
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of 1,2,4-azadiphosphole derivatives based on vanadium-catalyzed [2+2+1] cycloaddition reactions of azobenzenes with phosphaalkynes†
Wenbin Lianga,
Kazunari Nakajima *b and
Yoshiaki Nishibayashi*a
*b and
Yoshiaki Nishibayashi*a
aDepartment of Systems Innovation, School of Engineering, The University of Tokyo, Hongo, Bunkyo-ku, Tokyo 113-8656, Japan. E-mail: ynishiba@sys.t.u-tokyo.ac.jp
bFrontier Research Center for Energy and Resources, School of Engineering, The University of Tokyo, Hongo, Bunkyo-ku, Tokyo 113-8656, Japan. E-mail: nakajima@sys.t.u-tokyo.ac.jp
First published on 30th March 2020
Abstract
A new synthetic method is described to construct 1,2,4-azadiphosphole derivatives based on vanadium-catalyzed [2+2+1] cycloaddition reactions. Reactions of azobenzenes as nitrogen sources with phosphaalkynes as phosphorous counterparts in the presence of VCl2(thf)2 as a catalyst afford the corresponding 1,2,4-azadiphospholes.
Phosphorus heterocycles have provided important structural motifs to explore materials science and coordination chemistry.1 To access diverse skeletons, development of new and efficient synthetic methods is quite important. In addition to the conventional synthetic methods typically forming carbon–phosphorous single bonds, the use of phosphaalkynes as substrates is beneficial to construct phosphorous-containing π-systems.2 Stoichiometric reactivity of phosphaalkynes with transition metal complexes has been studied extensively,3 but catalytic reactions of phosphaalkynes under transition metal catalysis have been limited to several sporadic examples.4,5 Recently, our group has reported catalytic [2+2+2] cycloaddition reactions to produce phosphabenzenes and [3+2] cycloaddition reactions to produce 1,3-azaphospholes based on the use of phosphaalkynes as substrates.5 To investigate further utility of phosphaalkynes to prepare aromatic compounds containing phosphorus atoms, we have focused on the synthesis of 1,2,4-azadiphospholes as next targets.
Synthetic examples of 1,2,4-azadiphospholes have been limited to only a few reports.6,7 To the best of our knowledge, the first synthesis of the 1,2,4-azadiphosphole skeleton was reported in 1991, where a 1,2,4-azadiphosphole derivative was prepared based on a thermal dimerization of an amino-substituted phosphaalkyne.6a Later, other groups reported stoichiometric reactions of titanium- and vanadium-imide complexes as nitrogen sources with phosphaalkynes.6b–d Although these stoichiometric reactions provided effective synthetic methods of 1,2,4-azadiphospholes, transition metal-catalyzed synthesis of 1,2,4-azadiphospholes has never been achieved until now.
Recently, titanium-catalyzed [2+2+1] cycloaddition reactions have been reported by Tonks and co-workers to prepare pyrroles from reactions of azobenzenes with alkynes (Scheme 1a).8 In this reaction system, titanium-imide species generated from azobenzenes worked as a key intermediate. More recently, a similar pyrrole synthesis using vanadium-catalyzed reaction system has been reported by Tonks, Mashima, Tsurugi and co-workers.9
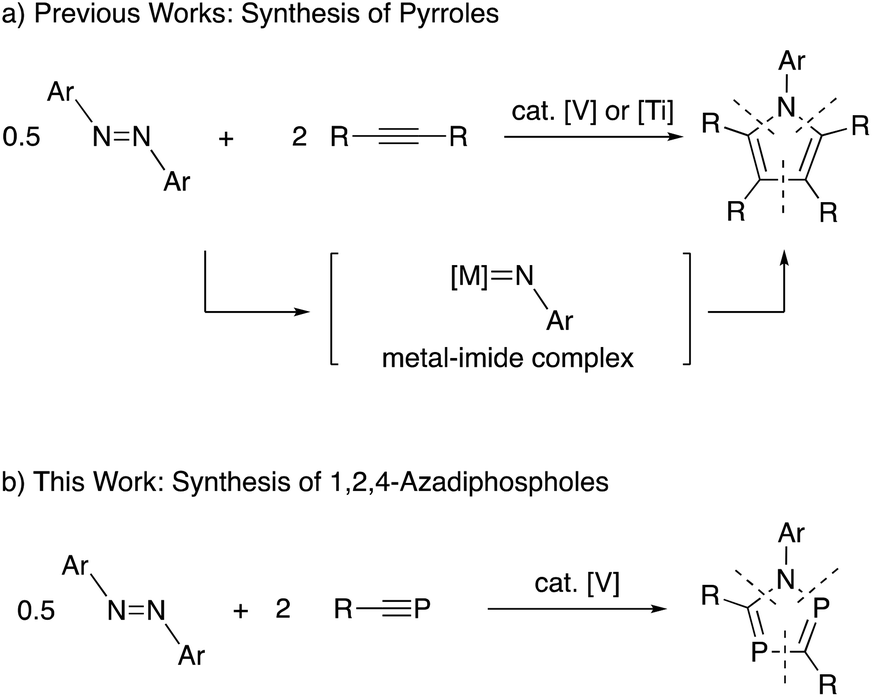 | ||
| Scheme 1 Catalytic [2+2+1] cycloaddition reactions with azobenzenes as nitrogen sources in heterocycle synthesis. | ||
Based on the research background, we have envisaged metal-catalyzed [2+2+1] cycloaddition reactions of azobenzenes with phosphaalkynes to produce 1,2,4-azadiphospholes (Scheme 1b). As a result, we have found that some vanadium complexes worked as effective catalysts toward the formation of 1,2,4-azadiphospholes. Herein, we report experimental results in detail.
First, investigation of reaction conditions was carried out with the use of azobenzene (1a) and 1-adamantylphosphaethyne (2a) as typical substrates. When the reaction of 1a (1 equiv.) with 2a (2 equiv.) in the presence of 20 mol% of VCl3(thf)3 was carried out in toluene at 110 °C for 16 hours, the desired 1,2,4-azadiphosphole (3a) was obtained in 48% yield (Table 1, entry 1). Replacing VCl3(thf)3 with VBr3(thf)3 and VCl3(py)3 was not effective in the current reaction system (Table 1, entries 2 and 3). Then, we investigated the use of VCl2(thf)2, VCl2(1,4-dioxane)2 and VI2(thf)4. Among these catalysts, VCl2(thf)2 showed the highest reactivity to afford the desired product 3a in 60% yield (Table 1, entries 4–6). Next, addition of ligands was examined. When bidentate phosphine ligands such as 1,1-bis(diphenylphosphino)methane (dppm) or 1,2-bis(diphenylphosphino)ethane (dppe) was used, only lower yields of the product were obtained (Table 1, entries 7 and 8) while no formation of the desired product was observed when 2,2′-bipyridine was used as a ligand (Table 1, entry 9). Separately, we confirmed no formation of 3a in the absence of vanadium complexes. Based on these results, we have chosen the use of VCl2(thf)2 as the optimal catalyst.
|
|||
|---|---|---|---|
| Entry | [V] catalyst | Ligand | Yield (%) |
| a Reactions of 1a (0.10 mmol) with 2a (0.20 mmol) in the presence of a catalyst (20 mol%) and a ligand (20 mol%) in toluene (2.0 mL) at 110 °C for 16 h. | |||
| 1 | VCl3(thf)3 | — | 48 |
| 2 | VBr3(thf)3 | — | 42 |
| 3 | VCl3(py)3 | — | 0 |
| 4 | VCl2(thf)2 | — | 60 |
| 5 | VCl2(1,4-dioxane)2 | — | 47 |
| 6 | VI2(thf)4 | — | 27 |
| 7 | VCl2(thf)2 | dppm | 27 |
| 8 | VCl2(thf)2 | dppe | 20 |
| 9 | VCl2(thf)2 | 2,2′-Bipyridine | 0 |

With the optimized reaction conditions in hand, the use of other substrates was investigated. The use of various functionalized azobenzene derivatives was examined (Scheme 2). When 3-methyl-, 3-ethyl- and 3-isopropyl-substituted azobenzenes were used, the corresponding products (3b, 3c and 3d) were obtained in 55%, 51% and 46% yield, respectively. Effect of alkyl groups at the 4-position of the benzene rings in azobenzene was also examined. Introduction of methyl, ethyl, isopropyl, tert-butyl, sec-butyl and n-butyl groups afforded the corresponding products (3e–3j) in good yields. The reaction system was tolerant toward oxygen functional groups such as 4-methoxyl and 4-methoxycarbonyl groups to give the desired products (3k and 3l) in 52% and 47% yields, respectively. Unfortunately, an azodicarboxylate ester was not applicable in this reaction system resulting in no formation of the corresponding product (3m). We also examined 2,2′-azobis(isobutyronitrile) as an aliphatic azo compound, but the corresponding product (3n) was not obtained at all.
 | ||
| Scheme 2 Reactions of azobenzene with 1-adamantylphosphaethyne 2a. aReactions of azobenzenes 1 (0.1 mmol) with 2a (0.2 mmol) in the presence of VCl2(thf)2 (20 mol%) in toluene (2.0 mL) at 110 °C for 16 h. b1H NMR yield with C6Me6 as an internal standard. | ||
Next, the use of various phosphaalkynes was investigated (Scheme 3). A phosphaakyne bearing 3,5-dimethyl-1-adamantyl moiety was successfully transformed into the corresponding product (3o) in 50% yield. The use of phosphaalkyne bearing a benzene ring was also successful to afford the corresponding product (3p) in 41% yield. Introduction of various alkoxyphenyl moieties into the phenyl ring was also investigated and the corresponding products were obtained in moderate yields (3q–3t). When the biphenyl substituent was introduced in phosphaalkyne, the corresponding product (3u) was obtained in 41% yield.
 | ||
| Scheme 3 Reactions of 1,2-di(4-tert-butyl)diazene 1h with phosphaalkynes 2. aReactions of 1h (0.1 mmol) with phosphaalkynes 2 (0.2 mmol) in the presence of VCl2(thf)2 (20 mol%) in toluene (2.0 mL) at 110 °C for 16 h. | ||
As mentioned above, recently vanadium-catalyzed [2+2+1] cycloaddition reactions of azobenzenes with alkynes to afford the pyrroles were reported.9 In this reaction system, bis-(trimethylsilyl)aniline was used as an additive along with VCl3(thf)3 catalyst, and a vanadium-bis(imide) complex was proposed to be a catalytically active species. Bis(trimethylsilyl)aniline supplies an imide moiety on the vanadium center through a redox neutral ligand exchange process and the second imide moiety comes from azobenzene substrates through an oxidative process, where the generated vanadium-bis(imide) complex is supposed to undergo further reactions to give the pyrroles.
According to the experimental result, we investigated the effect of bis(trimethylsilyl)aniline as an additive in the current reaction system (Scheme 4). When bis(trimethylsilyl)aniline (20 mol%) was added along with VCl2(thf)2 catalyst in the reaction of 1a with 2a, the yield of 3a did not improve. This result suggests that the introduction of imide moiety from bis(trimethylsilyl)aniline was not effective in this reaction system. Previously, vanadium-mono(imide) complexes were reported to react stoichiometrically with phosphaalkynes to give the corresponding 1,2,4-azadiphospholes.6b,c Based on the experimental result, we consider that a vanadium-mono(imide) species may work as a catalytically active species in the current reaction system.
 | ||
| Scheme 4 Effect of bis(trimethylsilyl)aniline as additive. | ||
Based on these observations, a plausible reaction pathway is shown in Scheme 5. First, vanadium complex A reacts with an azobenzene 1 to give vanadium-imide complex B. Then, [2+2] cycloaddition reaction occurs between B and a phosphaalkyne 2 to afford a four-membered ring intermediate C. Subsequent insertion of the second phosphaalkyne 2 forms a six-membered ring intermediate D. Finally, reductive elimination affords the desired 1,2,4-azadiphosphole 3 accompanied by the regeneration of the initial vanadium complex A.
 | ||
| Scheme 5 Plausible reaction pathway. | ||
In summary, we have succeeded in the development of the first vanadium-catalyzed [2+2+1] cycloaddition reactions of azobenzenes with phosphaalkynes to synthesize a variety of 1,2,4-azadiphospholes. We believe the present study has opened a new aspect of synthetic application for the utilization of phosphaalkynes as building blocks in the construction of phosphorous-containing heterocyclic skeletons.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The present project was supported by CREST, JST (grant JPMJCR1541). We acknowledge Grants-in-Aid for Scientific Research (Grants JP17H01201, JP15H05798, JP18K19093, and JP19K15556) from JSPS and MEXT.Notes and references
- (a) L. Nyulászi, Chem. Rev., 2001, 101, 1229 CrossRef PubMed; (b) T. Baumgartner and R. Réau, Chem. Rev., 2006, 106, 4681 CrossRef CAS PubMed; (c) L. Kollár and G. Keglevich, Chem. Rev., 2010, 110, 4257 CrossRef PubMed.
- (a) M. Regitz, Chem. Rev., 1990, 90, 191 CrossRef CAS; (b) F. Mathey, Angew. Chem., Int. Ed., 2003, 42, 1578 CrossRef CAS PubMed; (c) A. Chirila, R. Wolf, J. C. Slootweg and K. Lammertsma, Coord. Chem. Rev., 2014, 270–271, 57 CrossRef CAS.
- (a) J. F. Nixon, Chem. Rev., 1988, 88, 1327 CrossRef CAS; (b) J. F. Nixon, Coord. Chem. Rev., 1995, 145, 201 Search PubMed.
- (a) F. Geoffrey, N. Cloke, P. B. Hitchcock, J. F. Nixon and D. J. Wilson, Chem. Commun., 2000, 2387 Search PubMed; (b) F. Tabellion, C. Peters, U. Fischbeck, M. Regitz and F. Preuss, Chem.–Eur. J., 2000, 6, 4558 CrossRef CAS PubMed; (c) S. G. Ruf, M. Andreas, J. Steinbach, U. Bergsträßer and M. Regitz, Synthesis, 2000, 360 CrossRef CAS; (d) J. Dietz, T. Schmidt, J. Renner, U. Bergsträßer, F. Tabellion, F. Preuss, P. Binger, H. Heydt and M. Regitz, Eur. J. Org. Chem., 2002, 1664 CrossRef CAS; (e) M. Trincado, A. J. Rosenthal, V. Matthias and H. Grützmacher, Eur. J. Inorg. Chem., 2014, 1599 CrossRef CAS.
- (a) K. Nakajima, S. Takata, K. Sakata and Y. Nishibayashi, Angew. Chem., Int. Ed., 2015, 54, 7597 CrossRef CAS PubMed; (b) K. Nakajima, W. Liang and Y. Nishibayashi, Org. Lett., 2016, 18, 5006 CrossRef CAS PubMed; (c) W. Liang, K. Nakajima, K. Sakata and Y. Nishibayashi, Angew. Chem., Int. Ed., 2019, 58, 1168 CrossRef CAS PubMed.
- (a) A. S. Ionkin, S. N. Ignat’eva, I. A. Litvinov, V. A. Naumov and B. A. Arbuzov, Heteroat. Chem., 1991, 2, 577 CrossRef CAS; (b) F. G. N. Cloke, P. B. Hitchcock, J. F. Nixon, D. J. Wilson, F. Tabellion, U. Fischbeck, F. Preuss, M. Regitz and L. Nyulászi, Chem. Commun., 1999, 2363 RSC; (c) F. Tabellion, C. Peters, U. Fischbeck, F. Preuss, M. Regitz and F. Preuss, Chem.–Eur. J., 2000, 6, 4558 CrossRef CAS PubMed; (d) F. G. N. Cloke, J. C. Green, N. Hazari, P. B. Hitchcock, P. Mountford, J. F. Nixon and D. J. Wilson, Organometallics, 2006, 25, 3688 CrossRef CAS; (e) R. Suter, Z. Benkö and H. Grützmacher, Chem.–Eur. J., 2016, 22, 14979 CrossRef CAS PubMed.
- (a) M. K. Cyranski, P. R. Schleyer, T. M. Krygowski, H. Jiao and G. Hohlneicher, Tetrahedron, 2003, 59, 1657 CrossRef CAS; (b) N. H. Martin and J. D. Robinson, J. Mol. Graphics Modell., 2012, 38, 26 CrossRef CAS PubMed; (c) I. Alkorta and J. Elguero, Struct. Chem., 2016, 27, 1531 CrossRef CAS.
- (a) Z. W. Gilbert, R. J. Hue and I. A. Tonks, Nat. Chem., 2016, 8, 63 CrossRef CAS PubMed; (b) H.-C. Chiu and I. A. Tonks, Angew. Chem., Int. Ed., 2018, 57, 6090 CrossRef CAS PubMed; (c) Z. W. Davis-Gilbert, W. Wen, J. D. Goodpaster and I. A. Tonks, J. Am. Chem. Soc., 2018, 140, 7267 CrossRef CAS PubMed; (d) A. J. Pearce, X. Y. See and I. A. Tonks, Chem. Commun., 2018, 54, 6891 RSC; (e) Z. W. Davis-Gilbert, K. Kawakita, D. R. Blechschmidt, H. Tsurugi, K. Mashima and I. A. Tonks, Organometallics, 2018, 37, 4439 CrossRef CAS PubMed; (f) H.-C. Chiu, X. Y. See and I. A. Tonks, ACS Catal., 2019, 9, 216 CrossRef CAS PubMed.
- K. Kawakita, E. P. Beaumier, Y. Kakiuchi, H. Tsurugi, I. A. Tonks and K. Mashima, J. Am. Chem. Soc., 2019, 141, 4194 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1974229. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0ra02503h |
| This journal is © The Royal Society of Chemistry 2020 |