
Glutamate sensing in biofluids: recent advances and research challenges of electrochemical sensors
Jessica
Schultz
a,
Zakir
Uddin
b,
Gurmit
Singh
c and
Matiar M. R.
Howlader
 *a
*a
aDepartment of Electrical and Computer Engineering, McMaster University, 1280 Main Street West, Hamilton, ON L8S 4K1, Canada. E-mail: mrhowlader@ece.mcmaster.ca
bSchool of Rehabilitation Science, McMaster University, 1400 Main St W, Hamilton, ON L8S 1C7, Canada
cDepartment of Pathology and Molecular Medicine, McMaster University, 1280 Main Street West, Hamilton, ON L8S 4 K1, Canada
First published on 15th November 2019
Abstract
Glutamate is a nonessential amino acid and a putative neurotransmitter. When its consumption exceeds its synthesis, it becomes necessary to monitor its levels. Hence, a low-cost, sensitive and real-time monitoring of glutamate to quantify pain and detect neurodegenerative diseases is imperative to improve pharmacotherapy and early diagnosis for health care. While enzymatic electrochemical sensors are promising to address issues in lab-based detection techniques, non-enzymatic sensors are better due to their higher stability and lower cost. In this review, we aim to discuss the recent advances and remaining challenges of sensing glutamate in biofluids. First, we discuss the metabolic routes of glutamate, followed by its transmission processes to the biofluids. Second, we identify the connection of glutamate to pathologies as a potential biomarker. Third, we emphasize electrochemical sensors instantaneously detect glutamate in biofluids in real-time, quantifying pain and monitoring neurodegenerative diseases. The literature shows the concentration of glutamate in biofluids, such as plasma, cerebral spinal fluid, urine, and saliva are in the range of 5–100 μM, 0.5–2 μM, 8.5 (3.3–18.4) μM mM−1 creatinine, and 0.232 ± 0.177 μM respectively. While these concentration levels are sometimes lower than the detection limit of electrochemical sensors, functionalization of the nanomaterials currently being used such as NiO and Co3O4 with carbon nanotubes or beta-cyclodextrin may improve the sensing performance. Another key challenge in the research is to develop relationships between glutamate and biofluids. Finally, we have to advance electrochemical sensors that are compatible to detect glutamate in physiological conditions for long durations of time.
1. Introduction
Glutamate is the major excitatory neurotransmitter of the central nervous system (CNS) in vertebrate.1 Glutamate can be synthesized from glucose, an essential energy source for the human body, through the malate–aspartate shuttle following glycolysis.2 While glucose sensors have been widely developed, the development of glutamate sensors has lagged, despite the notable effects of glutamate in the CNS.3 Accurate detection and monitoring of extracellular glutamate levels is of critical importance as excess glutamate can lead to excitotoxicity and contribute to the pathogenesis of neurodevelopment disorders such as Autism or neurodegenerative diseases such as amyotrophic lateral sclerosis (ALS) and Alzheimer's disease (AD).4,5 Chronic pain and migraines have also been associated with elevated glutamate concentrations in brain and saliva, respectively.6,7Continuous and real-time monitoring of glutamate levels in the cerebrospinal fluid (CSF) and other biofluids in a continuous and real-time manner can provide a means to identify these diseases or quantify the level of pain. Current technologies being used to measure glutamate include laboratory-based methods such as high performance liquid chromatography (HPLC) and gas chromatography-mass spectrometry (GCMS).8,9 While these methods measure glutamate to a high degree of accuracy, they require expensive equipment, trained professionals and a longer measurement time. Additionally, in vivo sensors have been developed to detect glutamate levels in brain.10In vivo sensors are extremely invasive and can lead to further complications such as infections.11 Consequently, it is ideal to develop an instantaneous, low-cost and continuous monitoring technique that eliminates the need of an operator and is minimally invasive.
Electrochemical sensors can facilitate real-time, continuous monitoring of analytes such as glutamate. Currently, enzymatic electrochemical sensors measure glutamate using glutamate oxidase (GluOx) or dehydrogenase (GLDH).12,13 Enzymatic sensors offer high sensitivity, however they are often expensive and decrease in stability over time, resulting in a signal reduction causing inaccuracies in measurement.14 Typically, electrochemical sensors consist of a three-electrode system: working, reference and counter electrodes.15 The reference and counter electrodes are commonly fabricated with Ag/AgCl and Pt wire respectively. The working electrode of electrochemical glutamate sensor systems varies, for instance M. Jamal et al. used a modified glassy carbon electrode, whereas M. Hussain et al. fabricated a cobalt oxide (Co3O4) mesoporous nanosheet.16,17 Glassy carbon electrodes can be manipulated with nanoparticles such as Ni or Au nanoparticles, although Ag and Au are less desirable as they are expensive.18 There are numerous electrode fabrication techniques including solution processing, inkjet printing, and roll-to-roll bonding.19 Varying in cost and efficiency, fabrication techniques such as inkjet printing offer an efficient and low-cost method to produce numerous electrodes quickly and accurately.
Electrochemical sensing devices of analytes include amperometric sensors, potentiometric sensors, chemo-resistors, and field-effect transistors.20 Y. Huang et al. explored the fabrication of graphene to perform as a field-effect transistor to sense glutamate.21 The use of graphene, a two-dimensional (2D) nanomaterial, improved the performance of the device's electronic properties and detection. However, in combination with an enzyme, the stability was still suboptimal. In contrast, M. Jamal et al. developed a non-enzymatic three-electrode sensor consisting of nickel oxide (NiO) glassy carbon working electrode. While the sensor demonstrated comparable results to previous glutamate detecting electrochemical sensors, it lacked an ability to be used in physiological settings. During cyclic voltammetry, the sensor fabricated with NiO/GCE required an OH− during the oxidation of Ni, thus the solution requires highly alkalotic conditions. In fact, the conditions are above the normal pH of all biofluids.22–26
To the best of our knowledge, previous review papers on electrochemical sensors to detect glutamate focus on either specifically in vivo electrochemical detection of brain glutamate or only enzymatic or non-enzymatic electrochemical glutamate sensors.27,28 As previously mentioned, in vivo sensing is very invasive, which is why this review paper explores glutamate in biofluids and outside of the brain to provide a non-invasive solution. While non-enzymatic electrochemical sensors are promising, they are still lacking behind the enzymatic alternative. Hence, this review paper provides a comprehensive investigation in enzymatic sensors in addition to non-enzymatic sensors in an effort to improve non-enzymatic sensors through understanding why enzymatic sensors currently perform better. Additionally, a review on electrochemical sensors to detect glutamate has been published, however it lacks the background knowledge in glutamate and foundation for those in electrochemical sensing.
This paper outlines the role of glutamate in biofluids and recent advances of electrochemical sensors and the application of these biosensors to detect glutamate in the human body. Section 2 details the fundamental synthesis processes of glutamate and its role as a crucial amino acid in bodily fluids. The presence of glutamate in biological matrices is outlined in section 3. The importance and connections of glutamate as a biomarker for diseases along with current technologies used to detect glutamate are discussed in section 4. Section 5 explores electrochemical sensing of glutamate, sensing materials as well as fabrication techniques of sensing electrodes. Section 6 provides potential guidelines for future electrochemical sensors tasked with high-performance sensing of glutamate in biofluids.
2. Glutamate in the human body
Glutamate is an important neurotransmitter responsible for cortical and cognitive functionality.29 Glutamate binding with glutamate receptors allows sodium ions into the cell and potassium ions out of the cell.30 While this particular mechanism occurs in the CNS, glutamate and glutamate receptors, including ionotropic and metabotropic receptors, are detected throughout the human body in non-neuronal tissues.31 This section explores the synthesis and role of glutamate in the CNS, other tissues and organs in the human body.2.1 Glutamate in the central nervous system
Glutamate is the most abundant amino acid based neurotransmitter in the CNS.32 This amino acid is responsible for synapse induction and elimination, as well as cell migration, differentiation and death. Stimuli results in the release of glutamate from presynaptic neuron to the synaptic cleft. As depicted in Fig. 1, once in the synaptic cleft, glutamate interacts with glutamate receptors on the postsynaptic neuron or removed by glutamate transporters on glial cells.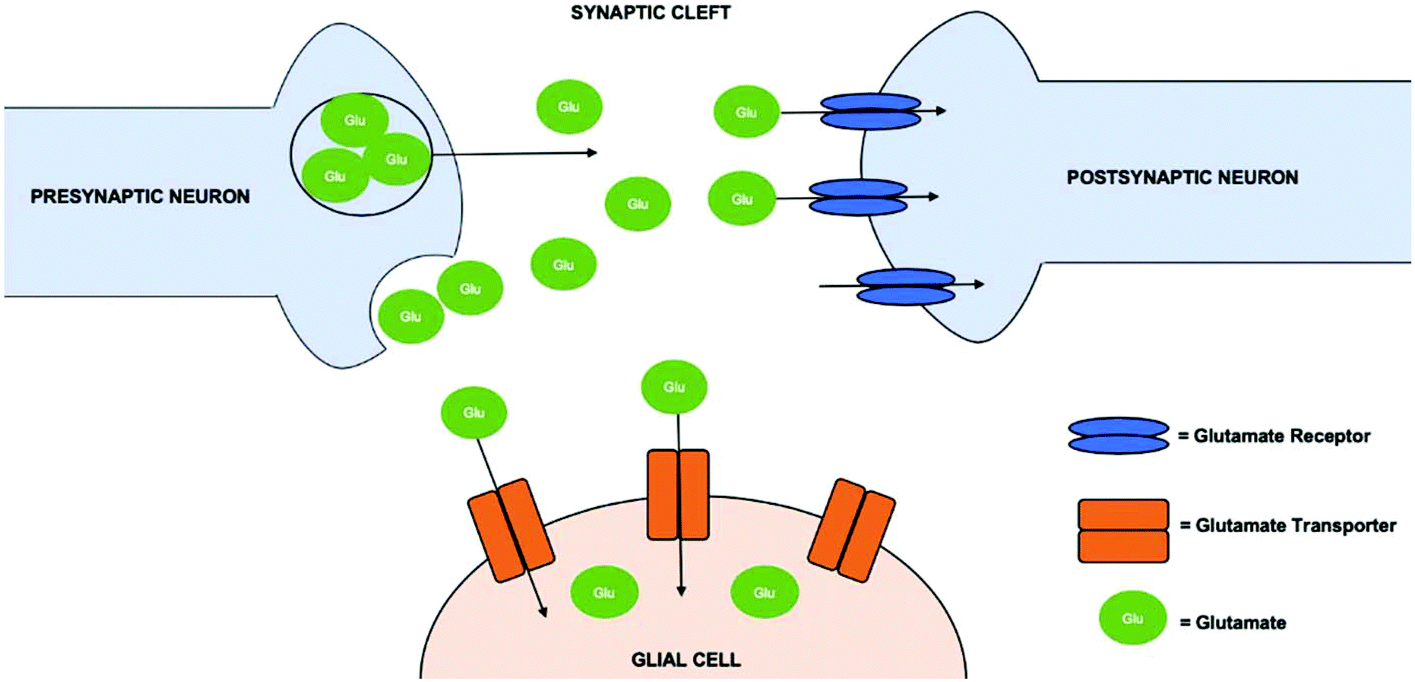 | ||
| Fig. 1 Overview of the mechanism of glutamate in the central nervous system. Glutamate transporters are present on the glial cells, which support the central nervous system. Glutamate receptors on the postsynaptic cell body enables them to interact after vesicles in the presynaptic cell body release glutamate into to the synaptic cleft. | ||
Typically, the highest concentration of glutamate is within cells, where a small fraction is outside the cells, in the cerebrospinal fluid (CSF) and plasma.33 There is no extracellular enzyme to metabolize glutamate, thus the only removal from the extracellular fluid (ECF) is through glutamate uptake.34 This uptake of glutamate, altering the concentration gradient, occurs through glutamate transporters.
Intracellular glutamate is generally inert, responsible for the regulation of glutamate transporters and propagating signals in the CNS.35 Glutamate uptake is accomplished through glutamate transport proteins, called excitatory amino acid transporters (EAAT),36 which utilize the electrochemical gradient across the plasma membrane to bring glutamate into the cell. EAATs transport 2–3 sodium ions, a proton and glutamate with an anti-porter carrying a potassium ion along the electrochemical gradient, across the plasma membrane. This results in a high concentration of glutamate inside of the cell. Similarly, simple diffusion is another mode of removal of glutamate, although it only occurs under short distances and if the extracellular concentration is very low.
Once glutamate is within the cell, the glutamate–glutamine cycle is the major pathway of glutamate transmission and recycling.37 Glutamate is taken up to the inside of the cell, converted to glutamine (inert), which is released to the extracellular fluid, taken up by neurons, and converted back to glutamate within neurons. When glutamate is converted inside of neurons, it can be metabolized by conversion to either glutamine or alpha-ketoglutarate. Glutamate is amidated to glutamine though ATP-dependent, glial specific glutamate synthase. Alternatively, glutamate is converted to alpha-ketoglutarate through either deamination by GLDH or by transmission of one of the transaminases. Alpha-ketoglutarate is metabolized through the tri-carboxylic acid cycle. This is important as excess glutamine or alpha-ketoglutarate can contribute to excitoxicity or excess lactic acid, respectively.
Similar to alpha-ketoglutarate, γ-aminobutyric acid (GABA) can be broken down to glutamate through the tri-carboxylic acid cycle. Consequently, glutamate is a metabolic precursor of GABA, the main inhibitory neurotransmitter.38 Prior to multiple metabolic pathways, glutamate forms GABA through alpha-decarboxylation by glutamic acid decarboxylase. Consequently, the concentration of the main inhibitory amino acid is contingent on the amount of glutamate. Fig. 2 summarizes the precursors and products of glutamate.
 | ||
| Fig. 2 Summary of precursors and products of glutamate, exemplifying the formation of glutamate and molecules synthesized from glutamate. | ||
Receptors for glutamate are found on the surface of cells, including dendrites, nerve terminals, neuronal cell bodies and glial cells.39 Glutamate receptors serve an important role: when glutamate is present in the extracellular fluid it binds to glutamate receptors, increasing the probability of an action potential occurring. Without the receptor, simply the release of glutamate would not be sufficient to start an action potential. This stimulation of glutamate receptors is dependent on the extracellular concentration of glutamate. The extracellular concentration must be kept low to avoid glutamate toxicity, which occurs when the receptors are overstimulated.
Glutamate receptors include ionotropic, ligand-gated ion channels, and metabotropic, G-protein coupled receptors. Ionotropic glutamate receptors are on the postsynaptic membrane and are activated by N-methyl-D-aspartate (NMDA), α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid (AMPA) and kainite receptors.40 Ionotropic receptors open calcium channels, causing the influx of calcium into neurons. In neurons, calcium is responsible for stimulating plasmatic proteolytic enzymes, causing detrimental effects. Metabotropic receptors signal second messenger systems when there is binding with glutamate and include three major groups of receptors.41 Abnormal stimulation of glutamate receptors results in abnormal ion concentrations, which can have adverse effects.
High glutamate levels in the extracellular space can result in excitotoxicity. The excess glutamate leads to an increase in glutamate receptor activation, which can lead to pathologies such as ischemia in the case of an increase of ionotropic glutamate receptors.42 It has been proposed that chronic activation of glutamate receptors can lead to pathologies such as AD and ALS.43,44
2.2 Glutamate in the peripheral tissues and organs
Glutamate is crucial for cell metabolism in the peripheral tissues and organs.45 While glutamate's role in the CNS is known and correlated to multiple pathologies, it is also detected in non-neuronal cells, along with glutamate receptors in peripheral tissues. Although this amino acid is detected in peripheral tissues, the metabolism and function of glutamate is sometimes unclear.Kidneys are an example of where glutamate receptors are found and affect their function.46 In the early findings, Berenbom et al. found that isotopic precursors of glutamate intravenously injected resulted in a slight increase of the isotope in the liver and kidneys.47 It also resulted in a large amount of urea excreted in urine. Their study proposed some of the initial connections between glutamate and the renal system. In the kidneys, glutamate is responsible for ammonia secretion and maintaining acid–base balance. Over activation of NMDA receptors impact the kidney functions, resulting in excitotoxicity in a similar manner to in the CNS. Alternatively, glutamate is a precursor for metabolites and required for synthesizing nucleic acids and proteins in the liver.48 Hepatocytes require intracellular glutamate for the metabolism of almost all amino acids. Glutamate contributes to the transdeamination of most amino acids and catabolism of some amino acids.49 In addition to the liver, glutamate is detected in the pancreas.
Pancreatic islets cells contain glutamate, responsible for cell function and viability.50 Islet endocrine cells behave in a similar manner to neurons in the CNS, expressing glutamate receptors. However, high extracellular glutamate levels are noticeable, to activate a large number of glutamate receptors on beta cells. Additionally, demonstrating similar patho-mechanisms with neurodegenerative diseases and diabetes mellitus, where both diseases present amyloid plaques and cell death (Fig. 3).
 | ||
| Fig. 3 Glutamate signalling in endocrine cell, exemplifying relationship between beta and alpha cells with blood capillary. Reproduced from ref. 50 with permission from Elsevier, Copyright 2016. | ||
Ionotropic glutamate receptors, most notably NMDA receptors, were detected in the trachea, major airways, and peripheral and lung samples of rats.51 This was determined through completing PCR on the isolated mRNA from each section. Particularly, it is also suggested glutamate contributes to physiology of lungs. Similarly, glutamate receptors are found on osteoblasts, with NMDA receptors occurring most frequently.52 This notion was further supported after NMDA receptors were inhibited; bone reabsorption was ceased. Hence, it was suggested glutamate is responsible for the regulation of bone remodelling.53
While glutamate is well-understood in the CNS, further research is necessary to continue exploring glutamate in other organs. Further research is also required to determine if there are connections between glutamate in the peripheral tissues and the central nervous system.
3. Glutamate's connection in biological matrices
Biofluids are biological matrices, specifically fluids that may contain biomarkers to indicate a disease or disorder. Biofluids can serve as a diagnostic tool in early detection or improve the management of current diseases.54 In comparison to tissue samples, biofluids cannot be localized to a specific cell environment, however they are easy to acquire, ideal for early detection, and useful for therapeutic monitoring.55 Early detection can lead to a better prognosis and outlook on the patient's life. While this is ideal, patients often don't recognize this due to barriers including the ones identified by Y. Lee et al.: complexity, cost, and discomfort. Biofluids that contain biomarkers and can be acquired easily, non-invasively and in a cost-effective manner would break down these barriers, hence improving the health of patients.3.1 Glutamate in the cerebrospinal fluid
Cerebrospinal fluid is a component of ECF in the CNS.56 As outlined in section 2.1, glutamate is found is the ECF of the CNS. The concentration of glutamate in the CSF is 0.5–2 μM.57 Elevated glutamate concentrations in the CSF by several hundred-fold has been correlated to multiple neurogenerative disorders. Elevated plasma and CSF glutamate levels have been detected in patients with glioma, in comparison to patients with meningioma.58In vivo electrochemical sensors have already been developed to capture CSF levels of glutamate in animal specimens. While glutamate is understood in this biofluid, it must be extracted from the brain or spinal cord, thus posing a high level of risk and discomfort to the patient.59 More information on diseases associated with the nervous system and CSF are detailed in section 4.3.2 Blood as a biofluid to detect glutamate
Blood has been extracted and analyzed for decades as it contains valuable information and biomarkers for diagnosis and monitoring.60 Whole blood consists of four components: platelets, erythrocytes, leukocytes, and plasma.61 Plasma is the liquid component of blood, responsible for its fluidity. Blood is ideal for its fast response rate to demonstrate irregularities, evident with glutamate levels measured in rats.62 While the exact time it takes for glutamate to indicate an abnormality in humans needs to be determined, glutamate has been detected in the blood as a potential biomarker. Further research is required to address these issues.Platelets synthesize, uptake and release glutamate from the blood in a similar manner to neurons.63 Glutamate concentrations are higher in the plasma at 5–100 μM L−1 and in whole blood at 150–300 μM L−1 in comparison to in the CSF. This could be due to the synthesis of glutamate from platelets in combination with the unidirectional movement of glutamate from the brain to the blood plasma. EAATs are responsible for transporting glutamate from the CSF across the abluminal membrane into the endothelial cells. The intracellular concentration of glutamate can increase in one of the following ways: through the transport of glutamine into the cell, which is converted into glutamate or the direct transport of glutamate into the cell. This generates an electrochemical gradient across the luminal membrane, consequently glutamate leaves the cell into the blood plasma through facilitated diffusion.
It is still debatable whether the concentration of glutamate in the blood plasma correlates to the concentration in CSF. This correlation is supported in a study by Campos et al., where glutamate scavengers were added to the blood after a stroke.64 Following this addition, the glutamate concentration in the CSF decreased. Blood scavengers act to indirectly decrease the blood glutamate concentration, resulting in the movement of glutamate down the concentration gradient from the brain to the blood. This also resulted in neurological improvement. Alternatively, this theory has been disputed in a study by Y. Shulman et al. where no correlation between glutamate in the medial prefrontal cortex or in the blood plasma of healthy males was found.65 The glutamate in the blood plasma was detected using HPLC, and the glutamate in the brain was measured using proton magnetic resonance spectroscopy. While the correlation between the concentration of glutamate in the blood and the CSF is inconclusive, blood offers an accurate detection of glutamate that can be analyzed independently.
Glutamate has been detected in blood and proposed as a biomarker for disorders and diseases such as acute lung injury (ALI), autism, and schizophrenia. ALI is diagnosed clinically as acute onset of bilateral pulmonary infiltrates with hypoxemia and lack of hydrostatic pulmonary edema.66 W. Bai et al. reported elevated blood glutamate levels in patients whom had a poor prognosis and presented ALI after a stroke.67 Another study by W. Bai et al. found blood glutamate concentrations increased following traumatic brain injury and indicated traumatic brain injury-induced acute lung injury (TBI-ALI).67 W. Bai et al. suggested glutamate plays a role in the pathogenesis of TBI-ALI and suggested it be used a predictor of TBI-ALI. Similarly, high glutamate concentrations in platelet-poor plasma were found in children with high functioning autism (HFA).68 The significant difference of platelet-poor plasma glutamate levels in children with HFA compared to normally developing children, suggested measuring glutamate levels for early detection of disorders. Alternatively, Madeira et al. found glutamine/glutamate ratios increased in the blood plasma of patients with recent onset schizophrenia and decreased in the blood of patients with chronic schizophrenia.69 Consequently, it was suggested blood glutamate levels are elevated during the inception of schizophrenia and decreased as the disorder progresses. Conclusively, blood glutamate levels can be measured and used as a diagnostic tool for HFA, TBI-ALI, stroke prognosis, or to detect the progression of schizophrenia (Fig. 4).
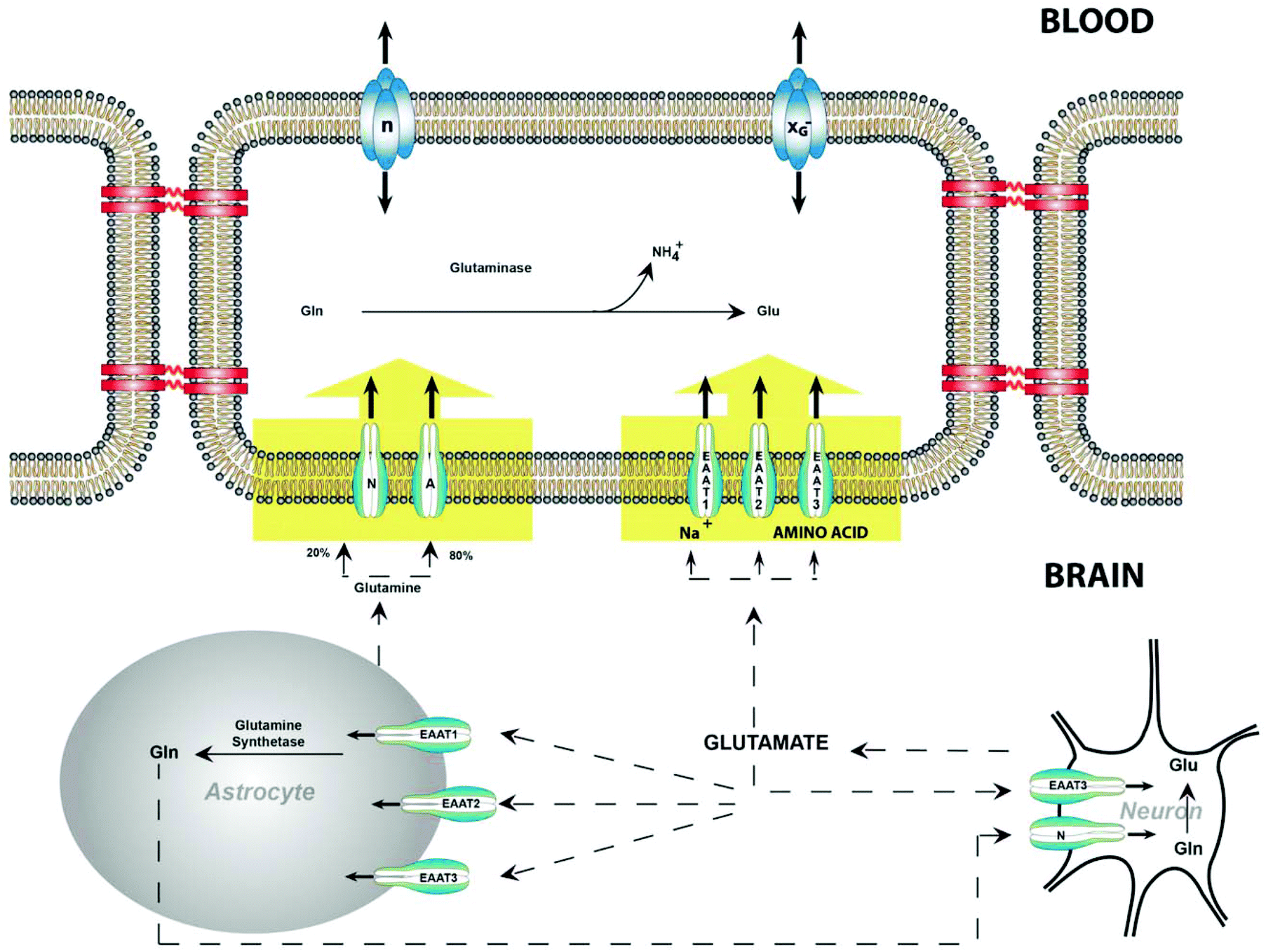 | ||
| Fig. 4 Retrieved from Hawkins et al.70 Glutamate mechanism between blood and brain. It is evident glutamate and glutamine play a role in brain in addition to blood. Reproduced from ref. 70 from Ivy Union Publishing, Open Licensed Copyright, 2013. | ||
3.3 Glutamate in the urine
Urine is a biofluid that is useful due to its sterility, lack of large molecules, and ease of acquiring large quantities.8 Accordingly, since glutamate is a small molecule, it can be excreted into the urine. In fact, glutamate is the main source of ammonia (NH3) to the kidneys which influences the pH of urine. Further support to this notion was found during a study of pink urine syndrome. Decreased amounts of glutamate correlated to pink urine syndrome, a phenomenon which results in pink sediments of uric acid.71 Glutamate levels also linked to maple syrup urine disease (a metabolic and excitotoxicity neuropathological disorder among children) due to accumulation of branched-chain type of amino acids and their corresponding ketoacids.72–74 The urine acid (decreased pH level) is elevated in maple syrup urine disease due to excretion of ketoacids.73,74 This evidence supported the notion that the decreased urinary glutamate resulted in a decreased pH level of urine. The amount of glutamate in urine was postulated to decrease due to the use of glutamate for NH3 synthesis. This NH3 homeostasis is regulated via glutamate–glutamine cycle. Consequently, glutamate levels in the urine could be an indicator of acid-based function of the renal system in kidney.In normal acid–base (pH) balance, the kidneys catabolize and extract a little of the plasma glutamine, although arterial blood plasma constitutes around 20–25% of the circulating free amino acids as glutamine.75 The homeostasis of plasma glutamine mainly reflects a balance between release from other tissues (e.g. muscle, lung, and adipose tissue) and uptake by the splanchnic vascular bed.76 However, in abnormal or pathological condition like metabolic acidosis, the kidney plays the major role of glutamine extraction and catabolism.75 This imbalance of acid and base (pH level) is sensitive to urinary excretion of glutamine and glutamate. For example, urinary glutamine/glutamate ratio has been identified as a promising biomarker for chronic intestinal pseudo-obstruction with very high specificity (92%) as the ratio is decreased as compared to normal.77
Glutamine levels in urine are mainly due to a renal gluconeogenic substrate and is also increased in type II diabetes.76 Glutamine metabolism manipulation is a potential novel target for preventing diabetes and obesity related pathological conditions.78 Similarly, patients with hyperthyroidism had elevated glutamate concentrations in plasma and urine.79 The concentration increased almost tenfold, suggesting glutamate as a promising indication of hyperthyroidism. Consequently, blood plasma biomolecules including urine have been suggested to understand the tissue-specific function of thyroid hormones.80
3.4 Glutamate in the saliva
Saliva is a serous and mucinous oral biofluid produced by salivary glands.81 Approximately 1–5 L of this clear, slightly acidic fluid is produced daily. Substances can transport from the blood to the saliva through transcellular or paracellular diffusion. Consequently, the functional composition of saliva reflects the blood. While many components are the same, concentrations of each component can vary. Saliva typically has smaller concentrations of glutamate than blood and CSF, thus very sensitive and precise methods must be developed for detection.Stimulated whole saliva is found through chewing on a paraffin tablet.82 This method induces masticatory action, resultantly affecting the quantity and pH of saliva. Masticatory action includes the release of digestive enzymes and proteins.83 This method of collecting saliva supports the notion that glutamate concentrations in the saliva correlated to levels in plasma. At the moment, saliva is easier to handle and safer (relative to blood) as it does not clot and has not been found to carry pathogens such as human immunodeficiency virus (HIV).84 Consequently, saliva samples could be a non-invasive indicator of glutamate levels in the blood or independently.
3.5 Glutamate and pH in biofluids
Biofluids have varying compositions and mechanisms, affecting their pH and glutamate concentration. In experimental procedures of electrochemical sensing, the pH of the analyte should mimic the pH of biofluids. Additionally, the performance of electrochemical sensors can be affected by the pH of the medium they are detecting in. Thus, the pH of biofluid is an important characteristic to take into consideration when biofluids are the medium glutamate is being detected in. The pH of saliva is usually slightly acidic and fluctuates based on the food or drink consumed.85 Similarly, the pH of CSF and arterial blood are related and waver according to the homeostatic regulation.86 As previously mentioned, the urinary pH is affected by ammonium and indirectly glutamate. Analogously, ammonium also impacts the pH of sweat and blood, where a linear correlation is suggested between the increased sweat production and presence of ionic ammonium.87 The pH can also impact the metabolism of glutamate and glutamine in the kidney, liver and brain.88 In particular, the concentration of glutamate in biofluids such as urine or CSF can be affected by a change in pH. For instance, in astrocytes an increased pH results in glutamate uptake, consequently a decrease in the concentration of glutamate in CSF. Typically, the concentration of glutamate in blood and CSF depends on the concentration gradient and cell metabolism, for instance it is synthesized within platelets. Above, Table 1 outlines characteristics of biofluids, important to consider in the development of an electrochemical sensor to detect glutamate in biofluids.4. Glutamate a powerful biomarker for abnormalities in the body
Glutamate is a potential biomarker for detecting and monitoring abnormalities in the body like neurodegenerative diseases, malignant diseases and acute to chronic pain. Biomarkers can offer insight to processes or responses occurring within the body.91 Extracting this information non-invasively is not only less intimidating to individuals being tested, but it eliminates the risks involved with invasively extracting information from the body. Many correlations have been made connecting glutamate to pathologies, suggesting glutamate as a biomarker. For instance, overstimulation of glutamate receptors can result in neuronal death.92 The neuronal death is hypothesized to be due to the excessive calcium influx, which result in catabolic enzymes. Similarly, the large release of glutamate to the extracellular fluid can result in extreme extracellular glutamate concentrations. While further development is needed to ensure samples are free of contamination and are maximally sensitive, biomarkers, such as glutamate, have the potential to serve as an important diagnostic tool in healthcare.Current technologies should be more focused on this issue of measuring glutamate in disease conditions using biofluids. However, most studies use conventional laboratory techniques to measure glutamate of biofluids and assess its viability as a biomarker. These technologies offer glutamate measurements with high sensitivity and wide detection range, ideal for measuring small concentrations to a high degree of accuracy. In fact, they often require a trained professional to operate the machinery that is not only expensive but often requires a large amount of time to achieve results. Table 2 evaluates the current technologies used to detect and measure glutamate. Key parameters assessed are the length of time to acquire results, the limit of detection and detection limit range. It is important to consider these parameters when considering potential healthcare applications.
| Detection technique | Detection limit range | Limit of detection | Measurement time | Ref. |
|---|---|---|---|---|
| a Experiment was conducted with D-glutamic acid. | ||||
| Instant enzyme-linked immunosorbent assay | <7.8–500 pg ml−1 | — | 2–3 h | 93 |
| Gas chromatography-mass spectrometry | 0.001–0.291 mg L−1 | 0.355 mg kg−1 | 5 min | 94 |
| Gas chromatography-mass spectrometry | — | 0.38–8.13 ng mL−1 | 7.06 min | 95 |
| Fluorometric probe | 5 pmol | — | ∼1 h | 96 |
| In vivo electrochemical sensor | 0.02–0.08 μM | — | — | 97 |
| Ratiometric and colorimetric chemisensor | — | 0.796 μM | — | 98 |
| High performance liquid chromatographya | 10 nM–1 μM | 1 nM | — | 99 |
| High performance liquid chromatography | 0.15625–5 μM | 0.8 pmol | 30 min | 100 |
| High performance liquid chromatography-fluorescence | 0.1–20 μg mL−1 | 0.025 μg mL−1 | 8.9 min | 9 |
4.1 Neurodegenerative diseases
Glutamate excitoxicity is linked to both acute and chronic neurodegenerative diseases.101 Specifically, glutamate receptors have chronically elevated input or EAATs are downregulated, thus the extracellular glutamate concentration stays high. The elevated extracellular glutamate levels cause continuous depolarization of the cells. As a result, chronic excitotoxicity has been evident in AD, Huntington's disease and ALS.Alzheimer's disease (AD) occurs due to the senile plaques of beta amyloid and aggregated intraneuronal neurofibrillary tangles. This neuronal death is hypothesized due to excess glutamate and increased receptor activation.102 It is proposed NMDA receptors are specifically responsible, for they are more susceptible to carrying calcium. The enhanced activation results in a surplus of intracellular calcium and resulting catabolic enzymes resulting in neuronal death in Alzheimer's disease. Glutamate can potentially be detected and measured in the CSF as a biomarker for AD. Madeira et al. utilized HPLC to measure glutamate and glutamine levels in CSF of normal and AD patients.103 The study reported significantly elevated glutamate and glutamine levels in the CSF of patients with AD, suggesting glutamate as a biomarker to indicate AD. The laboratory techniques used in this study provide results as quick as in 8.9 min, faster in comparison with other methods, but still not instantaneous.104 Ion suppression also affects the selectivity of HPLC when the molecule to be measured is not independent of other interfering molecules.105 In the case of biofluids, this could pose an issue as glutamate is within a biological matrix with other interferent molecules. Laboratory techniques require trained personnel, expensive equipment and time; thus, it is ideal to develop a method of detection that eliminates these factors.
Huntington's disease is due to the genetic mutation that leads to the neurodegeneration in the basal ganglia and cortex.106 It is hypothesized this degeneration is a result of glutamate excitoxicity, where neurotransmission ceases as the disease progresses. Specifically, it has been proposed that dopamine is unable to balance the glutamate neurotransmission, resulting in anomalies. Further neurodegeneration is evident in ALS.
Amyotrophic lateral sclerosis (ALS) is a paralytic disease resulting from motor neuronal death.44 Elevated glutamate levels in the CSF were found in ALS patients, resulting in excessive AMPA receptor activation. Due to high amount of AMPA receptors, it is hypothesized that glutamate excitoxicity is a contributor to motor neuron death in ALS. Moving forward, glutamate could be a biomarker to detect neurodegenerative diseases.
4.2 Malignant diseases
Glutamate and glutamate receptors are hypothesized to impact malignant diseases, such as glioma, breast carcinoma, prostate carcinoma, and melanoma.107–109 In glioma and meningioma, commonly malignant and benign brain tumors respectively, elevated glutamate concentrations contribute to tumour growth, proliferation and survival.58 When excitoxicity from excess extracellular glutamate leads to apoptosis, glioma cells have the opportunity to grow and proliferate. T. Takano and colleagues’ study further supports this: they reported when tumours, which released glutamate, were supplied with NMDA receptor agonists, the tumour growth slowed.110In breast cancer, glutamine, a precursor and product of glutamate, is found to contribute to tumour progression. Glutamate receptors, both metabotropic and ionotropic, are found on cancer cells and responsible for signal transmission. As a consequence, metabotropic glutamate receptors were explored and targeted for breast cancer treatment.111 Specifically, it was proposed metabotropic glutamate receptors contributed to the growth and signal transmission of triple negative breast cancer. Similarly, glutamine also affects the metabolism of prostate cancer, where increased glutamine levels were detected as the cancer progressed. Analogously, metabotropic glutamate receptors were targeted to treat melanoma. Gelb et al. found that glutamate was responsible for DNA synthesis and cell viability in two cell lines of melanoma.109
Glutamate and glutamine were measured in patients with metastatic head and neck squamous cell carcinoma (HNSCC) using isotype dilution GC-MS.112 Glutamate was significantly elevated in metastatic HNSCC tissues compared to normal tissues. Additionally, glutamate levels in unmatched saliva and plasma of patients with HNSCC were significantly increased in comparison to healthy individuals. This indicates glutamate could be a biomarker for HNSCC in certain biofluids. Although, the measurement technique has a low limit of detection at 0.38–8.13 ng mL−1, ideal in testing biofluids, it requires large equipment and long lengths of time.95 A study detected glutamate in urine with GC-MS took 7.06 min, not fast or compact enough for real-time health monitoring. In the future, a compact device with a small footprint and fast response time could detect excitoxicity as a biomarker and be utilized for health monitoring to indicate tumor presence or progression.
4.3 Acute to chronic pain
Nociceptors are sensory neurons to detect pain and communicate noxious stimuli to the peripheral nervous system through primary afferent neurons.113 Glutamate contributes to the nociceptive process as a part of physiological process of pain and playing an important role in the primitive protective function in our body which is helpful for detecting harm and preventing tissue damage. Patients suffering from chronic pain may experience exacerbated responses to both painful and non-painful stimuli.114 This phenomenon happens due to hyperexcitability in the CNS and it is known as central sensitization.115 Central sensitization occurs when the postsynaptic cell is more sensitive to incoming signals, resulting in elevated responses and pain. Glutamate receptors throughout the nervous system contribute to this pain response. It is known as the primary transmitter for information to the dorsal horn, the part of the CNS that processes sensory information.Glutamate level found positively correlated with individual pain sensitivity.116 Glutamate level plays a key role in pain mechanism by lowering neural threshold and/or increasing pain response.117–119 Increased sensitivity can include a lower threshold potential required when glutamate receptors are activated. Thus, the overactivation of these receptors could be the cause of pain associated with elevated glutamate in studies outlined below.
Glutamate has been detected in saliva and revealed a significant change in concentration during a study by G. D'Andrea et al.7 During migraines without aura, elevated GABA levels were detected in saliva. This is important as glutamate is a metabolic precursor of GABA, as mentioned in section 2.1. Additionally, antiglutamatergic drugs reduced the severity and duration of familial hemiplegic migraine. Hence, glutamate could potentially play a role in the severity and duration of migraines or pain associated with them. Similarly, Nam et al. reported significantly higher salivary glutamate levels in individuals with chronic migraine compared to individuals with episodic migraine or no migraine. Salivary glutamate levels were detected using an ELISA kit from Abnova, with a detection limit of 0.3 μg mL−1.120 While the limit of detection is ideal for testing glutamate levels in biofluids, Abnova's colorimetric ELISA is costly ($565) and takes a minimum of 2 hours when incubated on a shaker.121 While this ELISA is less expensive compared to other techniques, such as GC-MS, it is still costly in comparison to inexpensive electrochemical sensors. Additionally, ELISA does not provide a quick response time or wearable functionality for real-time health monitoring.
Elevated glutamate plasma levels are correlated with chronic widespread pain, such as fibromyalgia syndrome.122 While the cause of fibromyalgia is not determined, many studies suggested glutamate dysregulation has a role. In a study by Gerdle and colleagues, glutamate was injected into male masseter and splenius muscles, which led to pain for approximately 10 minutes.123 Thus, elevated glutamate levels could be a cause of pain in facial muscles. Chronic pain in cancer patients has also been hypothesized due to glutamate.124 Excess production of glutamate inside of cells, leads to release of glutamate through the cystine/glutamate antiporter. This results in high extracellular concentrations of glutamate and elevated nociceptive responses. Consequently, correlations both direct and indirect have suggested glutamate as a potential biomarker for pain.
5. Electrochemical sensing
Electrochemical sensing is an alternative to conventional laboratory-based techniques, offering instantaneous, low cost, real-time continuous monitoring. Electrochemical sensors consist of a receptor, to recognize and attach to the intended element, and a transducer, to convert the binding to a measured signal.125 The measured signal is then sent for signal processing. Biosensors are the biological manipulation of electrochemical sensors, where a biological analyte is detected on a bioreceptor.126 Examples of bioreceptors include enzymes, such as GluOx. Fig. 5 outlines the components of biosensors, exemplifying different bioreceptors and biological analytes. While there is lots of literature regarding electrochemical sensors and enzymatic sensors, this paper provides it all in one review regarding glutamate. Specifically, the following section provides a snapshot at electrochemical sensors to detect glutamate, ideal for novices in electrochemical sensing. Through detecting and measuring a biological analyte, sensors can serve as an analytical device in medical care. | ||
| Fig. 5 Illustration of the components of biosensors, outlining examples of each component. | ||
5.1. Evolution of electrochemical sensors to detect glutamate
Possible electrochemical sensor structures to measure glutamate include transistors and potentiometric sensors.127,128 An electrochemical sensor can consist of electrodes to detect an analyte indirectly. A three-electrode sensor consists of a working electrode to sense the intended electrochemical event; a reference electrode to maintain the constant equilibrium potential (acts as a dynamic baseline in order to compare the measured potentials of the other electrodes); a counter electrode to ensure the electrical circuit between electrodes is a closed loop.129 The electrodes are submerged in an electrolyte solution to ensure electrical neutrality as ions migrate with minimal resistance and connected to a data processor to relay the results. Potentiometric sensors can be fabricated with two- or three-electrode systems. The three-electrode method typically utilize Ag/AgCl for the reference electrode and Pt wire as the counter electrode.130 The electrodes can all be connected to a potentiostat for voltammetric or amperometric analysis. For instance, in amperometric sensors the potential is held at a constant value. An example of this system is illustrated in Fig. 6, where the electrodes are all suspended in an electrolyte. The working electrode of enzymatic and non-enzymatic sensors typically consists of metal or metal oxide, where a reduction–oxidation reaction can occur. Nanostructures are used as a low-cost solution to increase the electrocatalytic activity of the working electrode by increasing the electrochemically active surface area.131 A higher electrochemically active surface area decreases the noise produced and results in a higher sensitivity.132 Nanostructures, including nanoparticles and nanosheets, have a minimum of one dimension between 1 to 100 nm in size.133 Metal nanostructures are not hindered at lower threshold operating voltages, ideal for physiological environments where high voltages can be detrimental. | ||
| Fig. 6 Setup of electrochemical sensor with working, counter, and reference electrodes connected to a potentiostat. | ||
Biosensors have advanced tremendously in the past two decades since the initial “true” biosensor was brought to life by Leland C. Clark.134 They are exhibiting improved sensitivity and selectivity becoming comparable to conventional laboratory-based methods. Enzymatic sensors are generally developed from one of the first three generations of electrochemical sensors.135 The first generation utilizes an enzyme to catalyze a reaction. When the analyte is detected a reduction–oxidation (redox) reaction occurs, indirectly producing an electron through the production of hydrogen peroxide. The following reaction occurs with a first generation sensor utilizing GluOx as the enzyme.
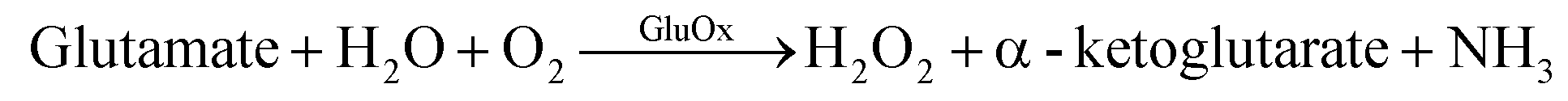 | (1) |
| H2O2 → H2 + O2 + 2e− | (2) |
In the second generation, the same concept occurs as the first generation, rather the redox reaction occurs with a mediator, which transfers an electron to the electrode.136 In the third generation a redox reaction occurs when the substrate is present, where the enzyme directly produces an electron to the electrode. Fig. 7 outlines these generations of enzymatic sensors, graphically demonstrating each process. S. Qin et al. developed in vivo first and second generation electrochemical biosensors and found the first generation sensors performed with a better sensitivity and faster response time.137 Second generation sensors posed issues with reproducibility due to the coating requirement.
 | ||
| Fig. 7 Graphical representation of the generations of enzymatic electrochemical glutamate sensors. (a) Demonstrates a first generation sensor with an enzyme and H2O2. (b) Exhibits a second generation sensor with an enzyme and a mediator that undergoes reduction–oxidation. (c) Showcases an enzymatic, mediator-free reaction. | ||
In the next generation of biosensors, outlined in Fig. 8, no enzyme is required.138 The substrate is indirectly measured by undergoing a redox reaction with a metal or metal oxide, which produces a product that is measured. This is ideal as it avoids enzymes and mediators, thus not altered in environmental or temperature changes. Depending on the metal being utilized this sensor is also less expensive, ideal for economic purposes. A summary of the generations of biosensors is evident in Table 3.
 | ||
| Fig. 8 Graphical representation of non-enzymatic electrochemical glutamate sensors. | ||
| Generation | Reactions | Ref. | Performance comparison | Ref. |
|---|---|---|---|---|
| 1st |

|
144 | Detection of hydrogen peroxide break down | 136 |
| H2O2 → H2 + O2 + 2e− | Simple fabrication | |||
| Good reproducibility | ||||
| Faster response time | ||||
| High sensitivity | ||||
| 2nd |

|
137 | Osmium mediator | 136 |

|
Lower oxidation potential (eliminate interfering current) | |||
| 2Os3+ + 2e− → 2Os2+ | ||||
| 3rd | — | Direct measurement of enzyme/substrate pairings | 145 | |
| 4th | Ni(OH)2 + OH− → NiOOH + H2O + e− | 16 | Enzyme-less | 16 |
| NiOOH + H2O + e− → Ni(OH)2 + OH− | Reduce complexities | |||
| Cost effective |
Current modifications of glutamate sensing electrodes in non-enzymatic electrochemical sensors include nanoparticles, nanosheets, and nanoarrays. Vertically aligned nanowire array modified with Ni particles, improves the electrocatalytic surface area.139 Further improvement is accomplished with nanosheets, which have enhanced electrocatalytic surface area greater than nanorods or nanowires. Analogous to two dimensional nanosheets, two-dimensional materials like graphene have become popular in modified glassy carbon electrodes for their enhanced electrocatalytic abilities.140 This can be attributed to graphene's increased surface area, conductivity and electrical mobility. Similarly, metal and metal oxide nanoparticles offer higher surface energy and conductivity to catalyze reactions and enhance electron transfer respectively.141 This is ideal for electrochemical sensors, which determine the concentration based on the quantity of electron transfer reactions. While nanoparticles typically have relatively simple fabrication and offer a cost-effective approach to improving detection, metals such as gold nanoparticles are costly.142 Single-walled carbon nanotubes (SWCNT) are similar to graphene, composed of carbon, however they are three-dimensional.
Claussen et al. manipulated the surface of the enzymatic electrode with SWCNT and Pt nanocubes and nanospheres.143 This modification was proposed to be responsible for increased electrochemical transduction and electrocatalytic properties. Resultantly, the sensor had a wide linear detection range and low limit of quantification, making it ideal for detecting glutamate in biofluids. Similarly, Jamal et al. developed a non-enzymatic sensor utilizing NiO nanoparticles and chitosan in an effort to simplify and reduce the cost of electrochemical glutamate sensors.16 NiO nanoparticles were used to increase the surface area and increase the sensitivity. Although, the sensor did not have a LDR sufficient to quantify glutamate in biofluids. Recently, Ahmad et al. developed a template polymer-based glutamate sensor with an improved limit of detection in comparison to the sensor with NiO. However, the LDR was not reported, so it is unknown whether it can quantify glutamate in biofluids.
5.2. Electrochemical glutamate sensor performance properties
In this section, we outline important terminologies to guide a wide range of readers in electrochemical sensing of glutamate. These are critical to determine the suitability and performance of sensors, properties, such as sensitivity, especially in the comparison between sensors. The performance of electrochemical glutamate sensors is evaluated on the following properties (Fig. 9): | ||
Fig. 9 Graphical representation of sensor performance properties. The x-axis is the concentration of glutamate and the y-axis is the signal measured, specifically current in electrochemical glutamate sensors. LoQ is limit of quantification and describes the limit of where the analyte is accurately measured in a linear response, or the lower bound of the linear detection range (LDR). Similarly, the limit of detection or LoD is the slope or 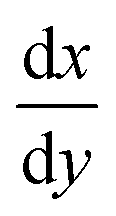 of the linear detection range is the sensitivity. of the linear detection range is the sensitivity. | ||
| LoD = μblank + kσblank | (3) |
5.3. Fabrication technologies
Biofluids contain multiple analytes, demanding many sensing electrodes onto a single substrate at low-cost. For such electrochemical sensors, fabrication technologies such as solution processing, inkjet printing or roll-to-roll bonding can be used.151–155 Inkjet printing has been used since being developed in the nineteenth century, recently utilized to fabricate electrical devices such as PCBs, sensors, and electrodes.156Inkjet printers work by either continuous inkjet (CIJ) printing or drop-on-demand (DOD) printing.156 DOD printers are simpler and more commonly used. Both printers consist of print heads, typically thermal or piezoelectric. In a thermal print head, a current is drawn through to superheat the ink in a resistive heater to the bubble nucleation temperature. At this temperature, the bubble performs as an insulator, protecting the ink from overheating. The bubble increases in volume, resulting in the ink depositing from the chamber to the nozzle. A transient pressure wave is created when the current and heat cease, causing the bubble to collapse. Momentum from the wave causes the droplet to depart from the nozzle onto the medium to be printed. Piezoelectric print heads are similar, only they consist of piezoelectric material that contract when a voltage is applied, causing the nozzle to release a droplet. Piezoelectric heads are not suitable if there is possible thermal degradation of ink or specific nucleation temperature. In general, inkjet printing is ideal for its low waste production, low-cost, and flexible product.
Nanoparticle inks have been used for their good electrical conductivity, making them suitable for electrode fabrication.157 Due to their high surface area to volume ratio, they will also melt at lower temperatures, thus are more efficient. Although, since they are in a solution, they are susceptible to agglomeration. Agglomeration can lead to an increased viscosity and build of nanoparticles at the nozzle. Some nanoparticles are also susceptible to oxidation, thus effecting the longevity of the device.
Inkjet printers are a series of continuous droplets onto a material. This should occur at a quickly, thus the droplet velocity should be fast to overcome drag (between 1 to 30 m s−1). With piezoelectric heads, this velocity is dependent on the kinetic energy in the piezoelectric material. Once an ink droplet has been dropped it depends on the type of ink whether the droplet is solidified or evaporated. Conductive inks and paper with inkjet printers could be used to fabricate electrochemical sensors.158 In combination with flexible substrates, inkjet printing provides a method to provide many low-cost wearable sensors, ideal for disposable devices in healthcare applications.159 Inkjet printing can also improve the sensitivity of sensors, by altering the pattern of adjacent ink drops.160 The sensitivity of an ammonia sensor was improved nearly threefold, by altering the drop spacing between adjacent ink drops to cause hold depletion and affect the carrier transport. Inkjet printing is an efficient, low-cost and easy way to consistently and accurately fabricate a large quantity of electrodes, improving the affordability and reproducibility of electrochemical sensors.
Roll-to-roll bonding has been used in the adhesion of silver nanowire (AgNW) on a plastic substrate.161 The process consisted of first being spraying the film with a solvent to dissolve the polymer layer from the substrate (which the AgNW was deposited on). Then the film was compressed between two pressing rollers. Afterwards the film was soaked in a salt solution and washed with distilled water. This ensured the Ag atoms were ionized and re-deposited. While it was not tested on electrochemical sensors specifically, it was tested on organic field-effect transistors and performed successfully as a source–drain electrode. Similarly, roll-to-roll gravure bonding has been used to develop a wearable electrochemical sensor for health monitoring.162 Roll-to-roll gravure bonding provided enhanced sensitivity, lower production times, and good reproducibility compared to methods such as screen printing.
Another fabrication method is through metal–polymer bonding. Previously, electrodes have been fabricated with copper and liquid crystal polymer (LCP).163 Bonding can be used to combine a variety different of material in a fast and facile approach. This wide range of materials is sought of to find the optimal combination of materials, for instance this method is compatible with materials such as metals, glass and LCP. LCP is ideal for its flexibility and high moisture barrier, ideal for exploring wearable sensors. Additionally, this method is ideal in developing simple, single material-based system. Redhwan et al. fabricated surface activated Cu/polymer bonded electrodes.164 In the case of rolled-annealed Cu, a compact grain structure and larger cross-sectional area leads to good conductivity and signal integrity. While rolled-annealed Cu has a weak adhesion property, surface-activated bonding provides a method to overcome this. Consequently, metal–polymer bonding provides a method to produce a flexible sensor and maintain high sensitivity by preserving the characteristics of metals. Their process allowed a flexible sensor to be fabricated in a simple and time effective manner, ideal for efficient and consistent mass production.
5.4. Enzymatic electrochemical sensors and recent development to detect glutamate
Currently, enzyme-based biosensors have been sought of for their excellent sensitivity and low detection limit. Table 4 outlines recent enzymatic electrochemical sensors, comparing the analytical performance of each sensor. Materials such as glutaraldehyde or nanoparticles were used to enhance the performance of glutamate sensors.136,165| Working electrode material | Enzyme | Detection limit (μM) | Linear detection range (μM) | Operating voltage (V) | Response time (s) | Sensitivity (nA μM−1) | Biofluid tested in | Ref. |
|---|---|---|---|---|---|---|---|---|
| Pt platinum, MEA microelectrode array, BSA bovine serum albumin, GluALD glutaraldehyde, mPD m-phenylenediamine, PoPD poly(o-phenylenediamine), AscOx ascorbate oxidase, CNF carbon nanofiber, SiO2 sodium dioxide or silicalite, PI polyimide, CFE carbon fiber electrode, PB prussian blue, PEI polyethyleneimine, PG pencil grpahite, ZnONR zinc oxide nanorods, PPy polypyrrole, GCE glassy carbon electrode, Naf 5% Nafion, Gldh-bacteria bacteria surface-displayed glutamate dehydrogenase, PEI-MWNT polyethyleneimine dispersed multi-walled carbon nanotube, PtNP platinum nanoparticle, SPCE screen printed carbon electrode, MB medola's blue, Chit chitosan, MWCNT multi-walled carbon nanotube, BDD boron-doped diamond, PPD poly(phenylenediamine), CeO2NP ceria oxide nanoparticles, TiO2NP titanium oxide nanoparticles, oPD o-phenylenediamine, Au gold, PANI polyaniline, Nano-PPCPE nano-porous pseudo carbon paste electrode, Cat catalase, Ti titanium, PAA porous anodic alumina, SWCNT single-walled carbon nanotubes, EDC N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydro-chloride, TGA activated thioglycolic acid, SAM self-assembled monolayer, mPD m-phenylenediamine, PU polyurethane. | ||||||||
| Pt-MEA/BSA/GluALD/mPD | GluOx | 0.16 ± 0.02 | 10–570 | 0.7 | — | 270 ± 28 cm−2 | Rat ECF | 194 |
| Pt/PoPD/AscOx/BSA | GluOx | 0.044 | 5–150 | 0.6 | 2 | 0.097 ± 0.001 | Rat ECF | 198 |
| Pt-CNF | GluOx | — | 1–100 | −0.15 | — | 266 cm−2 | Rat ECF | 190 |
| Pt/SiO2/BSA/GluALD | GluOx | 1 | 2.5–450 | 0.6 | 20–30 | 0.5 | — | 195 |
| Pt/PI | GluOx | 0.150 | — | 0.45 | — | 1 | — | 199 |
| CFE/PB/PoPD/PEI/GluALD | GluOx | 1.5 | 0–150 | 0.05 | 135 ± 228 cm−2 | — | 200 | |
| PG/ZnONR/PPy | GluOx | 0.00018 | 0.02–500 | 0.065 | <5 | — | — | 170 |
| GCE/Naf/Gldh-bacteria/PEI-MWNT | GLDH | 2 | 10–1000 & 2000–10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 000 |
0.52 | — | — | — | 12 |
| MEA/PtNP/mPD/GluALD/BSA | GluOx | 0.5 | 5–30 | 0.7 | <8 | 0.056 | Rat ECF | 196 |
| SPCE/MB/chit/MWCNT | GLDH | 3 | 7.5–105 | 0.1 | 20–30 | 0.39 | — | 168 |
| BDD/PtNP/PPD | GluOx | 0.35 | 0.5–50 | 0.5 | 4 | 24 | — | 13 |
| GCE/Pt-GNPs/PPD | GluOx | 0.75 | 0.2–100 | 0.5 | 4 | 174 | — | 13 |
| Pt/CeO2NP/TiO2NP/chit/oPD/BSA (oxygenated conditions) | GluOx | 0.594 | 5–90 | 0.6 | 2 | 0.793 | Rat CSF | 165 |
| Pt/CeO2NP/TiO2NP/chit/oPD/BSA (deoxygenated conditions) | GluOx | 0.493 | 5–50 | 0.6 | 5 | 0.395 | Rat CSF | 165 |
| Au/PPy/PANI/GluALD | GluOx | 0.0001 | 0.02–400 | −0.13 | 3 | 533 cm−2 | — | 193 |
| Pt/PPy/Naf/chit | GluOx | 2.5 ± 1.1 | 20–217 | 0.7 | <2 | 34.9 ± 4.8 cm−2 | Rat ECF | 136 |
| Pt/PPy/Naf/BSA/GluALD | GluOx | 6.5 ± 1.7 | 20–352 | 0.7 | <5 | 86.8 ± 8.8 cm−2 | Rat ECF | 136 |
| Pt/PI/mPD/BSA/GluALD | GluOx | 0.22 | <150 | 0.45 | 4.9 ± 1.9 | 2.16 ± 0.08 mm−2 | Rat ECF | 201 |
| Nano-PPCPE/cat/BSA | GluOx | 0.25 | 0.5–10 | — | — | — | — | 169 |
| MEA/Pt/PI/mPD/BSA/GluALD | GluOx | 0.6 | — | 0.7 | — | — | — | 177 |
| Pt/SiO2/Ti | GluOx | 50 | 50–10000 |
0.6 | — | 3.68 | — | 191 |
| Pt/PPD | GluOx | 3 | — | 0.4 | — | 7.129 | Human serum | 197 |
| Pt/PAA/SWCNT | GluOx | 0.0046 | 0.05–1600 | 0.35 | — | 72.4 | — | 143 |
| Au/EDC/TGA/SAM | GluOx | 0.072 | 0.1–10000 |
— | — | 17.89 cm−2 | — | 202 |
| Pt/PPy/MWCNT/PU | GluOx | 0.3 | <140 | — | 7 | 3.84 mm−2 | — | 179 |
| Pt-PPD/SiO2 | GluOx | 0.005 | 0.5–100 | 0.5 | 10 | 279.4 ± 2 cm−2 | Rat ECF | 203 |
5.4.1.1 Sensing electrode – metals. Electrodes can be fabricated from a multitude of materials, commonly consisting of platinum (Pt), carbon, gold (Au) or ceramic. Specifically, Pt is the most prevalent material in Table 4. Although outdated, a comparative analysis on Pt, Au, Pd, and GC electrodes for glutamate sensing was performed and found Pt was the best overall glutamate detection. Pt is sought of for its biocompatibility, used in a variety of biomedical devices.166 In a study of Pt electrodes fabricated on Si substrates, analogous to some of the electrodes in Table 4, they found Pt electrodes performed well for BioMEMS.167 The electrodes exhibited high chemical inertness and surface integrity in the presence of applied potentials, proving to be promising against degradation. They also found Pt to be biocompatible against human dermal fibroblasts and demonstrated electrochemical behaviour compatible with biomedical devices.
5.4.1.2 Sensing electrode – non-metals. Carbon is the second most common material for glutamate sensing electrodes in Table 4, apparent as screen-printed carbon electrode (SPCE), nano-porous pseudo carbon paste electrode (Nano-PPCPE), boron-doped diamond (BDD) and pencil graphite (PG).13,168–170 Carbon is a low-cost alternative in comparison to Au or Pt. Meanwhile, it still has ideal characteristics such as biocompatibility.171 Nano-PPCPE was used for its porous nature leading to an increased surface area.169 SPCEs are sought of because carbon ink has resistance to many solvents, low background current and wide potential windows, thus making it easy to modify.172 BDD was used, for boron makes the diamond conductive, together leading to a low background current, high response stability, and high stability against fouling.13
5.4.1.3 Sensing electrode modifications – metals. Analogously, nanostructures of metal and lanthanide oxides, including nanorods and nanoparticles of zinc oxide (ZnO), titanium oxide (TiO), and cerium oxide (CeO2) have been used to enhance the detection of glutamate. ZnO is sought of for its high isoelectric point, biocompatibility, and fast electron transfer kinetics.173 The morphology of nanostructures can affect the catalytic performance, evident in a study of CeO2 nanostructures.174 It was apparent the crystal plane exposed affected the amount of oxidation of CeO. It was found nanowires exposed the most reactive planes thus had the best oxidation performance, followed by nanorods and nanoparticles.
5.4.1.4 Sensing electrode modifications – non-metals. Electrodes have been modified with a multitude of metals, usually in the form of nanostructures to variations of carbon. Previous nanostructures used in Table 4 include nanoparticles and nanorods, made of metal oxides or platinum. While allotropes and variations of carbon used to detect glutamate include carbon nanotubes (CNT), carbon nanofiber (CNF), and graphene NP. Graphene is a two-dimensional material composed of a hexagonal lattice of carbon atoms.175 CNT can either be single-walled (SWCNT) or multi-walled (MWCNT) varying in the number of layers of rolled up graphene. Graphene is advantageous over CNT due to its lack of metallic impurities along with its facile and low-cost synthesis process.176
5.4.1.5 Sensing electrode modifications – organic polymers. While glutaraldehyde is ideal for its cross-linking ability to attach enzymes, Tseng et al. found electrodeposited chitosan enhanced the selectivity of the sensor compared to glutaraldehyde.136 Chitosan is a natural polymer derived from crustaceans, such as shrimp.178 It has an affinity for proteins and ability to immobilize enzymes, ideal for enzymatic electrochemical sensors. Additionally, the biopolymer has ideal properties for sensors in physiological conditions including being inert, biodegradable, and antibacterial. Analogous to chitosan, polypyrrole (PPy) is a conducting polymer used for its ability to reject interference molecules.179 Similarly, other polymers such as phenylenediamine have been used to form perm-selective layers.180
Variations of phenylenediamine specifically, meta-phenylenediamine (mPD), ortho-phenylenediamine (oPD or 1,3-diaminobenzene) to form poly(o-phenylenediamine) (PoPD) are commonly present among biosensors. Where mPD and oPD are isomers of phenylenediamine and oPD is the monomer used to synthesize PoPD.181,182 Another isomer used in electrochemical sensors is p-phenylenediamine, although it is not known to be used in electrochemical glutamate sensing. The three isomers were electrodeposited to fabricate a permselective polymer on Pt–Ir cylinders and compared in a characterization study for the detection of H2O2 and ascorbic acid (AA).180 Perm-selectivity is important for sensing analytes in biofluids where interference molecules are present, such as AA. At low concentrations of AA, lower than 200 μM, PmPD was found to have the best perm-selectivity for H2O2. Although, their study also found PmPD had poor stability, degrading in one day, in comparison to PoPD. As a result, where longevity is necessary PoPD was the most promising polymer. Similar to polymers, direct electrode modification with metals and nanostructures is also proven to improve the performance.
5.4.1.6 Sensing electrode modifications – enzymes. The selectivity and sensitivity of enzymatic sensors depend on the enzyme being used, as to how many substrates it had and the degree of specificity. Enzymatic sensors are susceptible to environmental changes, such as changes in temperature or pH, rendering them unstable.183 GluOx is commonly immobilized as the enzyme on the working electrode. GluOx is typically derived from Streptomyces sp. and has been used to detect glutamate in phosphate-buffered saline (PBS) at a pH of 7.4.184 This is ideal for its physiological compatibility. This enzyme had 100% specificity for L-glutamate and 0.6% for L-aspartate. A drawback with GLDH, another enzyme used to detect glutamate, is the requirement of a cofactor, such as NADPH used by Basu et al.185 Accordingly, biosensors which immobilize GLDH require additional materials and the performance is dependent on the presence of the cofactor. Additionally, B. Liang et al. found the purification of native GLDH was inefficient and time-consuming.12 Another downside of utilizing enzymes is their tendency to denature. For instance, a strand of GLDH from Pyrococcus furiosus begins thermal denaturation at 100 °C and is inactive below 40 °C. This could be troublesome for healthy adults’ oral temperature is between 36.1 °C to 37.2 °C, thus the enzyme would not be compatible in saliva.186 Enzymes also require additional materials such as BSA to immobilize the enzyme on a substrate. Bovine serum (BSA) albumin is commonly utilized as a proteic feeder for cross-linking enzymes.187 BSA is helpful in cases, like typically with glutamate where glutaraldehyde is used alongside BSA as it can lead to the inactivity of enzymes. Enzymatic electrochemical sensors to detect glutamate also lack reproducibility evident by S. Park and colleagues.188 In the future the stability, reproducibility and complexity of an electrochemical sensor could be improved with an enzyme-less sensor.
5.4.2.1 Selectivity. One issue with selectivity is apparent with GLDH, where the cofactor, NAD+ or NADP+, requires a high oxidation potential on bare electrodes.12,189 This can allow easily oxidizable species to interfere with the detection. B. Liang et al. explored this issue testing two electrodes in 1 mM NADPH, finding an oxidation peak at 0.78 V with Naf/GCE vs. 0.45 V with Naf/PEI-MWCNTs/GCE.12 The oxidation peak current of the modified electrode increased with an increase in NADPH, suggesting the PEI-MWCNTs enhanced the electrocatalytic activity. Similarly, Medola's Blue and oPD have been used as mediators to lower the potential.168 The same group suggested Gldh-bacteria and Nafion film as a reasoning for the good selectivity. 10 mM of other amino acids such as lysine, threonine, and serine and uric acid (UA) did not affect the current response of 0.5 mM glutamate. The specificity of Gldh-bacteria was attributed to the excellent selectivity compared to other amino acids, while the negatively charged Nafion film was attributed to the lack of interference from negatively charged species such as UA. N. Isoaho and colleagues developed an enzymatic glutamate sensor with Pt-grown carbon nanofibers.190 However, it could be impacted by interference molecules such as oxygen, which affected the reduction reaction at higher linear detection range.
5.4.2.2 Limit of detection (LoD). Bäcker et al. developed an enzymatic sensor with the highest LoD at 0.05 mM.191 The sensor was a chip with microfluidic channel, fabricated with Pt thin film on a SiO2 substrate, along with a Ti adhesion layer. This sensor also consisted of glucose and glutamine sensors, all detecting hydrogen peroxide. Multiple sensors on a single chip could lead to cross talk, which they tried to eliminate with catalase membranes to ensure hydrogen peroxide breaks down before entering a neighbouring sensor. They identified further improvement in optimizing the flow-injection analysis parameters (which performs the electrochemical characterization) could enhance the sensor performance. In contrast, other groups focused on exclusively glutamate sensors and found a better limit of detection (Fig. 10).
 | ||
| Fig. 10 A biosensor chip to simultaneously detect glutamate, glutamine and glucose. Reproduced from ref. 191 with permission from Elsevier, Copyright 2013. | ||
While MWCNTs are supposed to enhance the surface area and enhance the electrocatalytic activity, one trend in Table 4 is the relatively high LoD of electrodes modified with MWCNT at 2 or 3 μM.12,168,192 This could due to the multi-walled nature of the structure, as SWCNT were used by Claussen et al. and they reported a LoD of 0.0046 μM. One reason for this difference could be the varying structures between single versus multi-walled. Although this LoD is improved with SWCNT in comparison to MWCNT, there is still room for improvement to ensure the sensor can detect glutamate in all biofluids.
5.4.2.3 Linear detection range (LDR). The most promising sensor for detecting and quantifying glutamate in biofluids, was developed by Batra et al. and had the lowest LoD and lowest lower bound of the LDR at 0.1 nM and 0.02 μM respectively.193 This enzymatic sensor consisted of glutamate oxidase on a Au electrode modified with PPy nanoparticles (NP) on polyaniline (PANI). PPyNPs increase the surface area and are highly porous, responsible for enhanced conductivity. Meanwhile, PANI is conductive and biocompatible, ideal for biosensors. The combination of materials results in a quick electron-transfer rate and both PPyNPs and PANI contribute to an enhanced electrocatalytic effect in regard to the oxidation of H2O2. The sensor is ideal, as glutamate can be detected and quantified in almost all biofluids, as saliva may still pose a challenge.
Batra et al. continued their development with another enzymatic sensor using GluOx with ZnO nanorods (NR) and polypyrrole (PPy) electrodeposited on pencil graphite, as outlined in Fig. 11a.170 The one-dimensional ZnONRs are attributed to large surface area and fast electron transfer kinetics, while the polymer PPy was used for its semiconducting properties. Electrodeposited layers of these materials have good stability due to the low likelihood the layer will deteriorate over time. The combination of previously mentioned characteristics resulted in a sensor that operates at the lowest potential of 0.065 V, apparent in the cyclic voltammetry response of Fig. 11c. The use of metal oxide NR could be how they overcame the higher operating voltage of 0.7 V, where those sensors all lacked metal oxide NR.136,194 This sensor also tied with Batra's previous sensor for the lowest LoQ at 0.02 μM. While it was not the lowest LoD, their LoD was very low at 0.18 nM, again attributed to the combination of materials. However, again this sensor could not quantify or detect glutamate in saliva.
 | ||
| Fig. 11 Reproduced from ref. 170 and 193 with permission from Elsevier, Copyright 2016 and 2013. (a) The outline of the fabrication of the ZnONR/PPy/PG/GluOx working electrode. (b) The cyclic voltammetry response of Au/PPy/PANI/GluALD/GluOx electrode in 0.5 mM glutamate in 25 mL of 0.1 phosphate buffered saline (PBS) at a scan rate of 50 mV s−1. (c) The cyclic voltammetry response of ZnONR/PPy/PG/GluOx working electrode in 0.5 mM glutamate in 25 mL of 0.1 M sodium phosphate buffer at a scan rate of 50 mV s−1. | ||
5.4.2.4 Response time. Özel et al. developed an enzymatic sensor with GluOx on a Pt microelectrode with ceria and titania NPs dispersed within chitosan.165 This sensor was tested in oxygenated conditions and had one of the fastest response times at 2 s. Thus, this sensor exhibited a high diffusion rate at the surface. This could be due to the use of chitosan as Tseng et al. also reported a fast response rate <2 s and their sensor was fabricated with Pt, PPy, 5% Nafion, and chitosan.136 Contrastingly, the sensor developed by Soldatkina et al., had a 20–30 s response time and consisted of simply Pt and silicalite.195
5.4.2.5 Stability. Wei et al. found the MEA/PtNP/mPD/GluALD/BSA sensor's sensitivity decreased to 97.60 ± 3.08% after 2 hours of in vivo measurements, and to 46.32 ± 5.08% after 9 hours.196 After 26 days at 4 °C, the sensor's sensitivity only decreased to 93.39 ± 2.71%. Similarly, Özel et al. reported optimal stability after the sensor was kept dry at 4 °C, exhibiting 80% of its initial activity after 10 days and 55% of its initial activity after 20 days.165 Consequently, the stability of sensors in vivo requires improvement for the application of health monitoring, which could require monitoring longer than 2–9 hours.
5.4.2.6 Sensitivity. Özel et al. further developed the CeO2NP/TiO2NP/chit/Pt sensor for deoxygenated conditions with o-phenylenediamine (oPD) and bovine serum albumin (BSA).165 The sensitivity was the second highest in Table 4 at 395 pA μM−1. This high sensitivity could be attributed to the ceria NPs, which have a high oxygen mobility and large oxygen diffusion coefficient. As a result, oxygen can be stored or released within ceria. This is ideal for when oxygen is not present in the environment since oxygen is necessary for GluOx. Consequently, this sensor is not only accurate, but also able to perform in hypoxic conditions.
While multiple groups developed working electrodes with Pt-MEA/BSA/GluALD/mPD.194,196 Wei et al.'s in vivo sensor had the highest sensitivity of 56 pA μM−1. While Scoggin et al. used similar materials, they reported a sensitivity of 270 nA μM−1. The discrepancy could be due to the different origin of materials. For instance, Scoggin et al. used a ceramic based Pt-MEA, whereas Wei et al. used a silicon-on-insulator based MEA. In comparison to other electrodes fabricated with Pt, Wei et al. used MEA modified with Pt nanoparticles resulting in a more sensitive electrode rather than using Pt as the substrate.
5.4.2.7 Performance in biofluids. Biosensors are tested in biofluids to analyze the sensor's capability to detect glutamate in healthcare applications. J. Scoggin et al. developed a biosensor to detect astrocytic glutamate uptake.194 The probe had a sensitivity of 62.3 ± 6.1 nA μM−1 cm−2 in basal media and a sensitivity of 270 ± 28 nA μM−1 cm−2 in PBS. Consequently, the sensitivity improved in basal media, ideal for detecting glutamate in physiological conditions. The sensor distinguished glioma cells from normal astrocytes, for instance the sensor detected the clearance rate was significantly faster in glioma cells compared to astrocytes. Similarly, W. Wei et al. developed a MEA and implanted it into the striatum of a rat.196 Real-time glutamate concentrations and electrophysiological signals were recorded before and after the injection of KCl. The results were consistent with the forecasted behaviour, as glutamate increased the injection of KCl leading to a depolarizing voltage due to the excitation of glutamate-activated neurons. This probe was dual-mode, detecting electrophysiological signals along with glutamate concentrations, resulting in less brain-damage and thus more accurate data acquisition. Additionally, the MEA had precise spatial definition, ideal for detecting glutamate in layered brain tissues. The response time of the sensor was in seconds, nearly providing a real-time analysis.
Windmiller et al. fabricated a microneedle array biosensor with Pt/PDD, testing it in human serum and phosphate buffer.197 The microneedle array consists of microneedles, the working electrode, and microneedle covers to entrap enzymes in the area between the needle and cover. The simplicity of the sensor results in a greater surface area for enzyme immobilization. The sensor demonstrated a sensitivity of 7.129 nA μM−1 and a LoD of 3 μM in phosphate buffer, and a sensitivity of 8.077 nA μM−1 in human serum with LoD of 21 μM. Consequently, the performance is almost consistent between simulated conditions and real serum samples, attributed to the robustness of PDD. PDD was originally chosen for its high selectivity and stability, specifically at rejecting interfering molecules. This notion is supported with the high sensitivity and promising limit of detection in human serum, which contains potential interference molecules. This sensor is also less invasive than previously mentioned sensors, which require extraction CSF in a potentially harmful and invasive matter. Although, moving forward the invasiveness could be further reduced measuring glutamate levels in biofluids such as urine or saliva.
5.5. Recent non-enzymatic electrochemical sensor development to detect glutamate
Although currently there is a limited number of non-enzymatic glutamate sensors in literature, there is a variety of enzymatic electrochemical sensors and field-effect transistors to detect glutamate in literature. Moving towards the fourth generation of electrochemical sensors, the lack of enzymes could improve the stability and decrease the cost of the sensor. Below is a summary of previous non-enzymatic electrochemical sensors fabricated to measure glutamate.Table 5 exhibits the non-enzymatic electrochemical sensors fabricated to date. This table is brief, potentially due to the lack of emphasis on glutamate and lack of information revolving the topic. Non-enzymatic electrochemical sensors also require the recent advances in nanomaterials to enhance the performance and provide a competitive alternative to conventional methods. Therefore, in this section we comprehensively surveyed the nonenzymatic electrochemical sensors for glutamate detection.
| Working electrode material | Detection limit (μM) | Linear detection range (μM) | Operating voltage (V) | Response time (s) | Sensitivity | Ref. |
|---|---|---|---|---|---|---|
| NiO/chit/GCE | 272 | 1000–8000 | +0.55 | <5 | 11 μA μM−1 cm−2 | 16 |
| Co3O4NS/GCE | 0.00001 | 0.0001–1000000 |
— | — | 9.5 × 10−5 μA μM−1 cm−2 | 17 |
| Au/SPCE/GEPPGBP | 0.1 | 0.1–1 | — | — | — | 204 |
| NiNAE | 68 | 500–8000 | +0.52 | — | 65 μA μM−1 cm−2 | 139 |
| Pt/NiNAE | 83 | 500–8000 | +0.52 | — | 96 μA μM−1 cm−2 | 139 |
| PNIPAM/4-VP/MAA | 1.7 ± 0.2 | — | — | — | 0.86 ± 0.12° μM−1 | 205 |
| Ni(OH)2 + OH− → NiOOH + H2O + e− | (4) |
| NiOOH + H2O + e− → Ni(OH)2 + OH− | (5) |
Pt/NiNAE experienced a decreased catalytic current when 100 mM of glutamate was present.
M. Jamal et al. developed another enzyme-free sensor consisting of NiO glass carbon working electrode, Ag/AgCl reference electrode and Pt wire counter electrode.16 Nickel was chosen as it is one of the most abundant elements on earth, thus a cost effective and sustainable material. Glutamate oxidation occurred within 0.1 M NaOH solution. NiO underwent reduction–oxidation reactions as denoted in eqn (4) and (5). Cyclic voltammetry and amperometric experiments were used to assess and measure the electrode. Cyclic voltammetry reveals an enhanced oxidation peak at 0.63 V, attributed to the oxidation of glutamate.
M. Hussain et al. developed a non-enzymatic sensor to detect L-glutamic acid and uric acid simultaneously.17 The sensor consisted of a counter electrode made up of Pt wire and working electrode. Mesoporous Co3O4 nanosheets (NS) were synthesized for the glass carbon working electrode using a wet chemical technique. The process consisted of ultrasonication of water with reactant precursors, followed by pH adjustment, then heating and drying resulting in large molecules that were grinded into powders, followed by additional drying. Brunauer–Emmett–Teller analysis revealed high surface area, pore volume and pore diameter of the Co3O4 NSs. Once the product was verified with methods such as Fourier-transform infrared spectroscopy, 5% ethanolic Nafion was added to bind the GCE and with Co3O4 NSs. Glutamate was detected through the increase in current as a result from the release of two electrons after undergoing oxidation. Glutamate was converted to (E)-pent-2-enedioic acid, contributing to the increase in current. The sensor was analyzed using an I–V technique and the results evident in Fig. 12. This sensor is chemically inert, non-toxic, safe and stable in air.
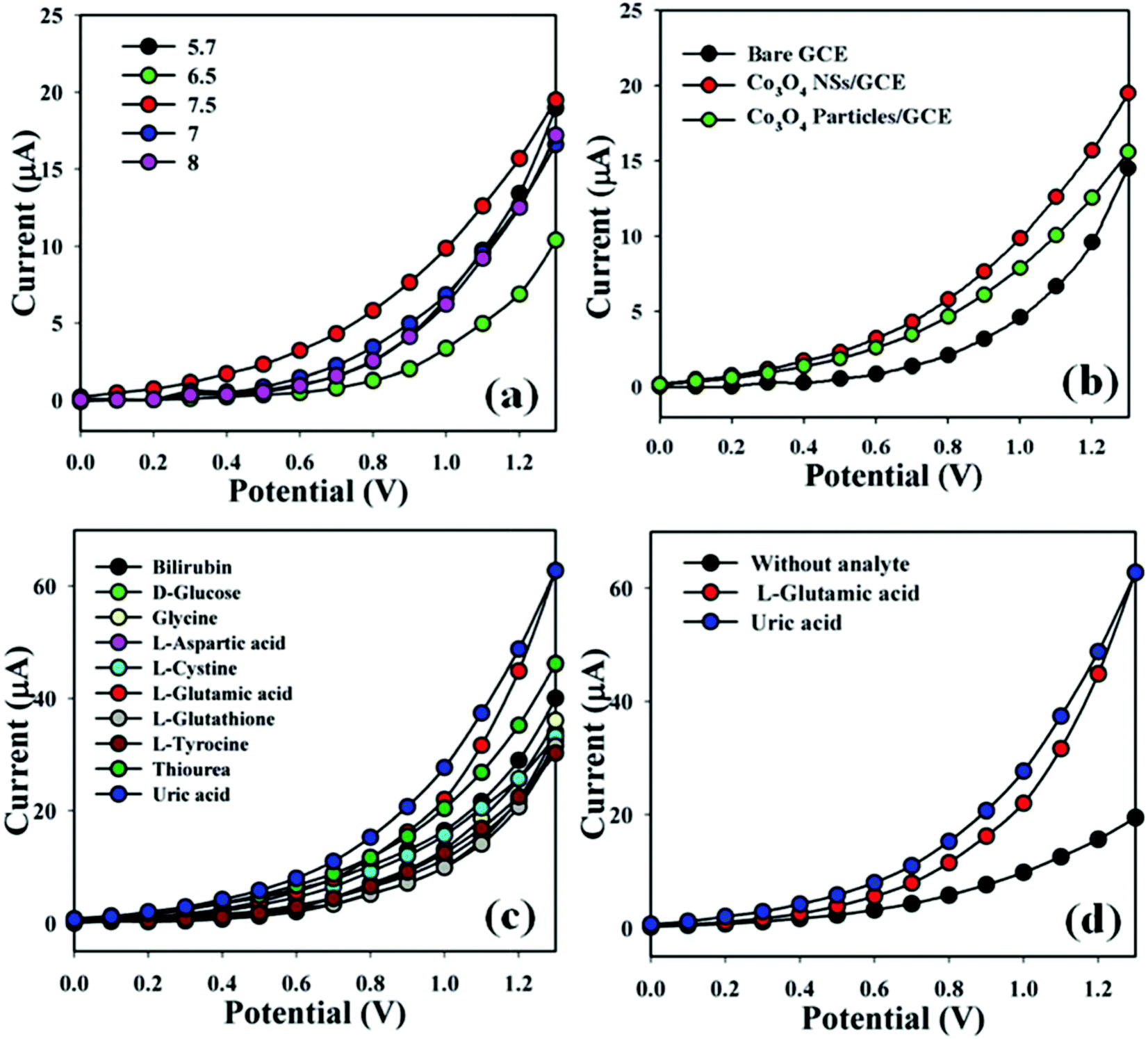 | ||
| Fig. 12 Experimental results of Co3O4NS/GCE as an electrochemical sensor to detect glutamate. (a) Current (I)–voltage (V) response on Co3O4NS/GCE in PBS with varying pH. (b) I–V response in PBS with of Co3O4NS/GCE, of Co3O4NP/GCE and bare GCE. (c) I–V response of Co3O4NS/GCE with interfering species. (d) I–V response with Co3O4NS/GCE in PBS with and without glutamate. Reproduced from ref. 17 with permission from The Royal Society of Chemistry, Copyright 2016. | ||
Ahmad et al. utilized templated polymer-based target receptors to detect glutamate.205 The templated polymer was fabricated with a single-chain poly-N-isopropylacrylamide (PNIPAM) as the backbone copolymerized with functional monomers, 4-vinypyridine (4-VP) and methacrylic acid (MAA). The polymers were synthesized with the reversible addition–fragmentation chain transfer, with a methylglutamic acid template. The polymers were bonded to a gold electrode with thiol bonding. The electrochemical system includes a Pt wire counter electrode and Ag/AgCl reference electrode. Cyclic voltammetry revealed the polymer receptor decreased reduction–oxidation peaks in the presence of glutamate. The gold electrode was also measured with and without glutamate present, revealing no change in the response to confirm the gold electrode doesn't interact with glutamate. Electrochemical impedance spectroscopy was also performed and produced a Nyquist plot. The Nyquist plot confirmed surface blockage as it revealed the charge transfer at the electrode is more kinetically controlled in the presence of glutamate. Additionally, the Nyquist plot revealed a decreased diffusion of the electrolyte through the polymer when glutamate binds at the surface, further confirming the conformation change.
Below, Table 6 summarizes the recently developed non-enzymatic sensors, focusing on the on fabrication and cost and to identify the feasibility and flaws of these sensors. Contrastingly to Tables 1 and 5, this is a qualitative analysis of the sensors. For instance, the ability to detect glutamate is determined based on the limit of detection of the working electrode of the non-enzymatic sensor and the concentration of glutamate in the according biofluid. The ability of a sensor to quantify glutamate is determined based on the linear detection range. Cost is also an important factor, as the cost is a main setback with enzymatic sensors. Fabrication is also important in terms of the future feasibility of the sensor.
| Working electrode material | Advantages | Disadvantages | Ref. |
|---|---|---|---|
| Pt platinum, GEPPGBP genetically engineered periplasmic glutamate binding protein, NAE nanowire array electrode, NS nanoparticle sheet, GCE glassy carbon electrode, SPCE screen printed carbon electrode, Chit chitosan, Au gold, PNIPAM poly-N-isopropylacrylamide, 4-VP 4-vinypyridine, MAA methacrylic acid. | |||
| NiNAE | 1. Ni is earth abundant: sustainable and cost effective | 1. Lack of ease of fabrication | 139 |
| 2. Can detect glutamate in serum | 2. Cannot detect glutamate in CSF, urine or saliva | ||
| 3. May detect glutamate in plasma | 3. Cannot quantify glutamate in serum, CSF, plasma, urine or saliva | ||
| Pt/NiNAE | 1. Ni is earth abundant: sustainable and cost effective | 1. Expensive Pt | 139 |
| 2. Can detect glutamate in serum | 2. Lack of ease of fabrication | ||
| 3. May detect glutamate in plasma | 3. Cannot detect glutamate in CSF, urine or saliva | ||
| 4. Cannot quantify glutamate in serum, CSF, plasma, urine or saliva | |||
| Co3O4NS | 1. Easy fabrication | 1. Expensive CO3O4 | 17 |
| 2. Can detect glutamate in whole blood, CSF, plasma, urine or saliva | 2. Lack of ease of fabrication | ||
| 3. Can quantify glutamate in whole blood, CSF, plasma, urine, and saliva | |||
| NiO/GCE | 1. Ni is earth abundant: sustainable and cost effective | 1. Cannot detect glutamate in CSF, serum, plasma, urine or saliva | 16 |
| 2. Cannot quantify glutamate in whole blood, CSF, plasma, urine or saliva | |||
| Au/SPCE/GEPPGBP | 1. May detect glutamate in CSF or saliva | 1. Expensive Au | 204 |
| 2. May quantify glutamate in CSF or saliva | 2. Cannot detect glutamate in plasma, serum, or urine | ||
| 3. Cannot quantify glutamate in plasma, serum, or urine | |||
| PNIPAM/4-VP/MAA | 1. May detect glutamate in CSF, serum, plasma, or urine | 1. Cannot detect glutamate in saliva | 205 |
6. Conclusions and future perspectives
In this review, we have summarized recent advances and research challenges of sensing glutamate in biofluids with particular attention to metabolic routes of glutamate and its electrochemical sensing. We have emphasized on low-cost, sensitive and real-time monitoring of glutamate associated with diseases for early diagnosis for health care. We have observed that non-enzymatic sensors are more cost effective and stable compared to the enzymatic alternative. Through the detection of glutamate in a fourth-generation biosensor, where a reduction–oxidation reaction occurs on a metal or polymer in the presence of the analyte, the concentration of the analyte is measured indirectly. However, the majority of previously developed non-enzymatic sensors have a LoD and/or LoQ too high to detect and/or quantify glutamate in biofluids. A similar issue arose with enzymatic glutamate sensors, while they could detect or quantify glutamate in some biofluids, most sensors did not have a LDR suitable for detecting glutamate in health monitoring. Either the LDR does not encompass the concentration of glutamate in a particular biofluid at all, or it does not include abnormal glutamate concentrations key for healthcare applications. A non-enzymatic sensor fabricated with Co3O4NSs had a LoD and LDR suitable to detect and quantify glutamate in every biofluid. The good limit of detection of the sensor could be attributed to the optimized fabrication process producing the NSs, which has been shown to decrease the LoD.Another common issue among enzymatic sensors was the stability. An implanted MEA probe only retained ∼46% of its initial sensitivity after 9 hours in vivo. This is problematic for real-time healthcare monitoring where sensors could remain on the patient for long durations of time. The stability of the sensor could be improved by eliminating the enzyme and using a non-enzymatic sensor. For instance, a non-enzymatic sensor retained 96% of its initial activity after 4 weeks. The excellent stability could be attributed to the use of nanowires, and loss of Ni revealing more electroactive sites to enhance the stability and electrocatalytic activity.
A non-enzymatic sensor fabricated with Co3O4NS had one of the highest sensitivities among all electrochemical glutamate sensors. This could be due to the significantly high surface area, pore volume and pore diameter of the Co3O4NSs. While this sensor is promising, the sensitivity of non-enzymatic sensors was still outperformed by enzymatic sensors. Non-enzymatic sensors could continue in the direction of developing polymers or biomimetic surfaces, similar to enzymes, to enhance the affinity and binding of glutamate. In particular, the development of electrochemical glutamate sensors could take any of the following directions:
i. Wider linear detection range. To accurately detect and quantify glutamate in various biofluids sensors must have a wide linear detection. The LDR should encompass the smallest concentration of glutamate, which is found in saliva at 0.232 ± 0.177 μM.82 A higher specific surface area with nanostructures could accomplish this, detecting glutamate regardless of the lack of molecules.
ii. Improvement of selectivity. Improving the selectivity of current non-enzymatic sensors is sought for healthcare applications where glutamate is within a biological matrix with interfering molecules. This could be accomplished through coating the working electrode with polymers analogous to the way glutaraldehyde is used in enzymatic sensors. Although, the impact on reproducibility and sensitivity would also need to be considered when this coating is added. Contrastingly, the detection of multiple species may be desired in the case of multi-analyte sensors.
iii. Greater sensitivity. Further functionalization methods could be applied to current non-enzymatic electrochemical sensors for detecting glutamate. This could include MWCNT functionalized with beta-cyclodextrin to increase the effective surface area and electron transfer rate.207 This is important for when the performance matters, for instance in healthcare settings where accuracy is crucial.
iv. Multi-analyte detection. Multi-analyte electrochemical sensors are the future of fast, non-invasive diagnostic healthcare tools. The sensor can be fabricated easily and accurately with inkjet printers, to detect and monitor multiple pathologies simultaneously or a single pathology accurately. For instance, a multi-analyte biosensor for glutamate and glutamine could be used to measure the glutamate/glutamine ratio and detect chronic intestinal pseudo-obstruction. Using biofluids, such as saliva will minimize the invasion of privacy and discomfort of patients, while maximizing the efficiency of hospitals and healthcare settings. Multi-analyte detection is important to detect potential interference molecules such as non-electroactive molecules that could alter the results of measured glutamate levels. Thus, measuring these species in parallel can distinguish the effects of these molecules.
v. Physiological compatibility. A majority of current non-enzymatic sensors use NaOH as an electrolyte, although it is highly basic at a pH of around 13. This is much higher than the pH of biofluids, outlined in Table 1. Similarly, enzymes commonly immobilized may begin to denature at physiological temperatures around 37 °C. By exploring electrode materials that are compatible with physiological conditions, including the pH and temperature, biosensors can be effectively and easily used to detect glutamate in biofluids.
vi. Wearable sensor. Currently, most electrochemical sensors are either implantable or rigid. In order to provide continuous, real-time and fast detection of biomarkers from biofluids in a non-invasive manner, high performance electrochemical sensors should be adapted to become wearable. Wearable sensors could consist of a wristband, tattoo, or band-aid like devices.208 The sensor could also include an antenna to transmit the results to another device, ideal for self-monitoring. This could include altering the fabrication process of current electrochemical glutamate sensors to incorporate flexible substrates such as LCP through roll-to-roll bonding to produce a flexible sensor. Alternatively, rather than metals or metal oxides the sensor could consist of polymers as the sensing material. A recently reported non-enzymatic sensor consisted of PNIPAM/4-Vp/MAA and resulted in a flexible sensor.205 Further functionalization and optimization of this sensor could make it suitable for detecting glutamate in biofluids.
Electrochemical glutamate sensors could have many applications, including a quantitative measure of pain through detection of glutamate in the saliva. Quantifying pain with a non-invasive device can improve healthcare by altering the dosage of pharmaceuticals delivered to patients based on the level of pain measured. A sensor to detect glutamate could also serve as a diagnostic tool detecting neurodegenerative diseases or cancer. An electrochemical sensor able to sense glutamate in non-invasive biofluids, such as saliva, is crucial to offering a simple, discomfort less, low-cost method for early detection. Consequently, an electrochemical glutamate sensor would improve the prognosis and lives of patients.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We greatly appreciate Dr Arif Ul Alam's contribution to the initial revisions of the manuscript. This research is supported by Discovery Grant from the Natural Science and Engineering Research Council of Canada, an infrastructure grant from the Canada Foundation for Innovation, a FedDev of Southern Ontario grant, and McMaster start-up grant.Notes and references
- N. C. Danbolt, Prog. Neurobiol., 2001, 65, 1–105 CrossRef CAS.
- G. Gheni, M. Ogura, M. Iwasaki, N. Yokoi, K. Minami, Y. Nakayama, K. Harada, B. Hastoy, X. Wu, H. Takahashi, K. Kimura, T. Matsubara, R. Hoshikawa, N. Hatano, K. Sugawara, T. Shibasaki, N. Inagaki, T. Bamba, A. Mizoguchi, E. Fukusaki, P. Rorsman and S. Seino, Cell Rep., 2014, 9, 661–673 CrossRef CAS.
- A. S. Herrera, M. Del, C. A. Esparza, G. M. Ashraf, A. A. Zamyatnin and G. Aliev, Cent. Nerv. Syst. Agents Med. Chem., 2015, 15, 32–41 CrossRef CAS.
- D. C. Rojas, J. Neural Transm., 2014, 121, 891–905 CrossRef CAS.
- J. Lewerenz and P. Maher, Front. Neurosci., 2015, 9, 1–20 Search PubMed.
- V. Pereira and C. Goudet, Front. Mol. Neurosci., 2019, 11, 1–23 CrossRef.
- G. D'Andrea, D. Giovanni, F. Granella, M. Cataldini, F. Verdelli and T. Balbi, J. Headache Pain, 2001, 2, S57–S60 CrossRef.
- S. Bouatra, F. Aziat, R. Mandal, A. C. Guo, M. R. Wilson, C. Knox, T. C. Bjorndahl, R. Krishnamurthy, F. Saleem, P. Liu, Z. T. Dame, J. Poelzer, J. Huynh, F. S. Yallou, N. Psychogios, E. Dong, R. Bogumil, C. Roehring and D. S. Wishart, PLoS One, 2013, 8, 1–28 CrossRef.
- E. R. S. Moraes, A. B. A. Grisolia, K. R. M. Oliveira, D. L. W. Picanço-Diniz, M. E. Crespo-López, C. Maximino, E. J. O. Batista and A. M. Herculano, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2012, 907, 1–6 CrossRef CAS PubMed.
- B. K. Day, F. Pomerleau, J. J. Burmeister, P. Huettl and G. A. Gerhardt, J. Neurochem., 2006, 96, 1626–1635 CrossRef CAS.
- A. Ring, H. Sorg, A. Weltin, D. J. Tilkorn, J. Kieninger, G. Urban and J. Hauser, Biomed. Eng./Biomed. Tech., 2017, 63, 421–426 Search PubMed.
- B. Liang, S. Zhang, Q. Lang, J. Song, L. Han and A. Liu, Anal. Chim. Acta, 2015, 884, 83–89 CrossRef CAS PubMed.
- J. Hu, S. Wisetsuwannaphum and J. S. Foord, Faraday Discuss., 2014, 172, 457–472 RSC.
- G. Rocchitta, A. Spanu, S. Babudieri, G. Latte, G. Madeddu, G. Galleri, S. Nuvoli, P. Bagella, M. I. Demartis, V. Fiore, R. Manetti and P. A. Serra, Sensors, 2016, 16, 780 CrossRef.
- M. W. Shinwari, D. Zhitomirsky, I. A. Deen, P. R. Selvaganapathy, M. J. Deen and D. Landheer, Sensors, 2010, 10, 1679–1715 CrossRef CAS.
- M. Jamal, S. Chakrabarty, H. Shao, D. McNulty, M. A. Yousuf, H. Furukawa, A. Khosla and K. M. Razeeb, Microsyst. Technol., 2018, 24, 4217–4223 CrossRef CAS.
- M. M. Hussain, M. M. Rahman, A. M. Asiri and M. R. Awual, RSC Adv., 2016, 6, 80511–80521 RSC.
- J. A. Harnisch, A. D. Pris and M. D. Porter, J. Am. Chem. Soc., 2001, 123, 5829–5830 CrossRef CAS.
- L. Gonzalez-Macia, A. Morrin, M. R. Smyth and A. J. Killard, Analyst, 2010, 135, 845–867 RSC.
- A. U. Alam, Y. Qin, S. Nambiar, J. T. W. Yeow, M. M. R. Howlader, N. X. Hu and M. J. Deen, Prog. Mater. Sci., 2018, 96, 174–216 CrossRef.
- Y. Huang, X. Dong, Y. Shi, C. M. Li, L. J. Li and P. Chen, Nanoscale, 2010, 2, 1485–1488 RSC.
- G. Schnaberth, H. Schubert and K. Summer, Wien. Klin. Wochenschr., 1975, 87, 294–297 CAS.
- A.-M. Kelly, Emerg. Med. J., 2001, 18, 340–342 CrossRef CAS.
- M. J. Bono and W. C. Reygaert, Urinary Tract Infection, StatPearls Publishing, Treasure Island (FL), 2018 Search PubMed.
- S. Baliga, S. Muglikar and R. Kale, J. Indian Soc. Periodontol., 2013, 17, 461 CrossRef.
- K. Sato, W. Kang and K. Saga, J. Am. Acad. Dermatol., 1989, 20, 537–563 CrossRef CAS.
- S. K. Hamdan and Z. moHd Zain, Malays. J. Med. Sci., 2014, 12–26 Search PubMed , Special Is.
- A. Shadlaghani, M. Farzaneh, D. Kinser and R. C. Reid, Sensors, 2019, 19, 447 CrossRef.
- K. A. Rahn, B. S. Slusher and A. I. Kaplin, Curr. Med. Chem., 2012, 19, 1335–1345 CrossRef CAS.
- E. D. Purves, G. J. Augustine and D. Fitzpatrick, et al., Excitatory and Inhibitory Postsynaptic Potentials, Sinauer Associates, Sunderland (MA), 2nd edn, 2001 Search PubMed.
- J. Du, X. H. Li and Y. J. Li, Eur. J. Pharmacol., 2016, 784, 42–48 CrossRef CAS.
- B. S. Meldrum, J. Nutr., 2000, 130, 1007S–1015S CrossRef CAS.
- K. Moussawi, A. Riegel, S. Nair and P. W. Kalivas, Front. Syst. Neurosci., 2011, 5, 1–9 Search PubMed.
- N. C. Danbolt, D. N. Furness and Y. Zhou, Neurochem. Int., 2016, 98, 29–45 CrossRef CAS.
- D. E. Featherstone, ACS Chem. Neurosci., 2010, 1, 4–12 CrossRef CAS.
- C. B. Divito and S. M. Underhill, Neurochem. Int., 2014, 73, 172–180 CrossRef CAS.
- J. Shen, Front. Neuroenerg., 2013, 5, 1–13 CAS.
- O. A. C. Petroff, Neuroscientist, 2002, 8, 562–573 CrossRef CAS PubMed.
- D. E. Pankevich, M. Davis and B. M. Altevogt, Glutamate-Related Biomarkers in Drug Development for Disorders of the Nervous System Workshop Summary, 2011 Search PubMed.
- E. Karakas, M. C. Regan and H. Furukawa, Trends Biochem. Sci., 2015, 40, 328–337 CrossRef CAS.
- R. Crupi, D. Impellizzeri and S. Cuzzocrea, Front. Mol. Neurosci., 2019, 12, 1–11 CrossRef.
- T. W. Lai, S. Zhang and Y. T. Wang, Prog. Neurobiol., 2014, 115, 157–188 CrossRef CAS.
- N. Riaz, S. L. Wolden, D. Y. Gelblum and J. Eric, J. Alzheimer's Dis., 2016, 118, 6072–6078 Search PubMed.
- E. Foran and D. Trotti, Antioxid. Redox Signal., 2009, 11, 1587–1602 CrossRef CAS.
- S. S. Gill and O. M. Pulido, Toxicol. Pathol., 2001, 29, 208–223 CrossRef CAS.
- S. E. Dryer, Nephrol., Dial., Transplant., 2015, 30, 1630–1638 CrossRef CAS.
- M. Berenbom and J. White, J. Biol. Chem., 1950, 5–10 CAS.
- S. D. Yelamanchi, S. Jayaram, J. K. Thomas, S. Gundimeda, A. A. Khan, A. Singhal, T. S. K. Prasad, A. Pandey, B. L. Somani and H. Gowda, J. Cell Commun. Signaling, 2016, 10, 69–75 CrossRef.
- M. E. Brosnan and J. T. Brosnan, Am. J. Clin. Nutr., 2009, 90, 857S–861S CrossRef CAS.
- S. Otter and E. Lammert, Trends Endocrinol. Metab., 2016, 27, 177–188 CrossRef CAS.
- K. G. Dickman, J. G. Youssef, S. M. Mathew and S. I. Said, Am. J. Respir. Cell Mol. Biol., 2004, 30, 139–144 CrossRef CAS.
- E. Hinoi, T. Takarada and Y. Yoneda, J. Pharmacol. Sci., 2004, 94, 215–220 CrossRef CAS.
- C. Chenu, C. M. Serre, C. Raynal, B. Burt-Pichat and P. D. Delmas, Bone, 1998, 22, 295–299 CrossRef CAS.
- Y.-H. Lee and D. T. Wong, Am. J. Dent., 2009, 22, 241–248 Search PubMed.
- X. Xu and T. D. Veenstra, Proteomics – Clin. Appl., 2008, 2, 1403–1412 CrossRef CAS PubMed.
- M. H. Maurer, Mass Spectrom. Rev., 2008, 29, 17–28 Search PubMed.
- R. A. Hawkins, Am. J. Clin. Nutr., 2009, 90, 867S–874S CrossRef CAS PubMed.
- G. Dimogerontas, A. Polissidis, P. Karkalousos, E. Konstan-tinidis, Z. Papadopoulou-Daifoti and C. Liapi, Int. J. Pathol. Clin. Res., 2016, 2, 1–9 CrossRef.
- R. W. Evans, Neurol. Clin., 1998, 16, 83–105 CrossRef CAS.
- A. Morabia and F. F. Zhang, Postgrad. Med. J., 2004, 80, 463–469 CrossRef CAS.
- C. L. Conley and R. S. Schwartz, Encycl. Br, 2019 Search PubMed.
- M. Gottlieb, Y. Wang and V. I. Teichberg, J. Neurochem., 2003, 87, 119–126 CrossRef CAS PubMed.
- A. Leibowitz, M. Boyko, Y. Shapira and A. Zlotnik, Int. J. Mol. Sci., 2012, 13, 10041–10066 CrossRef CAS PubMed.
- F. Campos, T. Sobrino, P. Ramos-Cabrer, B. Argibay, J. Agulla, M. Pérez-Mato, R. Rodríguez-González, D. Brea and J. Castillo, J. Cereb. Blood Flow Metab., 2011, 31, 1378–1386 CrossRef CAS PubMed.
- Y. Shulman, S. Grant, P. Seres, C. Hanstock, G. Baker and P. Tibbo, J. Psychiatry Neurosci., 2006, 31, 406–410 Search PubMed.
- E. R. Johnson and M. A. Matthay, J. Aerosol Med. Pulm. Drug Delivery, 2010, 23, 243–252 CrossRef PubMed.
- W. Bai, W. L. Zhu, Y. L. Ning, P. Li, Y. Zhao, N. Yang, X. Chen, Y. L. Jiang, W. Q. Yang, D. P. Jiang, L. Y. Chen and Y. G. Zhou, Sci. Rep., 2017, 7, 1–9 CrossRef CAS PubMed.
- C. Shimmura, S. Suda, K. J. Tsuchiya, K. Hashimoto, K. Ohno, H. Matsuzaki, K. Iwata, K. Matsumoto, T. Wakuda, Y. Kameno, K. Suzuki, M. Tsujii, K. Nakamura, N. Takei and N. Mori, PLoS One, 2011, 6, 2–7 CrossRef PubMed.
- C. Madeira, F. V. Alheira, M. A. Calcia, T. C. S. Silva, F. M. Tannos, C. Vargas-lopes, M. Fisher, N. Goldenstein, M. A. Brasil, S. Vinogradov, S. T. Ferreira and R. Panizzutti, Front. Psychiatry, 2018, 9, 1–8 Search PubMed.
- H. Ra, R. A. Hawkins, J. R. Viña, A. Mokashi, D. R. Peterson, R. O. Kane, A. Ian, M. R. Dejoseph and H. Rasgado-flores, Am. J. Neurosci. Res., 2013, 1, 1–25 Search PubMed.
- S. Ogawa, J. Takiguchi, M. Shimizu, K. Nako, M. Okamura, Y. Kinouchi and S. Ito, Tohoku J. Exp. Med., 2016, 239, 103–110 CrossRef CAS PubMed.
- M. Yudkoff, Neurochem. Res., 2017, 42, 10–18 CrossRef CAS PubMed.
- P. R. Blackburn, J. M. Gass, F. P. E. Vairo, K. M. Farnham, H. K. Atwal, S. Macklin, E. W. Klee and P. S. Atwal, Appl. Clin. Genet., 2017, 10, 57–66 CrossRef CAS PubMed.
- R. G. Tavares, C. E. Santos, C. I. Tasca, M. Wajner, D. O. Souza and C. S. Dutra-Filho, J. Neurol. Sci., 2000, 181, 44–49 CrossRef CAS PubMed.
- L. Taylor and N. P. Curthoys, Biochem. Mol. Biol. Educ., 2004, 32, 291–304 CrossRef CAS PubMed.
- M. Stumvoll, G. Perriello, C. Meyer and J. Gerich, Kidney Int., 1999, 55, 778–792 CrossRef CAS PubMed.
- J.-K. Yan, K.-J. Zhou, J.-H. Huang, Q.-Q. Wu, T. Zhang, C.-C. Wang and W. Cai, Orphanet J. Rare Dis., 2017, 12, 62 CrossRef PubMed.
- W. Ren, Y. Xia, S. Chen, G. Wu, F. W. Bazer, B. Zhou, B. Tan, G. Zhu, J. Deng and Y. Yin, Adv. Nutr., 2019, 10, 321–330 CrossRef PubMed.
- R. Bélanger, N. Chandramohan, R. Misbin and R. S. Rivlin, Metabolism, 1972, 21, 855–865 CrossRef.
- M. Pietzner, T. Kacprowski and N. Friedrich, J. Endocrinol., 2018, 238, R13–R29 CAS.
- J. Feher, in Quantitative Human Physiology, 2nd edn, 2017, pp. 771–784 Search PubMed.
- H. Jasim, A. Carlsson, B. Hedenberg-Magnusson, B. Ghafouri and M. Ernberg, Sci. Rep., 2018, 8, 3220 CrossRef PubMed.
- J. L. Jensen, A. Karatsaidis and P. Brodin, Eur. J. Oral Sci., 1998, 106, 892–896 CrossRef CAS PubMed.
- A. Wang, C. P. Wang, M. Tu and D. T. W. Wong, Diagnostics, 2016, 6, 45 CrossRef PubMed.
- S. P. Humphrey and R. T. Williamson, J. Prosthet. Dent., 2001, 85, 162–169 CrossRef CAS PubMed.
- B. K. Siesjö, Kidney Int., 1972, 1, 360–374 CrossRef PubMed.
- S. W. Brusilow and E. H. Gordes, Am. J. Physiol., 1968, 214, 513–517 CrossRef CAS PubMed.
- I. Nissim, Am. Physiol. Soc., 1999, 277, F493–F497 CAS.
- J. F. Stover, K. Lowitzsch and O. S. Kempski, Neurosci. Lett., 1997, 238, 25–28 CrossRef CAS PubMed.
- N. Psychogios, D. D. Hau, J. Peng, A. C. Guo, R. Mandal, S. Bouatra, R. Krishnamurthy, R. Eisner, B. Gautam, N. Young, C. Knox, E. Dong, P. Huang, Z. Hollander, T. L. Pedersen, R. Steven, F. Bamforth, R. Greiner, B. Mcmanus, J. W. Newman, T. Goodfriend and D. S. Wishart, PLoS One, 2011, 6, 1–23 CrossRef PubMed.
- K. Strimbu and J. A. Tavel, Curr. Opin. HIV AIDS, 2010, 5, 463–466 CrossRef PubMed.
- R. Hawkins and J. Viña, Biology, 2016, 5, 37 CrossRef PubMed.
- Overview of ELISA, https://www.thermofisher.com/ca/en/home/life-science/protein-biology/protein-biology-learning-center/protein-biology-resource-library/pierce-protein-methods/overview-elisa.html.
- M. J. Nozal, J. L. Bernal, M. L. Toribio, J. C. Diego and A. Ruiz, J. Chromatogr. A, 2004, 1047, 137–146 CrossRef CAS PubMed.
- H. J. Shin, N. H. Park, W. Lee, M. H. Choi, B. C. Chung and J. Hong, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2017, 1051, 97–107 CrossRef CAS PubMed.
- PicoProbe™ glutamate assay kit (fluorometric).
- Neurophysiological Products for Preclinical In Vivo Research, Pinnacle Technology Inc., Lawrence, Kansas, 2019, p. 1 Search PubMed.
- A. Ghorai, J. Mondal, R. Chandra and G. K. Patra, Anal. Methods, 2015, 7, 8146–8151 RSC.
- L. Xin, L. Jie, C. W. Liu and S. L. Zhao, Chin. J. Anal. Chem., 2007, 35, 1151–1154 Search PubMed.
- S. Zhang, Y. Takeda, S. Hagioka, K. Takata, H. Aoe, H. Nakatsuka, M. Yokoyama and K. Morita, Brain Res. Protoc., 2005, 14, 61–66 CrossRef CAS PubMed.
- A. Lau and M. Tymianski, Pflugers Arch. Eur. J. Physiol., 2010, 460, 525–542 CrossRef CAS PubMed.
- M. R. Hynd, H. L. Scott and P. R. Dodd, Neurochem. Int., 2004, 45, 583–595 CrossRef CAS PubMed.
- C. Madeira, C. Vargas-lopes, C. O. Brandão, T. Reis, J. Laks, R. Panizzutti and S. T. Ferreira, Front. Psychiatry, 2018, 9, 1–8 Search PubMed.
- C. Agius, S. von Tucher, B. Poppenberger and W. Rozhon, Molecules, 2018, 23, 1389 CrossRef PubMed.
- T. M. Annesley, Clin. Chem., 2003, 49, 1041–1044 CrossRef CAS PubMed.
- V. M. André, C. Cepeda and M. S. Levine, CNS Neurosci. Ther., 2010, 16, 163–178 CrossRef PubMed.
- Y. J. Cha, E. S. Kim and J. S. Koo, Int. J. Mol. Sci., 2018, 19, 907 CrossRef PubMed.
- N. M. Zacharias, C. McCullough, S. Shanmugavelandy, J. Lee, Y. Lee, P. Dutta, J. McHenry, L. Nguyen, W. Norton, L. W. Jones and P. K. Bhattacharya, Sci. Rep., 2017, 7, 1–11 CrossRef CAS PubMed.
- T. Gelb, S. Pshenichkin, O. C. Rodriguez, H. A. Hathaway, E. Grajkowska, J. O. DiRaddo, B. Wroblewska, R. Yasuda, C. Albanese, B. B. Wolfe and J. T. Wroblewski, Oncogene, 2015, 34, 2711–2720 CrossRef CAS PubMed.
- T. Takano, J. H. Lin, G. Arcuino, Q. Gao, J. Yang and M. Nedergaard, Nat. Med., 2001, 7, 1010–1015 CrossRef CAS PubMed.
- C. L. Speyer, J. S. Smith, M. Banda, J. A. DeVries, T. Mekani and D. H. Gorski, Breast Cancer Res. Treat., 2012, 132, 565–573 CrossRef CAS PubMed.
- P. Kamarajan, T. M. Rajendiran, J. Kinchen, M. Bermúdez, T. Danciu and Y. L. Kapila, J. Proteome Res., 2017, 16, 1315–1326 CrossRef CAS PubMed.
- K. M. Wozniak, Y. W. C. Rojas and B. S. Slusher, Curr. Med. Chem., 2012, 12, 1323–1334 CrossRef PubMed.
- J. Sandkühler, Physiol. Rev., 2009, 89, 707–758 CrossRef PubMed.
- C. J. Woolf, Pain, 2011, 152, S2–15 CrossRef PubMed.
- M. Zunhammer, U. Bingel, L. M. Schweizer, V. Witte, T. Schmidt-Wilcke and R. E. Harris, Pain, 2016, 157, 2248–2256 CrossRef CAS PubMed.
- L. Rosendal, B. Larsson, J. Kristiansen, M. Peolsson, K. Søgaard, M. Kjær, J. Sørensen and B. Gerdle, Pain, 2004, 112, 324–334 CrossRef CAS PubMed.
- A. Shimada, E. E. Castrillon, L. Baad-Hansen, B. Ghafouri, B. Gerdle, K. Wåhlén, M. Ernberg, B. E. Cairns and P. Svensson, Eur. J. Pain, 2016, 20, 1502–1512 CrossRef CAS PubMed.
- E. E. Castrillon, M. Ernberg, B. E. Cairns, K. Wang, B. J. Sessle, L. Arendt-Nielsen and P. Svensson, J. Orofac. Pain, 2010, 24, 350–360 Search PubMed.
- J. H. Nam, H. S. Lee, J. Kim, J. Kim and M. K. Chu, Cephalalgia, 2018, 38, 1485–1492 CrossRef PubMed.
- Glutamate ELISA kit, http://www.abnova.com/products/products_detail.asp?catalog_id=KA1909.
- T. L. Pyke, P. G. Osmotherly and S. Baines, Clin. J. Pain, 2017, 33, 944–954 CrossRef PubMed.
- B. Gerdle, B. Larsson, F. Forsberg, N. Ghafouri, L. Karlsson, N. Stensson and B. Ghafouri, Clin. J. Pain, 2014, 30, 409–420 Search PubMed.
- J. Fazzari, K. Linher-Melville and G. Singh, Curr. Neuropharmacol., 2016, 15, 620–636 CrossRef PubMed.
- A. N. Kozitsina, T. S. Svalova, N. N. Malysheva, A. V. Okhokhonin, M. B. Vidrevich and K. Z. Brainina, Biosensors, 2018, 8, 1–34 CrossRef PubMed.
- C. Karunakaran, R. Rajkumar and K. Bhargava, Biosens. Bioelectron., 2015, 1–68 Search PubMed.
- D. Braeken, D. R. Rand, A. Andrei, R. Huys, M. E. Spira, S. Yitzchaik, J. Shappir, G. Borghs, G. Callewaert and C. Bartic, Biosens. Bioelectron., 2009, 24, 2384–2389 CrossRef CAS PubMed.
- N. Bakeman, I. M. Isa, N. M. Ali, M. Ahmad and S. A. Ghani, Int. J. Electrochem. Sci., 2012, 7, 4574–4584 CAS.
- N. Elgrishi, K. J. Rountree, B. D. McCarthy, E. S. Rountree, T. T. Eisenhart and J. L. Dempsey, J. Chem. Educ., 2018, 95, 197–206 CrossRef CAS.
- Y. Qin, H. J. Kwon, M. M. R. Howlader and M. J. Deen, RSC Adv., 2015, 5, 69086–69109 RSC.
- C. Zhu, G. Yang, H. Li, D. Du and Y. Lin, Anal. Chem., 2015, 87, 230–249 CrossRef CAS PubMed.
- J. Ustarroz, M. Kang, E. Bullions and P. R. Unwin, Chem. Sci., 2017, 8, 1841–1853 RSC.
- X. Cheng, Nanolithography, 2014, 348–375 Search PubMed.
- L. C. Clark, Biosens. Bioelectron., 2006, 21, 1403–1404 CrossRef.
- K. E. Toghill and R. G. Compton, Int. J. Electrochem. Sci., 2010, 5, 1246–1301 CAS.
- T. T. C. Tseng, C. F. Chang and W. C. Chan, Molecules, 2014, 19, 7341–7355 CrossRef PubMed.
- S. Qin, M. van der Zeyden, W. H. Oldenziel, T. I. F. H. Cremers and B. H. C. Westerink, Sensors, 2008, 8, 6860–6884 CrossRef CAS PubMed.
- S. A. Shabbir, S. Tariq, M. G. B. Ashiq and W. A. Khan, Biosci. Rep., 2019, 1–13 Search PubMed.
- M. Jamal, M. Hasan, A. Mathewson and K. M. Razeeb, Biosens. Bioelectron., 2013, 40, 213–218 CrossRef CAS PubMed.
- S. Guo, D. Wen, Y. Zhai, S. Dong and E. Wang, ACS Nano, 2010, 4, 3959–3968 CrossRef CAS PubMed.
- X. Luo, A. Morrin, A. J. Killard and M. R. Smyth, Electroanalysis, 2006, 18, 319–326 CrossRef CAS.
- M. Holzinger, A. Le Goff and S. Cosnier, Front. Chem., 2014, 2, 1–10 CAS.
- J. C. Claussen, M. S. Artiles, E. S. McLamore, S. Mohanty, J. Shi, J. L. Rickus, T. S. Fisher and D. M. Porterfield, J. Mater. Chem., 2011, 21, 11224–11231 RSC.
- C. P. Mcmahon and R. D. O'Neill, Anal. Chem., 2005, 77, 1196–1199 CrossRef CAS PubMed.
- P. Bollella, L. Gorton, R. Ludwig and R. Antiochia, Sensors, 2017, 17, 1912 CrossRef PubMed.
- A. D. McNaught and A. Wilkinson, Compendium of Chemical Terminology, Blackwell Scientific Publications, Oxford, 2nd edn, 1997 Search PubMed.
- X. Zhang, H. Ju and J. Wang, Electrochemical Sensors, Biosensors and Their Biomedical Applications, Elsevier, Amsterdam, 2007, ch. 1, pp. 11–12 Search PubMed.
- A. Shrivastava and V. Gupta, Chron. Young Sci., 2011, 2, 21 CrossRef.
- D. K. Maurya, A. Sardarinejad and K. Alameh, Coatings, 2014, 756–771 CrossRef.
- M. Jamal, S. Chakrabarty, M. A. Yousuf, A. Khosla and K. M. Razeeb, Microsyst. Technol., 2018, 24, 4193–4206 CrossRef CAS.
- Y. Qin, A. U. Alam, M. M. R. Howlader, N.-X. Hu and M. J. Deen, Adv. Funct. Mater., 2016, 26, 4923–4933 CrossRef CAS.
- Y. Qin, A. U. Alam, S. Pan, M. M. R. Howlader, R. Ghosh, N. X. Hu, H. Jin, S. Dong, C. H. Chen and M. J. Deen, Sens. Actuators, B, 2018, 255, 781–790 CrossRef CAS.
- A. U. Alam, Y. Qin, M. M. R. Howlader, N. X. Hu and M. J. Deen, Sens. Actuators, B, 2018, 254, 896–909 CrossRef CAS.
- M. M. Alam, A. U. Alam and M. M. R. Howlader, 2019 6th Int. Work. Low Temp. Bond. 3D Integr., 2019, 76–76.
- M. M. R. Howlader, P. R. Selvaganapathy, M. J. Deen and T. Suga, IEEE J. Sel. Top. Quantum Electron., 2011, 17, 689–703 CAS.
- G. Cummins and M. P. Y. Desmulliez, Circuit World, 2012, 38, 193–213 CrossRef CAS.
- L. Cao, X. Bai, Z. Lin, P. Zhang, S. Deng, X. Du and W. Li, Materials, 2017, 10, 1004 CrossRef PubMed.
- R. P. Tortorich, H. Shamkhalichenar and J.-W. Choi, Appl. Sci., 2018, 8, 288 CrossRef.
- E. Bihar, S. Wustoni, A. M. Pappa, K. N. Salama, D. Baran and S. Inal, npj Flexible Electron., 2018, 2, 30 CrossRef.
- J. Wang, B. Yang, F. Gao, P. Song, L. Li, Y. Zhang, C. Lu, M. C. Goh and Y. Du, Nanoscale Res. Lett., 2019, 14, 11 CrossRef PubMed.
- S. J. Lee, Y. H. Kim, J. K. Kim, H. Baik, J. H. Park, J. Lee, J. Nam, J. H. Park, T. W. Lee, G. R. Yi and J. H. Cho, Nanoscale, 2014, 6, 11828–11834 RSC.
- M. Bariya, Z. Shahpar, H. Park, J. Sun, Y. Jung, W. Gao, H. Y. Y. Nyein, T. S. Liaw, L.-C. Tai, Q. P. Ngo, M. Chao, Y. Zhao, M. Hettick, G. Cho and A. Javey, ACS Nano, 2018, 12, 6978–6987 CrossRef CAS.
- T. Z. Redhwan, A. U. Alam, Y. M. Haddara and M. M. R. Howlader, Jpn. J. Appl. Phys., 2018, 57, 02BB03 CrossRef.
- T. Z. Redhwan, A. U. Alam, Y. Qin, M. M. R. Howlader and Y. M. Haddara, Sens. Actuators, B. Search PubMed , submitted.
- R. E. Özel, C. Ispas, M. Ganesana, J. C. Leiter and S. Andreescu, Biosens. Bioelectron., 2014, 52, 397–402 CrossRef PubMed.
- A. Cowley and B. Woodward, Platinum Met. Rev., 2011, 55, 98–107 CrossRef.
- T. Geninatti, G. Bruno, B. Barile, R. L. Hood, M. Farina, J. Schmulen, G. Canavese and A. Grattoni, Biomed. Microdevices, 2015, 17, 1–11 CrossRef CAS PubMed.
- G. Hughes, R. M. Pemberton, P. R. Fielden and J. P. Hart, Sens. Actuators, B, 2015, 216, 614–621 CrossRef CAS.
- Y. Deng, W. Wang, L. Zhang, Z. Lu, S. Li and L. Xu, J. Biomed. Nanotechnol., 2013, 9, 318–321 CrossRef CAS PubMed.
- B. Batra, M. Yadav and C. S. Pundir, Biochem. Eng. J., 2016, 105, 428–436 CrossRef CAS.
- S. Sharma and M. Madou, Bioinspired, Biomimetic Nanobiomater., 2012, 1, 252–265 CrossRef CAS.
- S. Cinti, F. Arduini, M. Carbone, L. Sansone, I. Cacciotti, D. Moscone and G. Palleschi, Electroanalysis, 2015, 27, 2230–2238 CrossRef CAS.
- R. Wahab, S. G. Ansari, Y. S. Kim, M. Song and H. S. Shin, Appl. Surf. Sci., 2009, 255, 4891–4896 CrossRef CAS.
- Tana, M. Zhang, J. Li, H. Li, Y. Li and W. Shen, Catal. Today, 2010, 148, 179–183 CrossRef.
- A. H. C. Neto, F. Guinea, N. M. R. Peres, K. S. Novoselov and A. K. Geim, Rev. Mod. Phys., 2009, 81, 109–162 CrossRef.
- M. Pumera, A. Ambrosi, A. Bonanni, E. L. K. Chng and H. L. Poh, TrAC, Trends Anal. Chem., 2010, 29, 954–965 CrossRef CAS.
- J. J. Burmeister, V. A. Davis, J. E. Quintero, F. Pomerleau, P. Huettl and G. A. Gerhardt, ACS Chem. Neurosci., 2013, 4, 721–728 CrossRef CAS.
- M. Rinaudo, Prog. Polym. Sci., 2006, 31, 603–632 CrossRef CAS.
- M. Ammam and J. Fransaer, Biosens. Bioelectron., 2010, 25, 1597–1602 CrossRef CAS.
- S. J. Killoran and R. D. O'Neill, Electrochim. Acta, 2008, 53, 7303–7312 CrossRef CAS.
- S. Samanta, P. Roy and P. Kar, Macromol. Res., 2016, 24, 342–349 CrossRef CAS.
- H. M. Badawi, W. Förner and S. A. Ali, Spectrochim. Acta, Part A, 2013, 112, 388–396 CrossRef CAS PubMed.
- V. G. H. Eijsink, S. GÅseidnes, T. V. Borchert and B. Van Den Burg, Biomol. Eng., 2005, 22, 21–30 CrossRef CAS PubMed.
- S. M. Z. Hossain, H. Shinohara, F. Wang and H. Kitano, Anal. Bioanal. Chem., 2007, 389, 1961–1966 CrossRef CAS PubMed.
- A. K. Basu, P. Chattopadhyay, U. Roychudhuri and R. Chakraborty, Biosens. Bioelectron., 2006, 21, 1968–1972 CrossRef CAS.
- P. H. Dakappa and C. Mahabala, Crit. Rev. Biomed. Eng., 2015, 43, 385–399 CrossRef.
- S. Shah, A. Sharma and M. N. Gupta, Anal. Biochem., 2006, 351, 207–213 CrossRef CAS PubMed.
- S. Park, H. Boo and T. D. Chung, Anal. Chim. Acta, 2006, 556, 46–57 CrossRef CAS.
- D. Botman, W. Tigchelaar and C. J. F. Van Noorden, J. Histochem. Cytochem., 2014, 62, 802–812 CrossRef.
- N. Isoaho, E. Peltola, S. Sainio, J. Koskinen and T. Laurila, RSC Adv., 2018, 8, 35802–35812 RSC.
- M. Bäcker, D. Rakowski, A. Poghossian, M. Biselli, P. Wagner and M. J. Schöning, J. Biotechnol., 2013, 163, 371–376 CrossRef PubMed.
- S. Chakraborty and C. R. Raj, Electrochem. Commun., 2007, 9, 1323–1330 CrossRef CAS.
- B. Batra, S. Kumari and C. S. Pundir, Enzyme Microb. Technol., 2014, 57, 69–77 CrossRef CAS PubMed.
- J. L. Scoggin, C. Tan, N. H. Nguyen, U. Kansakar, M. Madadi, S. Siddiqui, P. U. Arumugam, M. A. DeCoster and T. A. Murray, Biosens. Bioelectron., 2019, 126, 751–757 CrossRef CAS PubMed.
- O. V. Soldatkina, O. O. Soldatkin, B. O. Kasap, D. Y. Kucherenko, I. S. Kucherenko, B. A. Kurc and S. V. Dzyadevych, Nanoscale Res. Lett., 2017, 12, 260 CrossRef CAS PubMed.
- W. Wei, Y. Song, L. Wang, S. Zhang, J. Luo, S. Xu and X. Cai, Microsyst. Nanoeng., 2015, 1, 1–6 Search PubMed.
- J. R. Windmiller, G. Valdés-Ramírez, N. Zhou, M. Zhou, P. R. Miller, C. Jin, S. M. Brozik, R. Polsky, E. Katz, R. Narayan and J. Wang, Electroanalysis, 2011, 23, 2302–2309 CrossRef CAS.
- M. Ganesana, E. Trikantzopoulos, Y. Maniar, S. T. Lee and B. J. Venton, Biosens. Bioelectron., 2019, 130, 103–109 CrossRef CAS PubMed.
- A. Weltin, J. Kieninger and G. A. Urban, Proceedings, 2017, 1, 521 CrossRef.
- P. Salazar, M. Martín, R. D. O'Neill and J. L. González-Mora, Sens. Actuators, B, 2016, 235, 117–125 CrossRef CAS.
- A. Weltin, J. Kieninger, B. Enderle, A. K. Gellner, B. Fritsch and G. A. Urban, Biosens. Bioelectron., 2014, 61, 192–199 CrossRef CAS PubMed.
- M. M. Rahman, T. Yamazaki, T. Ikeda, M. Ishida and K. Sawada, TRANSDUCERS 2009 - 15th Int. Conf. Solid-State Sensors, Actuators Microsystems, 2009, 88–91.
- F. Tian, A. V. Gourine, R. T. R. Huckstepp and N. Dale, Anal. Chim. Acta, 2009, 645, 86–91 CrossRef CAS PubMed.
- Y.-P. Yang, A. Manfredi and S. Daunert, ECS Meet. Abstr., 2015, 42, 2195 Search PubMed.
- H. M. N. Ahmad, B. Si, G. Dutta, J. R. Csoros, W. R. Seitz and E. Song, TRANSDUCERS 2019 20th International Conference on Solid-State Sensors, Actuators and Microsystems & Eurosensors XXXIII (TRANSDUCERS & EUROSENSORS XXXIII), IEEE, Berlin, 2019 Search PubMed.
- T. Gao, F. Liu, D. Yang, Y. Yu, Z. Wang and G. Li, Anal. Chem., 2015, 87, 5683–5689 CrossRef CAS PubMed.
- M. Motooka and S. Uno, Sensors, 2018, 18, 440 CrossRef PubMed.
- J. Zhao, H. Guo, J. Li, A. J. Bandodkar and J. A. Rogers, Trends Chem., 2019, 1, 559–571 CrossRef.
| This journal is © The Royal Society of Chemistry 2020 |