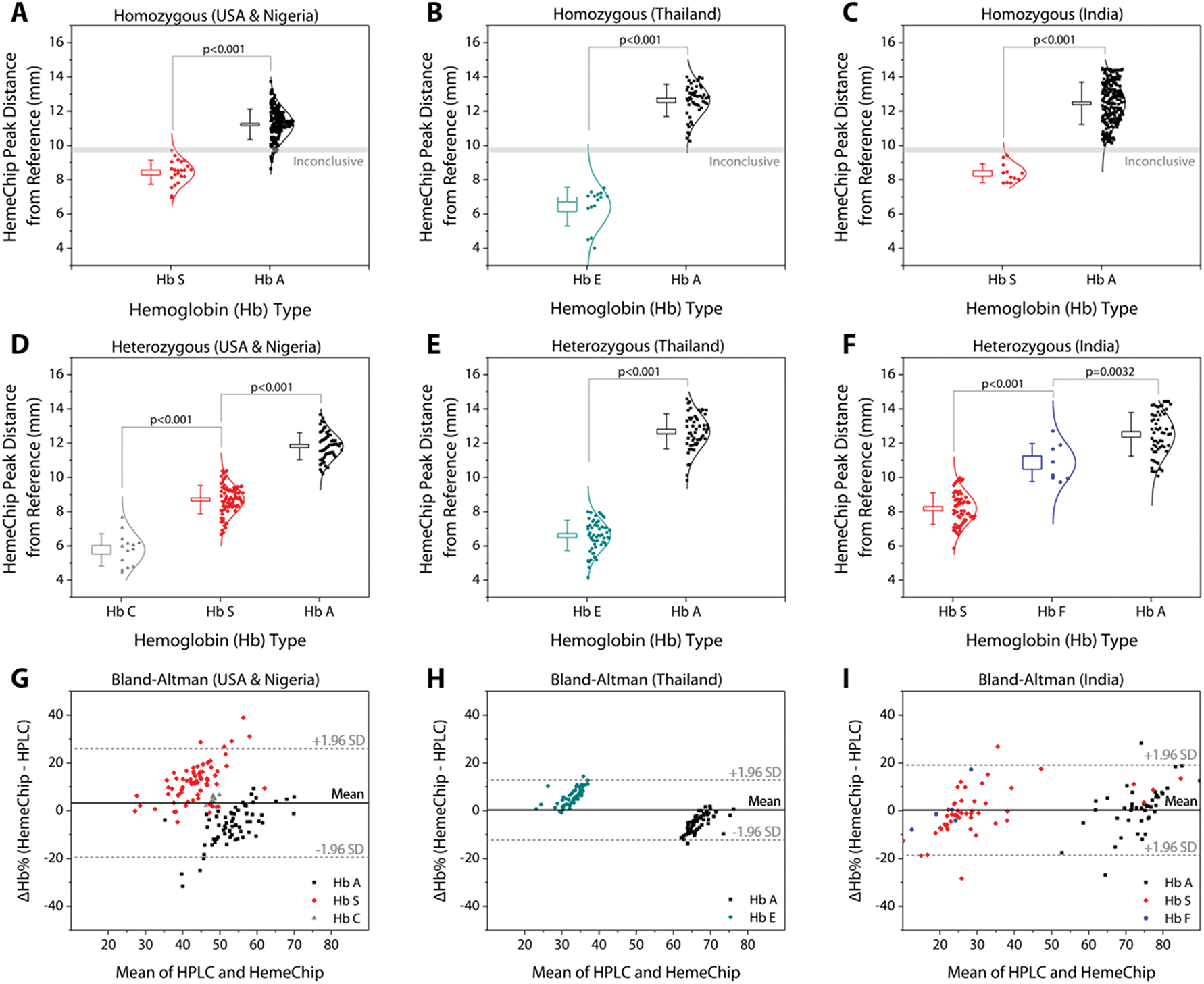 | ||
| Fig. 6 HemeChip hemoglobin band separation and quantification. The top row are data distribution plots that show the hemoglobin band separation for homozygous subjects tested in: (A) the USA (9 HbSS) and Nigeria (13 Hb SS, 212 Hb AA), (B) Thailand (15 Hb EE, 50 Hb AA), and (C) India (11 Hb SS, 219 Hb AA). The middle row are data distribution plots for heterozygous subjects tested in: (D) the USA (7 Hb AS, 14 Hb SC) and Nigeria (60 Hb AS), (E) Thailand (57 Hb AE), and (F) India (60 Hb AS, 7 Hb SF, 1 Hb AF). The horizontal lines between hemoglobin positions represent statistically significant differences based on one-way Analysis of Variance (ANOVA) with Tukey post-hoc test for multiple comparisons, where applicable. In whisker plots, brackets represent standard deviation of the mean, and boxes represent standard error of the mean. Lower row are Bland-Altman analysis plots comparing HemeChip with reference standard method (HPLC) for hemoglobin percentage quantification for the heterozygous subjects tested in the USA and in Nigeria (G), Thailand (H), and India (I). In Bland-Altman plots, solid line indicates the mean difference and dashed lines indicate 95% limits of agreement defined as mean difference ± 1.96 times standard deviation (SD). HemeChip reports hemoglobin C/E/A2. Test location was used to differentiate co-migrating hemoglobins C and E. | ||
| Total | Valid tests | Uninterpretable tests | Inconclusive tests | |
|---|---|---|---|---|
| 768 subjects were tested in clinical studies with HemeChip and the reference standard method. 315 subjects (6 weeks to 5 years old) were tested in Kano, Nigeria by local users. 124 subjects (7 weeks to 63 years old) were tested in Bangkok, Thailand by local users. 298 subjects (8 months to 65 years old) were tested in Jagdalpur, Chhattisgarh, India by local users. 31 subjects (20 to 59 years old) were tested in Cleveland, Ohio, United States, by the research team. | ||||
| Subjects and tests | 768 | 732 | 24 | 12 |
| Percentage | 100% | 95.3% | 3.1% | 1.6% |
| Category | Hemoglobin type | Correct | Incorrect | Accuracy |
|---|---|---|---|---|
| a Nine subjects with normal hemoglobin (Hb AA) were identified as SCD by HemeChip. b Three subjects with compound heterozygous Sβ-thalassemia+/0 were identified as SCD by HemeChip. HemeChip reports hemoglobin C/E/A2. Test location was used to differentiate co-migrating hemoglobins C and E. | ||||
| SCD-SS | Hb SS | 33 | 0 | 100% |
| SCD-SC | Hb SC | 14 | 0 | 100% |
| SCD trait | Hb AS | 129 | 0 | 100% |
| Hb E disease | Hb EE | 15 | 0 | 100% |
| Hb E trait | Hb AE | 57 | 0 | 100% |
| Normal (no abnormal Hb) | Hb AA | 472 | 9a | 98.1% |
| SCD-Sβ+, SCD-Sβ0 | Hb SAA2, Hb SFA2 | 0 | 3b | — |
| All categories | — | 720 | 12 | 98.4% |
| SCD-SS vs. others | SCD-SC vs. others | SCD trait vs. others | Hb E disease vs. others | Hb E trait vs. others | |
|---|---|---|---|---|---|
| HemeChip reports hemoglobin C/E/A2. Test location was used to differentiate co-migrating hemoglobins C and E. | |||||
| True positive, TP | 33 | 14 | 129 | 15 | 57 |
| True negative, TN | 687 | 706 | 591 | 705 | 663 |
| False positive, FP | 9 | 0 | 0 | 0 | 0 |
| False negative, FN | 0 | 0 | 0 | 0 | 0 |
| Sensitivity, TP/(TP + FN) | 100.0% | 100.0% | 100.0% | 100.0% | 100.0% |
| Specificity, TN/(TN + FP) | 98.7% | 100.0% | 100.0% | 100.0% | 100.0% |
| PPV, TP/(TP + FP) | 78.6% | 100.0% | 100.0% | 100.0% | 100.0% |
| NPV, TN/(TN + FN) | 100.0% | 100.0% | 100.0% | 100.0% | 100.0% |
Results
HemeChip separates, images, and tracks hemoglobin variants real-time during electrophoresis
The fundamental principle behind the HemeChip technology is hemoglobin electrophoresis, in which different hemoglobin variants can be separated based on electric charge differences when subjected to an electric field in the presence of a carrier substrate.66,67 The HemeChip test works with a standard finger-prick or a heel-prick blood sample that is collected according to the WHO guidelines for drawing blood,87 which typically yields about 25 μL per drop.88 The test also works with venous blood samples. For ease of handling, 20 μL of blood is collected and diluted with 40 μL of lysing solution. Next, less than 1 μL (0.56 μL ± 0.17 μL, N = 5) of lysed blood sample is transferred into the cartridge for electrophoresis (Fig. 1D). Actual blood volume utilized per test is approximately 0.2 μL. The HemeChip separates hemoglobins A, F, S, C/E/A2 on cellulose acetate paper that is subjected to an electric field (Fig. 1D). Tris/Borate/EDTA (TBE) buffer is used to provide the necessary ions for electrical conductivity at a pH of 8.4 in the cellulose acetate paper.66,67 The pH induced net negative charges of the hemoglobins cause them to travel from the negative to the positive electrode when placed in an electric field (Fig. 1D and Video S1†). Differences in hemoglobin mobilities due to their negative electric charge allow hemoglobin separation to occur (Fig. 1D) with visible results at the end of a HemeChip test (Fig. 1E). Among the four major hemoglobin types that we tested, hemoglobins C/E/A2 co-migrate and they are the slowest. Hemoglobin A is the fastest moving hemoglobin in alkaline solution. A unique feature of HemeChip is that it utilizes a blue control marker (xylene cyanol) that is pre-mixed with the blood sample before application into the cartridge (Fig. 1D & 2A). Towards the end of the test, the blue control marker reaches the end of the cellulose acetate paper strip (Fig. 1D) and disappears into the buffer pool, leaving behind the separated hemoglobin variant bands (Fig. 2B). At this point, the hemoglobin bands are automatically identified by a custom software based on their respective locations on the cellulose acetate paper strip relative to the sample application point (Fig. 4). Hemoglobins in the blood sample form visible bands at the end of the test due to the natural bright red color of the protein (Fig. 1E & 2B). This feature of naturally red, visible hemoglobin, combined with optically clear HemeChip cartridge (Fig. S3†) in transmission imaging mode (Fig. 2B) within the reader's imaging chamber (Fig. 3A & B), negates the need for picrosirius red staining, which is typically utilized in benchtop cellulose acetate hemoglobin electrophoresis.66,67HemeChip automatically identifies hemoglobin variants and determines their relative percentages
The blue control marker mobility on the cellulose acetate paper strip is tracked real-time by the image processing and decision algorithm (Fig. S5†). The mobility of the control marker is then used to confirm that the test is running as expected (Fig. 4 and Video S1†). Briefly, the algorithm searches and finds the sample application mark (Fig. 4, t = 0 min), and the intensity curve is generated for the red hemoglobin bands and the blue control marker. The intensity curve is evaluated to detect and track the movement of the peaks throughout the process (Fig. 4). The peaks are identified based on their final locations at the end of the test (Fig. 4, t = 8 min). The distance between each peak and the application point and the relative percentage of the areas under the peaks are evaluated to determine whether they fit into the categories pre-defined in the software for hemoglobin C/E/A2, hemoglobin S, hemoglobin F, and hemoglobin A. The amount of hemoglobin is presented as a percentage that is relative to that of the other hemoglobin bands in the sample. In the case of a single hemoglobin variant detected, such as Hb AA (Fig. 5A) or Hb SS (Fig. 5B), the HemeChip software reports a percentage value greater of >90%, which agrees with the results reported by the reference standard method (HPLC). If there is more than one peak identified, then the area under each of the peaks is calculated and the relative percentages are reported, for example in the cases of Hb AS (Fig. 5C), Hb SC (Fig. 5D), Hb SS blood containing fetal hemoglobin (Hb F) (Fig. 5E), and Hb AE (Fig. 5F). Hemoglobin A and hemoglobin S separation is significantly greater (p < 0.001) than hemoglobin F and hemoglobin S separation (Fig. S16†), which can be used to distinguish between Hb SF and Hb AS.HemeChip accurately identifies hemoglobin bands and quantifies hemoglobin variants in blood
Band positions for hemoglobin C/E/A2, hemoglobin S, hemoglobin F, and hemoglobin A were reported for homozygous and heterozygous subjects (Fig. 6). The band separation between the hemoglobin types was significantly different (Fig. 6A–F). For homozygous subjects tested in Nigeria (Fig. 6A), one Hb SS and eleven Hb AA test results were in the ‘Inconclusive’ zone (9.59 mm to 9.89 mm) between A and S bands (Table 1). Nine Hb AA subjects tested appeared below the ‘Inconclusive’ zone, resulting in false-positives or incorrect identification as Hb SS (Table 2). No test results were in the inconclusive zone for the subjects tested in Thailand (Fig. 6B) or in India (Fig. 6C). Heterozygous subjects tested include Hb AS and Hb SC in Nigeria and the USA (Fig. 6D), Hb AE in Thailand (Fig. 6E), and Hb AS, Hb SF, and Hb AF in India (Fig. 6F). In heterozygous subjects tested, appearance of two bands in different locations (Fig. 5C–F) negates the need for an ‘Inconclusive’ zone. Bland-Altman analysis showed an agreement between quantitative HemeChip results and HPLC results for the hemoglobin percentage measurements (Fig. 6G–I). For subjects tested in the USA and in Nigeria, Bland-Altman analysis showed a mean bias of +3.27% for HemeChip in comparison to HPLC with a systematic over-estimation of %Hb S at higher mean values and under-estimation of %HbA in lower mean values (Fig. 6G). For subjects tested in Thailand, Bland-Altman analysis showed a mean bias of +0.27% for HemeChip compared to HPLC (Fig. 6H). For subjects tested in India, Bland-Altman analysis showed a mean bias of +0.25% for HemeChip in comparison to HPLC (Fig. 6I).Clinical testing of HemeChip in the United States, Nigeria, Thailand, and India
We report the clinical study results and HemeChip diagnostic accuracy according to the STARD guidelines.82 A detailed description of the study design, participants, test methods, and analyses are available in the Methods section. In clinical validation studies, we tested a total of 768 subjects with both HemeChip and reference standard methods, in the United States, Nigeria, Thailand, and India (Table 1). We defined ‘Valid’, ‘Uninterpretable’, and ‘Inconclusive’ tests according to published recommendations in literature89 and the STARD guidelines.82 A ‘Valid’ test was defined as a test that performed as expected according to objective standards, such as the normal movement of the blue control marker (Fig. 4). Examples of ‘Valid’ test results are presented in ESI (Fig. S10–13†). An ‘Uninterpretable’ test was defined as a test that failed to produce any results and that did not meet the minimum set of objective criteria which constitute a valid or adequate test. Reasons for ‘Uninterpretable’ tests include the poor migration of the blue control marker, electrical connectivity issues, or faulty cartridges. An example ‘Uninterpretable’ test is shown in ESI (Fig. S14†). An ‘Inconclusive’ test was defined as a test that performed adequately according to an objective set of standards, such as the satisfactory movement of the blue control marker and appearance of hemoglobin bands, but the test algorithm could not make an identification of hemoglobin types present. Reasons for ‘Inconclusive’ tests include appearance of a band or bands at or close to the borderline region between two adjacent detection windows (Fig. 6A). An example ‘Inconclusive’ test is shown in ESI (Fig. S15†). An ‘Uninterpretable’ or ‘Inconclusive’ test can be recognized by the image analysis algorithm during or at the end of the test. In addition to the ‘Valid’, ‘Uninterpretable’, and ‘Inconclusive’ tests reported here, a number of tests were not included in the data analysis due to the following reasons: missing reference standard results, sample labeling errors, and the tests performed for user training or algorithm development. Among the total 768 test results reported here, 732 tests were ‘Valid’ (95.3%), 24 tests were ‘Uninterpretable’ (3.1%), and 12 were ‘Inconclusive’ (1.6%) (Table 1). An ‘Uninterpretable’ or ‘Inconclusive’ test does not result in a diagnostic decision.89 As such, when an ‘Uninterpretable’ or ‘Inconclusive’ test is encountered, the test can be repeated or other methods of testing can be utilized, which are common practices in diagnostics,89 as well as in hemoglobin testing.66,67 Therefore, based on recommended practices in the literature89 and the STARD guidelines,82 the uninterpretable and inconclusive test results were reported separately in this study (Table 1).Results of clinical studies with HemeChip indicate high diagnostic accuracy in hemoglobin testing
In clinical studies, HemeChip test results included the following (Table 2): SCD-SS (Hb SS), SCD-SC (Hb SC), SCD Trait (Hb AS), Hb E Disease (Hb EE), Hb E Trait (Hb AE), and Normal (no abnormal Hb, or Hb AA). HemeChip identified SCD-SS, SCD-SC, SCD Trait, Hb E Disease, and Hb E Trait with 100% accuracy (Table 2). Nine subjects with no abnormal Hb (Hb AA) were identified as SCD-SS. Three subjects with compound heterozygous Sβ-thalassemias (2 subjects with SCD-Sβ+, 1 subject with SCD-Sβ0) were identified as SCD by HemeChip (Table 2). Sensitivity and negative predictive value (NPV) were 100% for all hemoglobin types tested (Table 3). Specificity was 98.7% for SCD-SS vs. others, and 100% for all other types (Table 3). Positive predictive value was 78.6% for SCD-SS vs. others, and 100% for all other types (Table 3). For studies reporting diagnostic accuracy, one of the recommendations in the published guidelines is that, inconclusive results are excluded from binary statistics (i.e., sensitivity and specificity analyses), but an additional summary statistic is reported that accounts for them.89 Based on these recommendations, if the twelve inconclusive results are considered as incorrect, then the overall diagnostic accuracy of HemeChip would be 96.8%. When the inconclusive results are reported separately according to the published guidelines,82,89 the overall diagnostic accuracy of HemeChip is 98.4% in identifying SCD-SS, SCD-SC, SCD Trait, Hb E Disease, Hb E Trait, and Normal (Table 2).Discussion
HemeChip combines the benefits of standard hemoglobin electrophoresis with the benefits of a point-of-care test. HemeChip technology has a number of fundamental differences and unique features when compared to standard hemoglobin electrophoresis techniques. For example, HemeChip replaces the benchtop laboratory setup66,67 with a portable reader, and replaces the hemoglobin controls66,67 with a blue control marker (Fig. 4). HemeChip provides hemoglobin type identification and quantitative results of relative hemoglobin percentages (Fig. 5). The overall simplicity of the HemeChip enables any user to quickly and accurately screen for SCD and other hemoglobin disorders. The user is guided through the step-by-step process with on-screen animated instructions, which minimizes errors (Fig. S6 and Video S2†). HemeChip does not require a dedicated lab environment and battery operation allows use in remote places lacking electrical power. These critical features distinguish HemeChip from currently available laboratory methods (Table S1†) and other emerging point-of-care technologies for hemoglobin testing (Table S2†).Clinical studies with HemeChip in the USA, sub-Saharan Africa, Central India, and Southeast Asia demonstrated 100% sensitivity and specificity for SCD-SC, SCD Trait, Hb E Disease, and Hb E Trait (Table 3); and 100% sensitivity and 98.7% specificity for SCD-SS. The 100% sensitivity achieved in these clinical studies suggests that no individuals with SCD-SS, SCD-SC, SCD Trait, Hb E Disease, and Hb E Trait would be missed by HemeChip hemoglobin testing. Common practice in hemoglobin testing is that all positive test results (e.g., SCD-SS) are confirmed with a secondary method prior to final diagnostic decision and treatment initiation.90 Therefore, all SCD-SS positive tests would likely result in a secondary confirmatory test that should rule out any false positives.
Affordability has been one of the primary objectives of HemeChip design. The projected sale price per test, including all consumables, distribution, and support cost, is estimated at $2.00 USD. A rugged, portable battery-powered reader can be priced as low as $700 USD, and designed to last at least 5 years or 10000 tests (Fig. S17†). The portable reader would add about 7 cents to the $2.00 USD test price. The projected HemeChip end user price of $2.07 USD per test (including the reader cost), thus clearly offers substantial cost savings over current practice and it is significantly lower than the estimated price threshold in a recent study.91 Deploying a robust, portable, battery operated platform as part of a point-of-care test has several advantages over alternatives (Table S2†). For example, the reader includes a touchscreen display that guides and helps the user with the test procedure, reduces the training time and minimizes user errors. The portable reader also allows digital data entry (Fig. S17†), including patient demographics. The reader also enables secure, encrypted data storage and wireless transmission to the cloud, which would enable tracking and follow-up of patients using electronic records and mobile networks.29 The reader displays the results on the screen in a clear and objective way (Fig. S10–S15†), removing user interpretation errors (Fig. S18–S32†). In comparison, point-of-care lateral-flow assays lack a portable reader for objective analysis or electronic record keeping (Table S2†). Therefore, these methods rely on the user to visually interpret the results and manually record the clinical data, which is prone to errors. In fact, in clinical research databases, user misinterpretation and data entry errors have been reported to range between 2.3% to 26.9%.92
Hemoglobin electrophoresis techniques share a common limitation. Some hemoglobin types appear in the same electrophoretic window, as they exhibit the same or similar electrophoretic mobility in a given condition. For example, in capillary zone electrophoresis, it can be challenging to quantify hemoglobin A2 in the presence of hemoglobin C, due to partial overlap between the two zones for these two hemoglobin types,93,94 which may be improved using curve fitting methods. Hemoglobin G and hemoglobin D are difficult to separate because they have identical migration in gel electrophoresis, in capillary electrophoresis, and they have overlapping elution times in HPLC. This sharing of detection window or peak overlapping is also a challenge for the reference standard method, HPLC as well as its alternatives.93,95,96 For example, in HPLC, hemoglobin S elutes in the S window with 28 other hemoglobin variants, including 10 other β-chain variants.97 A similar issue occurs in the A2 window for HPLC, where hemoglobin E and 18 other hemoglobin variants elude in the same window, and 13 of these 18 variants are β-chain variants.97 Hemoglobins C, E, and A2, co-migrate in paper based hemoglobin electrophoresis.66,67 Hemoglobins C, A2, and E are all detectable, but it is not possible to differentiate them.66,67 Therefore, instead of reporting Hb C or E or A2 individually, HemeChip reports hemoglobin C/E/A2. Since HemeChip provides quantitative results, the relative percentages of these hemoglobins can be used to make a distinction (Fig. S18–S32†). For example, hemoglobin A2 is typically found at lower percentages (3–10%) in most subjects,67,98,99 whereas hemoglobin C is typically much higher (greater than 40% for Hb SC, greater than 90% for Hb CC).100 When hemoglobin C and hemoglobin A2 are both present in the same blood sample, the HemeChip shows the hemoglobin C percentage plus hemoglobin A2 percentage.101 Clinical decision-making is not affected by this overlap, for Hb SC, Hb AC, or Hb CC results, but it is a potential limitation for detecting Hb Sβ-thalassemia and Hb Eβ-thalassemia. The algorithm has not yet been trained to identify β-thal variants, however, it categorizes them as SCD. In our clinical studies, HemeChip identified three Hb Sβ-thal subjects as SCD (Table 2). Some hemoglobin variants display distinct geographical prevalence and distribution. For example, hemoglobin C is highly prevalent in West Africa,102 while hemoglobin E is the most prevalent in the Mediterranean region, Southeast Asia, and in the Indian subcontinent.7,34–36,43 Test location, the ethnicity of the subject, the hemoglobin percentage (Fig. S18–S32†), and clinical history can be used to help differentiate between co-migrating hemoglobin types.
Fetal hemoglobin expression represents 50–95% of total hemoglobin expression in newborn babies and declines significantly over the course of the first 6 months of life.103,104 Hemoglobin F at high levels might mask the hemoglobin variants in newborns.105 Screening for hemoglobin variants can be integrated into established, highly effective immunization programs in low income countries.29 Babies are typically vaccinated around 6–8 weeks of age,29,40 and by this age, hemoglobin F levels decrease significantly as hemoglobin F is replaced by adult hemoglobins, which would be easier to detect. Non-β chain hemoglobinopathies, such as α-thalassemia, are also major world-wide health problems in children, especially in Asia. We are eager to extend our technology to this population in future studies. This technology is expected to detect additional hemoglobins, such as hemoglobin Bart's or hemoglobin H, which are elevated in alpha thalassemia.
In summary, HemeChip enables, for the first time, cost-effective identification of common hemoglobin variants at the point-of-need. The HemeChip reader guides the user step-by-step through the test procedure with animated on-screen instructions to minimize user errors. Hb identification and quantification is automatically performed, and Hb types and percentages are displayed in an easily understandable, objective, and quantitative way. HemeChip is a versatile, mass-producible microchip electrophoresis platform technology that may address unmet needs in biology and medicine, when rapid, decentralized hemoglobin or protein analysis is needed.
Public health significance and future perspective
Despite SCD's historical recognition of being the first discovered molecular disease, the technological advances for this disease have been slow.106 The promise of the development of accurate, low-cost, point-of-care diagnostics, especially for low-income countries, has come closer than ever before to being met with new technologies, such as HemeChip. HemeChip and its diagnostic promise will address the global health burden, help close the gap of information in our current state of science and healthcare delivery around hemoglobin diseases, and build space for further innovation and advancement for SCD and other hemoglobinopathies. Through multi-lateral partnerships with local governments, pharmaceutical companies, the technological industry, and university systems, the translation, implementation, and dissemination of affordable screening and diagnostic technologies throughout countries with limited resources and infrastructure has the potential to change the paradigm of SCD, hemoglobin disorders, and global health.Author contributions
MNH, AF, YA, RU, CMP, JAL, UAG conceived the idea and performed the initial development of the technology. MNH, AF, YA, RU, AA contributed to the proof-of-concept experiments and initial development. PT, AJR, NJK, TO, GMO, FH, BJ, SG, JAL, and SKO helped with the planning and execution of clinical studies in Nigeria, including human subject research protocol development, subject recruitment, blood sample collection, and testing. PT, AKV, PKB, RS, and AD helped with the planning and execution of clinical studies in India, including human subject research protocol development, subject recruitment, blood sample collection, and testing. PT, JZX, SR, TN, TS, and VV helped with the planning and execution of clinical studies in Thailand, including human subject research protocol development, subject recruitment, blood sample collection, and testing. GW helped with the adaptation of HemeChip design for high volume manufacturing. MSC edited the manuscript and authored the public health significance section. MNH, AF, RA, and UAG performed the data analysis, prepared the tables, figures, figure captions, and supplementary information. MNH, AF, RA, UAG drafted and edited the manuscript. All authors read and/or edited the manuscript.Conflicts of interest
MNH, AF, RA, YA, RU, CMP, JAL, and UAG are inventors of intellectual property (patent applications: PCT/US2015/042907 and US20170227495A1) licensed by Hemex Health Inc. for commercialization. These inventors and Case Western Reserve University have financial interest in Hemex Health Inc., including licensed intellectual property, stock ownership, and consulting. PT is an employee of Hemex Health. Competing interests of Case Western Reserve University employees are overseen and managed by the Conflict of Interests Committee according to a Conflict of Interest Management Plan.Acknowledgements
The development of this technology has been supported by the Clinical and Translational Science Collaborative of Cleveland, UL1TR002548 from the National Center for Advancing Translational Sciences component of the National Institutes of Health (NIH) and NIH Roadmap for Medical Research. Other funding sources include Case-Coulter Translational Research Partnership Program, NIH Center for Accelerated Innovation at Cleveland Clinic funded by U54HL119810, Ohio Third Frontier Technology Validation and Start-up Fund, Consortia for Improving Medicine with Innovation & Technology (CIMIT), National Institutes of Health Fogarty International Center (R21TW010610), National Heart Lung and Blood Institute Small Business Innovation Research Program (R44HL140739), National Heart Lung and Blood Institute R01HL133574, OT2HL152643, and National Science Foundation CAREER Award #1552782. This work is supported in part by Vodafone Americas Foundation™ Wireless Innovation Project™ and the Bill & Melinda Gates Foundation (Award OPP1034619 to Stephen K. Obaro). Scientific illustration was made by Grace Gongaware and videos were made by Courtney Fleming, supported by a National Science Foundation CAREER Award #1552782 and Glennan Fellowship from University Center for Innovation in Teaching and Education. Clinical studies in Thailand were supported by the National Institutes of Health, Fogarty International Center Global Health Research Fellowship (D43TW009337) and the Cooley's Anemia Foundation Research Fellowship awarded to Julia Z. Xu. Ran An acknowledges NIH T32 CWRU – Cardiovascular Research Training Program (T32HL134622). This article's contents are solely the responsibility of the authors and do not necessarily represent the official views of the National Institutes of Health.References
- B. Modell and M. Darlison, Global epidemiology of haemoglobin disorders and derived service indicators, Bull. W. H. O., 2008, 86, 480–487 CrossRef PubMed.
- D. T. Jamison, W. Bank and D. C. P. Project, Disease Control Priorities in Developing Countries, Oxford University Press, 2006 Search PubMed.
- D. J. Weatherall and J. B. Clegg, Inherited haemoglobin disorders: an increasing global health problem, Bull. W. H. O., 2001, 79(8), 704–712 CAS.
- T. N. Williams, Sickle Cell Disease in Sub-Saharan Africa, Hematol. Oncol. Clin. North Am., 2016, 30(2), 343–358 CrossRef PubMed.
- R. Colah, M. Mukherjee and K. Ghosh, Sickle cell disease in India, Curr. Opin. Hematol., 2014, 21(3), 215–223 CrossRef PubMed.
- F. B. Piel, R. E. Howes, A. P. Patil, O. A. Nyangiri, P. W. Gething, S. Bhatt, T. N. Williams, D. J. Weatherall and S. I. Hay, The distribution of haemoglobin C and its prevalence in newborns in Africa, Sci. Rep., 2013, 3, 1671–1671 CrossRef PubMed.
- S. Fucharoen and D. J. Weatherall, The hemoglobin E thalassemias, Cold Spring Harbor Perspect. Med., 2012, 2(8), a011734 Search PubMed.
- D. C. Rees, T. N. Williams and M. T. Gladwin, Sickle-cell disease, Lancet, 2010, 376(9757), 2018–2031 CrossRef CAS.
- H. F. Bunn, Pathogenesis and treatment of sickle cell disease, N. Engl. J. Med., 1997, 337(11), 762–769 CrossRef CAS PubMed.
- M. J. Stuart and R. L. Nagel, Sickle-cell disease, Lancet, 2004, 364(9442), 1343–1360 CrossRef.
- J. Makani, S. F. Ofori-Acquah, O. Nnodu, A. Wonkam and K. Ohene-Frempong, Sickle cell disease: new opportunities and challenges in Africa, Sci. World J., 2013, 2013, 193252 CAS.
- G. M. Brittenham, A. N. Schechter and C. T. Noguchi, Hemoglobin S polymerization: primary determinant of the hemolytic and clinical severity of the sickling syndromes, Blood, 1985, 65(1), 183–189 CrossRef CAS PubMed.
- C. Dong, R. S. Chadwick and A. N. Schechter, Influence of sickle hemoglobin polymerization and membrane properties on deformability of sickle erythrocytes in the microcirculation, Biophys. J., 1992, 63(3), 774–783 CrossRef CAS.
- D. Manwani and P. S. Frenette, Vaso-occlusion in sickle cell disease: pathophysiology and novel targeted therapies, Blood, 2013, 122(24), 3892–3898 CrossRef CAS PubMed.
- Y. Alapan, Y. Matsuyama, J. A. Little and U. A. Gurkan, Dynamic deformability of sickle red blood cells in microphysiological flow, Technology, 2016, 4(2), 71–79 CrossRef CAS PubMed.
- Y. Alapan, J. A. Little and U. A. Gurkan, Heterogeneous red blood cell adhesion and deformability in sickle cell disease, Sci. Rep., 2014, 4, 7173 CrossRef CAS PubMed.
- M. Kim, Y. Alapan, A. Adhikari, J. A. Little and U. A. Gurkan, Hypoxia-enhanced adhesion of red blood cells in microscale flow, Microcirculation, 2017, 24(5), e12374 CrossRef PubMed.
- G. A. Barabino, M. O. Platt and D. K. Kaul, Sickle cell biomechanics, Annu. Rev. Biomed. Eng., 2010, 12, 345–367 CrossRef CAS PubMed.
- Y. Alapan, C. Kim, A. Adhikari, K. E. Gray, E. Gurkan-Cavusoglu, J. A. Little and U. A. Gurkan, Sickle cell disease biochip: a functional red blood cell adhesion assay for monitoring sickle cell disease, Transl. Res., 2016, 173, 74–91 CrossRef CAS PubMed.
- Y. Alapan, A. Fraiwan, E. Kucukal, M. N. Hasan, R. Ung, M. Kim, I. Odame, J. A. Little and U. A. Gurkan, Emerging point-of-care technologies for sickle cell disease screening and monitoring, Expert Rev. Med. Devices, 2016, 13(12), 1073–1093 CrossRef CAS PubMed.
- R. P. Hebbel, Beyond hemoglobin polymerization: the red blood cell membrane and sickle disease pathophysiology, Blood, 1991, 77(2), 214–237 CrossRef CAS PubMed.
- D. J. Schaer, P. W. Buehler, A. I. Alayash, J. D. Belcher and G. M. Vercellotti, Hemolysis and free hemoglobin revisited: exploring hemoglobin and hemin scavengers as a novel class of therapeutic proteins, Blood, 2013, 121(8), 1276–1284 CrossRef CAS PubMed.
- L. A. Verduzco and D. G. Nathan, Sickle cell disease and stroke, Blood, 2009, 114(25), 5117–5125 CrossRef CAS PubMed.
- J. B. Caboot and J. L. Allen, Hypoxemia in Sickle Cell Disease: Significance And Management, Paediatr. Respir. Rev., 2014, 15(1), 17–23 Search PubMed.
- D. R. Hargrave, A. Wade, J. P. M. Evans, D. K. M. Hewes and F. J. Kirkham, Nocturnal oxygen saturation and painful sickle cell crises in children, Blood, 2003, 101(3), 846 CrossRef CAS PubMed.
- M. H. Steinberg, Management of Sickle Cell Disease, N. Engl. J. Med., 1999, 340(13), 1021–1030 CrossRef CAS PubMed.
- C. Booth, B. Inusa and S. K. Obaro, Infection in sickle cell disease: a review, Int. J. Infect. Dis., 2010, 14(1), e2–e12 CrossRef PubMed.
- S. K. Obaro and P. Y. Iroh Tam, Preventing Infections in Sickle Cell Disease: The Unfinished Business, Pediatr. Blood Cancer, 2016, 63(5), 781–785 CrossRef PubMed.
- J. Mburu and I. Odame, Sickle cell disease: Reducing the global disease burden, Int. J. Lab. Hematol., 2019, 41(S1), 82–88 CrossRef PubMed.
- S. Kapoor, J. A. Little and L. H. Pecker, Advances in the Treatment of Sickle Cell Disease, Mayo Clin. Proc., 2018, 93(12), 1810–1824 CrossRef CAS PubMed.
- E. Vichinsky, D. Hurst, A. Earles, K. Kleman and B. Lubin, Newborn screening for sickle cell disease: effect on mortality, Pediatrics, 1988, 81(6), 749–755 CAS.
- T. Frempong and H. A. Pearson, Newborn screening coupled with comprehensive follow-up reduced early mortality of sickle cell disease in Connecticut, Conn. Med., 2007, 71(1), 9–12 Search PubMed.
- R. L. Nagel, M. E. Fabry and M. H. Steinberg, The paradox of hemoglobin SC disease, Blood Rev., 2003, 17(3), 167–178 CrossRef.
- S. Warghade, J. Britto, R. Haryan, T. Dalvi, R. Bendre, P. Chheda, S. Matkar, Y. Salunkhe, M. Chanekar and N. Shah, Prevalence of hemoglobin variants and hemoglobinopathies using cation-exchange high-performance liquid chromatography in central reference laboratory of India: A report of 65779 cases, J. Lab. Physicians, 2018, 10(1), 73–79 CrossRef CAS PubMed.
- D. Masiello, M. M. Heeney, A. H. Adewoye, S. H. Eung, H. Y. Luo, M. H. Steinberg and D. H. Chui, Hemoglobin SE disease: a concise review, Am. J. Hematol., 2007, 82(7), 643–649 CrossRef CAS PubMed.
- J. Z. Xu, S. Riolueang, W. Glomglao, K. Tachavanich, T. Suksangpleng, S. Ekwattanakit and V. Viprakasit, The origin of sickle cell disease in Thailand, Int. J. Lab. Hematol., 2019, 41(1), e13–e16 CrossRef PubMed.
- E. Wastnedge, D. Waters, S. Patel, K. Morrison, M. Y. Goh, D. Adeloye and I. Rudan, The global burden of sickle cell disease in children under five years of age: a systematic review and meta-analysis, J. Glob. Health, 2018, 8(2), 021103 CrossRef PubMed.
- T. N. Williams, Sickle Cell Disease in Sub-Saharan Africa, Hematol. Oncol. Clin. North Am., 2016, 30(2), 343–358 CrossRef PubMed.
- F. B. Piel, The Present and Future Global Burden of the Inherited Disorders of Hemoglobin, Hematol. Oncol. Clin. North Am., 2016, 30(2), 327–341 CrossRef PubMed.
- B. Aygun and I. Odame, A global perspective on sickle cell disease, Pediatr. Blood Cancer, 2012, 59(2), 386–390 CrossRef PubMed.
- Sickle Cell Anemia, Report by the Secreteriat, World Health Organization Fifty-Ninth World Health Assembly, 2006, p. A59/9 Search PubMed.
- S. D. Grosse, I. Odame, H. K. Atrash, D. D. Amendah, F. B. Piel and T. N. Williams, Sickle cell disease in Africa: a neglected cause of early childhood mortality, Am. J. Prev. Med., 2011, 41(6 Suppl 4), S398–S405 CrossRef PubMed.
- R. Colah, A. Gorakshakar and A. Nadkarni, Global burden, distribution and prevention of beta-thalassemias and hemoglobin E disorders, Expert Rev. Hematol., 2010, 3(1), 103–117 CrossRef PubMed.
- I. Odame, Perspective: We need a global solution, Nature, 2014, 515, S10 CrossRef PubMed.
- F. B. Piel, S. I. Hay, S. Gupta, D. J. Weatherall and T. N. Williams, Global burden of sickle cell anaemia in children under five, 2010–2050: modelling based on demographics, excess mortality, and interventions, PLoS Med., 2013, 10(7), e1001484 CrossRef PubMed.
- J. M. Benson and B. L. Therrell, History and Current Status of Newborn Screening for Hemoglobinopathies, Semin. Perinatol., 2010, 34(2), 134–144 CrossRef.
- B. J. Bain, Neonatal/newborn haemoglobinopathy screening in Europe and Africa, J. Clin. Pathol., 2009, 62(1), 53 CrossRef CAS PubMed.
- A. Streetly, R. Latinovic, K. Hall and J. Henthorn, Implementation of universal newborn bloodspot screening for sickle cell disease and other clinically significant haemoglobinopathies in England: screening results for 2005–7, J. Clin. Pathol., 2009, 62(1), 26 CrossRef CAS.
- J. Bardakdjian-Michau, M. Bahuau, D. Hurtrel, C. Godart, J. Riou, M. Mathis, M. Goossens, C. Badens, R. Ducrocq, J. Elion and J. M. Perini, Neonatal screening for sickle cell disease in France, J. Clin. Pathol., 2009, 62(1), 31–33 CrossRef CAS PubMed.
- B. L. Therrell, C. D. Padilla, J. G. Loeber, I. Kneisser, A. Saadallah, G. J. C. Borrajo and J. Adams, Current status of newborn screening worldwide: 2015, Semin. Perinatol., 2015, 39(3), 171–187 CrossRef PubMed.
- A. Streetly, R. Sisodia, M. Dick, R. Latinovic, K. Hounsell and E. Dormandy, Evaluation of newborn sickle cell screening programme in England: 2010–2016, Arch. Dis. Child., 2018, 103(7), 648 Search PubMed.
- C. M. Wanjiku, F. Njuguna, F. C. Asirwa, C. Njuguna, C. Roberson and A. Greist, Validation and Feasibility of a Point of Care Screening Test for Sickle Cell Disease in a Resource Constrained Setting—a New Frontier, Blood, 2018, 132(Suppl 1), 2229 CrossRef.
- F. Tluway and J. Makani, Sickle cell disease in Africa: an overview of the integrated approach to health, research, education and advocacy in Tanzania, 2004–2016, Br. J. Haematol., 2017, 177(6), 919–929 CrossRef PubMed.
- P. T. McGann, M. G. Ferris, P. Macosso, V. de Oliveira, U. Ramamurthy, A. R. Luis, L. Bernardino and R. E. Ware, A Prospective Pilot Newborn Screening and Treatment Program for Sickle Cell Anemia in the Republic of Angola, Blood, 2012, 120(21), 480 CrossRef.
- B. E. Clark and S. L. Thein, Molecular diagnosis of haemoglobin disorders, Clin. Lab. Haematol., 2004, 26(3), 159–176 CrossRef CAS PubMed.
- P. T. McGann and C. Hoppe, The pressing need for point-of-care diagnostics for sickle cell disease: A review of current and future technologies, Blood Cells, Mol., Dis., 2017, 67, 104–113 CrossRef PubMed.
- J. Makani, S. E. Cox, D. Soka, A. N. Komba, J. Oruo, H. Mwamtemi, P. Magesa, S. Rwezaula, E. Meda, J. Mgaya, B. Lowe, D. Muturi, D. J. Roberts, T. N. Williams, K. Pallangyo, J. Kitundu, G. Fegan, F. J. Kirkham, K. Marsh and C. R. Newton, Mortality in sickle cell anemia in Africa: a prospective cohort study in Tanzania, PLoS One, 2011, 6(2), e14699 CrossRef CAS PubMed.
- S. Obaro, Preventable deaths in sickle-cell anaemia in African children, Lancet, 2010, 375(9713), 460 CrossRef ; author reply 461.
- L. Tshilolo, G. Tomlinson, T. N. Williams, B. Santos, P. Olupot-Olupot, A. Lane, B. Aygun, S. E. Stuber, T. S. Latham, P. T. McGann and R. E. Ware, Hydroxyurea for Children with Sickle Cell Anemia in Sub-Saharan Africa, N. Engl. J. Med., 2019, 380(2), 121–131 CrossRef CAS PubMed.
- E. Voskaridou, D. Christoulas, A. Bilalis, E. Plata, K. Varvagiannis, G. Stamatopoulos, K. Sinopoulou, A. Balassopoulou, D. Loukopoulos and E. Terpos, The effect of prolonged administration of hydroxyurea on morbidity and mortality in adult patients with sickle cell syndromes: results of a 17-year, single-center trial (LaSHS), Blood, 2010, 115(12), 2354–2363 CrossRef CAS PubMed.
- M. H. Steinberg, W. F. McCarthy, O. Castro, S. K. Ballas, F. D. Armstrong, W. Smith, K. Ataga, P. Swerdlow, A. Kutlar, L. DeCastro and M. A. Waclawiw, The risks and benefits of long-term use of hydroxyurea in sickle cell anemia: A 17.5 years follow-up, Am. J. Hematol., 2010, 85(6), 403–408 CAS.
- W. C. Wang, R. E. Ware, S. T. Miller, R. V. Iyer, J. F. Casella, C. P. Minniti, S. Rana, C. D. Thornburg, Z. R. Rogers, R. V. Kalpatthi, J. C. Barredo, R. C. Brown, S. A. Sarnaik, T. H. Howard, L. W. Wynn, A. Kutlar, F. D. Armstrong, B. A. Files, J. C. Goldsmith, M. A. Waclawiw, X. Huang and B. W. Thompson, Hydroxycarbamide in very young children with sickle-cell anaemia: a multicentre, randomised, controlled trial (BABY HUG), Lancet, 2011, 377(9778), 1663–1672 CrossRef CAS.
- S. V. Deshpande, S. S. Bhatwadekar, P. Desai, T. Bhavsar, A. Patel, A. Koranne, A. Mehta and S. Khadse, Hydroxyurea in Sickle Cell Disease: Our Experience in Western India, Indian J. Hematol. Blood Transfus., 2016, 32(2), 215–220 CrossRef CAS PubMed.
- I. Lagunju, B. J. Brown, A. O. Oyinlade, A. Asinobi, J. Ibeh, A. Esione and O. O. Sodeinde, Annual stroke incidence in Nigerian children with sickle cell disease and elevated TCD velocities treated with hydroxyurea, Pediatr. Blood Cancer, 2019, 66(3), e27252 CrossRef.
- R. O. Opoka, C. M. Ndugwa, T. S. Latham, A. Lane, H. A. Hume, P. Kasirye, J. S. Hodges, R. E. Ware and C. C. John, Novel use Of Hydroxyurea in an African Region with Malaria (NOHARM): a trial for children with sickle cell anemia, Blood, 2017, 130(24), 2585–2593 CrossRef CAS PubMed.
- B. J. Bain, Haemoglobinopathy Diagnosis, Wiley, 2005 Search PubMed.
- B. J. Bain, B. Wild, A. Stephens and L. Phelan, Variant Haemoglobins: A Guide to Identification, Wiley, 2011 Search PubMed.
- J. M. Reilly, IPI storage guide for acetate film: instructions for using the wheel, graphs, and table: basic strategy for film preservation, Image Permanence Institute, Rochester Institute of Technology, Rochester, NY, 1993 Search PubMed.
- O. Desanctis, L. Gomez, N. Pellegri, C. Parodi, A. Marajofsky and A. Duran, Protective Glass Coatings on Metallic Substrates, J. Non-Cryst. Solids, 1990, 121(1–3), 338–343 CrossRef CAS.
- T. Hanawa, 1 - Overview of metals and applications, in Metals for Biomedical Devices, ed. M. Niinomi, Woodhead Publishing, 2010, pp. 3–24 Search PubMed.
- M. Guerra-Balcazar, D. Morales-Acosta, F. Castaneda, J. Ledesma-Garcia and L. G. Arriaga, Synthesis of Au/C and Au/Pani for anode electrodes in glucose microfluidic fuel cell, Electrochem. Commun., 2010, 12(6), 864–867 CrossRef CAS.
- W. Schrott, M. Svoboda, Z. Slouka and D. Snita, Metal electrodes in plastic microfluidic systems, Microelectron. Eng., 2009, 86(4–6), 1340–1342 CrossRef CAS.
- L. L. Qiang, S. Vaddiraju, J. F. Rusling and F. Papadimitrakopoulos, Highly sensitive and reusable Pt-black microfluidic electrodes for long-term electrochemical sensing, Biosens. Bioelectron., 2010, 26(2), 682–688 CrossRef CAS PubMed.
- M. M. Maharbiz, W. J. Holtz, R. T. Howe and J. D. Keasling, Microbioreactor arrays with parametric control for high-throughput experimentation, Biotechnol. Bioeng., 2004, 85(4), 376–381 CrossRef CAS PubMed.
- M. B. Fox, D. C. Esveld, A. Valero, R. Luttge, H. C. Mastwijk, P. V. Bartels, A. van den Berg and R. M. Boom, Electroporation of cells in microfluidic devices: a review, Anal. Bioanal. Chem., 2006, 385(3), 474–485 CrossRef CAS PubMed.
- S. W. Lee and Y. C. Tai, A micro cell lysis device, Sens. Actuators, A, 1999, 73(1–2), 74–79 CrossRef CAS.
- M. Fox, E. Esveld, R. Luttge and R. Boom, A new pulsed electric field microreactor: comparison between the laboratory and microtechnology scale, Lab Chip, 2005, 5(9), 943–948 RSC.
- A. St John and C. P. Price, Existing and Emerging Technologies for Point-of-Care Testing, Clin. Biochem. Rev., 2014, 35(3), 155–167 Search PubMed.
- S. Sharma, J. Zapatero-Rodriguez, P. Estrela and R. O'Kennedy, Point-of-Care Diagnostics in Low Resource Settings: Present Status and Future Role of Microfluidics, Biosensors, 2015, 5(3), 577–601 CrossRef PubMed.
- M. Biehl and T. Velten, Gaps and challenges of Point-of-Care technology, IEEE Sens. J., 2008, 8(5–6), 593–600 CAS.
- E. Prichard, Practical Laboratory Skills Training Guides (Complete Set), 2003, p. P001 Search PubMed.
- P. M. Bossuyt, J. B. Reitsma, D. E. Bruns, C. A. Gatsonis, P. P. Glasziou, L. Irwig, J. G. Lijmer, D. Moher, D. Rennie, H. C. W. de Vet, H. Y. Kressel, N. Rifai, R. M. Golub, D. G. Altman, L. Hooft, D. A. Korevaar and J. F. Cohen, STARD 2015: an updated list of essential items for reporting diagnostic accuracy studies, Br. Med. J., 2015, 351, h5527 CrossRef PubMed.
- N. M. Buderer, Statistical methodology: I. Incorporating the prevalence of disease into the sample size calculation for sensitivity and specificity, Acad. Emerg. Med., 1996, 3(9), 895–900 CrossRef CAS.
- A. S. Adewoyin, Management of sickle cell disease: a review for physician education in Nigeria (sub-saharan Africa), Anemia, 2015, 2015, 791498 CrossRef PubMed.
- D. L. Jain, V. Sarathi, D. Upadhye, R. Gulhane, A. H. Nadkarni, K. Ghosh and R. B. Colah, Newborn screening shows a high incidence of sickle cell anemia in Central India, Hemoglobin, 2012, 36(4), 316–322 CrossRef CAS PubMed.
- M. Nuinoon, K. Kruachan, W. Sengking, D. Horpet and U. Sungyuan, Thalassemia and hemoglobin e in southern thai blood donors, Adv. Hematol., 2014, 2014, 932306–932306 Search PubMed.
- W.H. Organization, World Health Organization guidelines on drawing blood: best practices in phlebotomy, World Health Organization, 2010 Search PubMed.
- H. R. Peck, D. M. Timko, J. D. Landmark and D. F. Stickle, A survey of apparent blood volumes and sample geometries among filter paper bloodspot samples submitted for lead screening, Clin. Chim. Acta, 2009, 400(1–2), 103–106 CrossRef CAS PubMed.
- B. Shinkins, M. Thompson, S. Mallett and R. Perera, Diagnostic accuracy studies: how to report and analyse inconclusive test results, Br. Med. J., 2013, 346, f2778 CrossRef PubMed.
- K. Ryan, B. J. Bain, D. Worthington, J. James, D. Plews, A. Mason, D. Roper, D. C. Rees, B. de la Salle and A. Streetly, Significant haemoglobinopathies: guidelines for screening and diagnosis, Br. J. Haematol., 2010, 149(1), 35–49 CrossRef CAS PubMed.
- M. Mvundura, C. Kiyaga, M. Metzler, C. Kamya, J. M. Lim, C. Maiteki-Sebuguzi and P. S. Coffey, Cost for sickle cell disease screening using isoelectric focusing with dried blood spot samples and estimation of price thresholds for a point-of-care test in Uganda, J. Blood Med., 2019, 10, 59–67 CrossRef PubMed.
- S. I. Goldberg, A. Niemierko and A. Turchin, Analysis of data errors in clinical research databases, AMIA Annu. Symp. Proc., 2008, 242–246 Search PubMed.
- D. F. Keren, D. Hedstrom, R. Gulbranson, C. N. Ou and R. Bak, Comparison of Sebia Capillarys Capillary Electrophoresis With the Primus High-Pressure Liquid Chromatography in the Evaluation of Hemoglobinopathies, Am. J. Clin. Pathol., 2008, 130(5), 824–831 CrossRef CAS PubMed.
- N. Borbely, L. Phelan, R. Szydlo and B. Bain, Capillary zone electrophoresis for haemoglobinopathy diagnosis, J. Clin. Pathol., 2013, 66(1), 29–39 CrossRef CAS PubMed.
- M. Nusrat, B. Moiz, A. Nasir and M. Rasool Hashmi, An insight into the suspected HbA2′ cases detected by high performance liquid chromatography in Pakistan, BMC Res. Notes, 2011, 4, 103–103 CrossRef CAS PubMed.
- P. Sharma and R. Das, Cation-exchange high-performance liquid chromatography for variant hemoglobins and HbF/A2: What must hematopathologists know about methodology?, World J. Methodol., 2016, 6(1), 20–24 CrossRef PubMed.
- J. D. Hoyer and R. M. Scheidt, Identification of hemoglobin variants by HPLC, Clin. Chem., 2005, 51(7), 1303–1304 CrossRef CAS PubMed ; author reply 1305.
- M. S. Figueiredo, The importance of hemoglobin A2 determination, Rev. Bras. Hematol. Hemoter., 2015, 37(5), 287–289 CrossRef.
- A. Giambona, C. Passarello, D. Renda and A. Maggio, The significance of the hemoglobin A2 value in screening for hemoglobinopathies, Clin. Biochem., 2009, 42(18), 1786–1796 CrossRef CAS.
- H. F. Bunn, C. T. Noguchi, J. Hofrichter, G. P. Schechter, A. N. Schechter and W. A. Eaton, Molecular and cellular pathogenesis of hemoglobin SC disease, Proc. Natl. Acad. Sci. U. S. A., 1982, 79(23), 7527–7531 CrossRef CAS PubMed.
- J. S. Krauss, P. A. Drew, M. H. Jonah, M. Trinh, S. Shell, L. Black and C. R. Baisden, Densitometry and microchromatography compared for determination of the hemoglobin C and A2 proportions in hemoglobin C and hemoglobin SC disease and in hemoglobin C trait, Clin. Chem., 1986, 32(5), 860–862 CrossRef CAS.
- B. Kreuels, C. Kreuzberg, R. Kobbe, M. Ayim-Akonor, P. Apiah-Thompson, B. Thompson, C. Ehmen, S. Adjei, I. Langefeld, O. Adjei and J. May, Differing effects of HbS and HbC traits on uncomplicated falciparum malaria, anemia, and child growth, Blood, 2010, 115(22), 4551–4558 CrossRef CAS PubMed.
- C. Thomas and A. B. Lumb, Physiology of haemoglobin, Continuing Education in Anaesthesia, Crit. Care Pain, 2012, 12(5), 251–256 Search PubMed.
- A. D. Metaxotou-Mavromati, H. K. Antonopoulou, S. S. Laskari, H. K. Tsiarta, V. A. Ladis and C. A. Kattamis, Developmental changes in hemoglobin F levels during the first two years of life in normal and heterozygous beta-thalassemia infants, Pediatrics, 1982, 69(6), 734–738 CAS.
- J. Watson, The significance of the paucity of sickle cells in newborn Negro infants, Am. J. Med. Sci., 1948, 215(4), 419–423 CrossRef CAS.
- R. E. Ware, Technological Advances in Sickle Cell Disease, Blood Cells, Mol., Dis., 2017, 67, 102–103 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9an02250c |
| This journal is © The Royal Society of Chemistry 2020 |