
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhotocurrent generation by a photosystem I-NiO photocathode for a p-type biophotovoltaic tandem cell†
Yuya Takekuma a,
Nobuhiro Ikedaa,
Keisuke Kawakamib,
Nobuo Kamiyac,
Mamoru Nangoc and
Morio Nagata*a
a,
Nobuhiro Ikedaa,
Keisuke Kawakamib,
Nobuo Kamiyac,
Mamoru Nangoc and
Morio Nagata*a
aDepartment of Industrial Chemistry, Graduate School of Engineering, Tokyo University of Science, 12-1 Ichigaya-funagawara, Shinjuku-ku, Tokyo 162-0826, Japan. E-mail: nagata@ci.tus.ac.jp
bResearch Center for Artificial Photosynthesis (ReCAP), Osaka City University, 3-3-138 Sugimoto, Sumiyoshi-ku, Osaka 558-8585, Japan
cThe OCU Advanced Research Institute for Natural Science & Technology (OCARINA), Osaka City University, 3-3-138 Sugimoto, Sumiyoshi-ku, Osaka 558-8585, Japan
First published on 21st April 2020
Abstract
Photosynthesis is a process used by algae and plants to convert light energy into chemical energy. Due to their uniquely natural and environmentally friendly nature, photosynthetic proteins have attracted attention for use in a variety of artificial applications. Among the various types, biophotovoltaics based on dye-sensitized solar cells have been demonstrated in many studies. Although most related works have used n-type semiconductors, a p-type semiconductor is also a significant potential component for tandem cells. In this work, we used mesoporous NiO as a p-type semiconductor substrate for Photosystem I (PSI) and demonstrated a p-type PSI-biophotovoltaic and tandem cell based on dye-sensitized solar cells. Under visible light illumination, the PSI-adsorbed NiO electrode generated a cathodic photocurrent. The p-type biophotovoltaic cell using the PSI-adsorbed NiO electrode generated electricity, and the IPCE spectrum was consistent with the absorption spectrum of PSI. These results indicate that the PSI-adsorbed NiO electrode acts as a photocathode. Moreover, a tandem cell consisting of the PSI-NiO photocathode and a PSI-TiO2 photoanode showed a high open-circuit voltage of over 0.7 V under illumination to the TiO2 side. Thus, the tandem strategy can be utilized for biophotovoltaics, and the use of other biomaterials that match the solar spectrum will lead to further progress in photovoltaic performance.
Introduction
Photosynthesis is a process used by algae and plants to convert light energy into chemical energy. In order to reduce the consumption of fossil fuels and curb global warming, the efficient use of sunlight by mimicking photosynthesis or directly using photosynthetic proteins is attracting attention. Photosynthetic proteins, which use highly complex and optimized processes that are impossible to reproduce, have been reportedly used in a variety of artificial applications such as biophotovoltaics, semi-artificial photosynthesis, biosensors, biosupercapacitors, and photosynthetic microbial fuel cells.1–7 As biomaterials, various photosynthetic protein-pigment complexes such as photosystem I (PSI), photosystem II (PSII), reaction centers/light-harvesting antenna complexes (RC-LH1), light-harvesting complexes (LHCII), etc. have been researched for artificial utilization. These biomaterials are naturally abundant and pose no threat to the environment. In addition, the reaction center of a protein-pigment complex exhibits near-unity quantum efficiency attributable to spatial charge separation by multiple sequential electron-transfer pathways within the protein, which is a very attractive feature for researchers in the photoenergy-conversion field.Generally, these biomaterials have been deposited onto various electrodes such as gold, graphene, and metal-oxide semiconductors to create photocathodes and photoanodes.8 These electrodes are used alone or in a combination of two electrodes.9–13 As one application, biophotovoltaics have been demonstrated using these biohybrid electrodes.1,14 Especially, biophotovoltaics based on dye-sensitized solar cells (DSSCs)15 have been investigated using the metal-oxide semiconductors, TiO2, Fe2O3 and ZnO as the electrode.14,16–22 DSSCs generally consist of a dye-sensitized photoanode, Pt counter electrode, and electrolyte. In 2012, Mershin et al. reported PSI-based DSSCs on nanostructured TiO2 and ZnO electrodes using a Co(II/III) redox mediator.16 Similarly, Kondo et al. demonstrated PSI- and PSII-based DSSCs using a TiO2 electrode,17 and in 2015, Yu et al. achieved the most effective PSI- and LHCII-based DSSC to date using two different particle sizes of TiO2.18 Furthermore, polyaniline/TiO2 (ref. 19) and hematite semiconductor electrodes20 have also been investigated as photoanodes for biophotovoltaic-based DSSCs. Although most studies have used n-type metal-oxide semiconductors as the photoanode, a p-type semiconductor would also be significant because it could be used as the photocathode component in a tandem cell.23,24 The tandem cell has attracted interest because high photovoltaic performances can be achieved by simply replacing a Pt counter electrode with a photocathode. In biophotovoltaics, a tandem cell is also an effective strategy for increasing the photoresponse and improving the photovoltaic performance.25 For example, Ravi et al. developed a biohybrid tandem cell consisting of two stacked sub-cells connected in parallel,25 which led to photocurrent enhancement by complementary absorption of the two RC-LH1 units.
Here, we used mesoporous NiO as a p-type semiconductor substrate for PSI and demonstrated PSI-biophotovoltaics based on p-type DSSCs and a tandem cell. The NiO electrode has often been used for p-type DSSCs.24,26,27 The power conversion mechanism of p-type DSSCs begin with excitation of the dye, after which the hole is transferred from the dye to the valence band of the semiconductor, while the electron reduces the redox agent in the electrolyte; this is the reverse direction compared with n-type DSSCs. In PSI, upon photoexcitation of the chlorophyll dimer, called P700, the excited electron on P700 is transferred to the primary electron acceptor Chl a (A0, Fig. 1), the secondary acceptor phylloquinone (A1), and finally the acceptor [4Fe–4S] iron–sulfur clusters (FX, FA, and FB).28 This multi-step, sequential, unidirectional electron transfer has a near unity charge-separation quantum efficiency.28,29 We fabricated a PSI-adsorbed NiO electrode and biophotovoltaics based on p-type DSSCs and confirmed that the PSI-adsorbed NiO electrode acts as the photocathode. Moreover, a tandem biophotovoltaic cell was demonstrated consisting of the PSI-NiO photocathode and the well-known photoanode PSI-TiO2. Our fabricated tandem cell exhibited an open-circuit voltage of over 0.7 V, which is comparable to that of other biophotovoltaics exhibiting high photovoltages.30,31
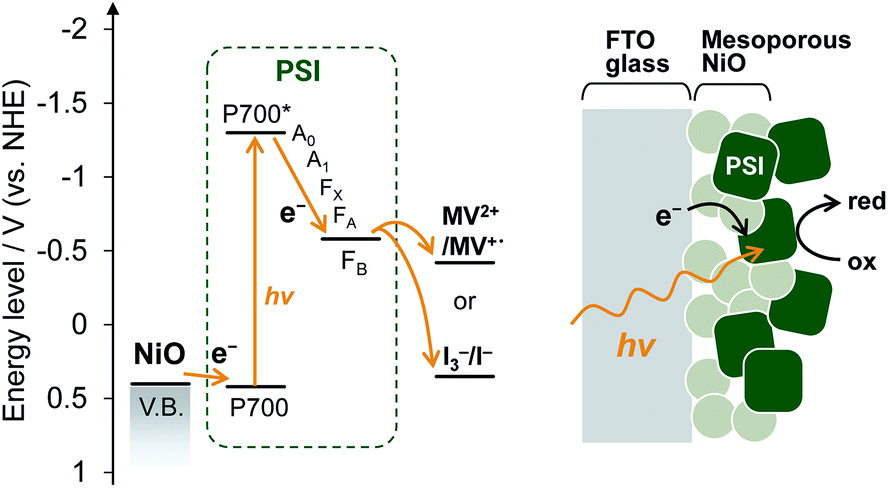 | ||
| Fig. 1 Energy levels and schematic representation of the PSI-adsorbed NiO photocathode. | ||
Results & discussion
PSI-adsorbed NiO electrode
Mesoporous NiO-coated FTO glass was fabricated based on a previous method32 with slight modifications. A scanning electron microscope (SEM) image of the NiO electrode is shown in Fig. S1.† The SEM image shows that there are 50–500 nm NiO particles, which form larger pores than the PSI trimer diameter (ca. 21 nm). These results indicate that PSI can enter the NiO pores.For deposition onto the NiO electrode, PSI core complexes were purified from Thermosynechococcus vulcanus according to previous methods33–35 with slight modifications. PSI was then deposited onto the NiO electrode by a vacuum-assisted drop-casting method.36 To confirm the presence of PSI, the UV-visible spectrum of the PSI-adsorbed NiO electrode was measured and is shown Fig. S2.† In the UV-visible spectrum, two distinct peaks at 680 nm and 440 nm are present corresponding to the Q-band and Soret-band absorption of chlorophyll in PSI, respectively, which indicated the presence of PSI on the NiO electrode. In addition, the amount of adsorbed PSI was calculated to be 4.55 ± 0.42 μgChl cm−2 by measuring the absorption of chlorophyll desorbed from the NiO electrode using an acetone solution. This value is larger than the adsorbable amount of PSI on a 1 cm2 plane (calculated as 0.097 μgChl). Collectively, these results indicate that PSI was adsorbed not only onto the NiO surface but also inside the mesopores of the NiO film, as shown in Fig. 1.
Electrochemical and photoelectrochemical experiments
The electrochemical properties and photocurrent response of the PSI-adsorbed NiO electrode were investigated. These experiments employed a Pt counter electrode and Ag/AgCl (saturated KCl) reference electrode in an aqueous solution containing 0.1 M phosphate buffer (pH 7) and 0.1 M NaClO4. The cyclic voltammograms of NiO electrodes with and without PSI are shown in Fig. 2a. The bare NiO electrode exhibited a quasi-reversible electrochemistry and distinct peaks in both the anodic and cathodic scans. These peaks are attributable to the redox reaction of surface Ni.37 Upon scanning the cathode, the surface NiII first oxidizes to NiIII and then to NiIV. In the PSI-adsorbed NiO electrode, these peaks decreased. In addition, the cyclic voltammogram indicated a slower oxidation reaction than that of the bare NiO electrode, and there appeared to be a peak shift toward a higher potential. These results indicate that PSI was adsorbed onto the NiO surface.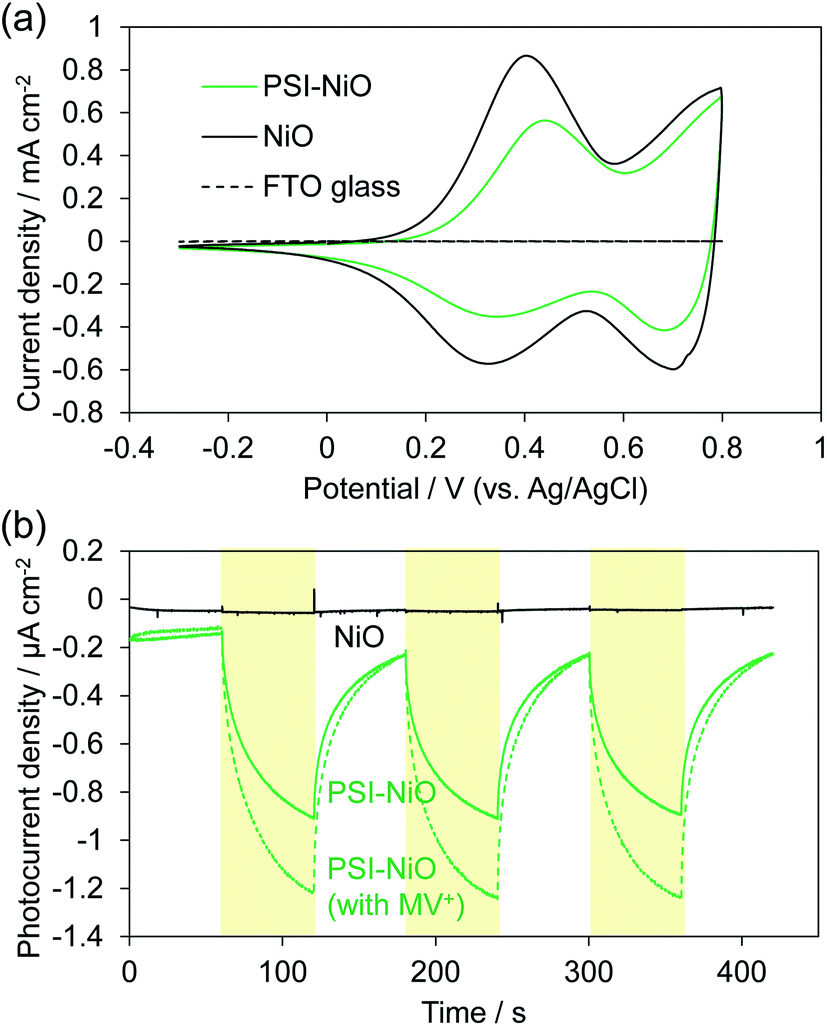 | ||
| Fig. 2 (a) Cyclic voltammograms of NiO (black) and PSI-NiO (green) electrodes in the dark in a 0.1 M phosphate aqueous buffer (pH 7) solution. The potential of the working electrode was scanned from −0.3 to 0.8 V vs. Ag/AgCl at a rate of 50 mV s−1. (b) Photoelectrochemical measurements of NiO (black), PSI-NiO without methyl viologen (MV2+) (green solid), and PSI-NiO with MV2+ (green dash) at 0 mV vs. Ag/AgCl in a 0.1 M phosphate aqueous buffer (pH 7) solution containing 0.1 M NaClO4. The experiment was performed under AM 1.5G illumination with a 420 nm long-pass filter. The intensity was ca. 85 mW cm−2. | ||
Fig. 2b shows the photocurrent response of the NiO electrodes with and without PSI. The photocurrent at 0 mV vs. Ag/AgCl was measured under illumination by a Xe lamp with a 420 nm long-pass filter to exclude the photocurrent of NiO. In the PSI-NiO electrode, a cathodic photocurrent of −0.75 ± 0.01 μA cm−2 occurred, whereas the photocurrent from NiO was hardly observed. The electron flow can be explained as follows: the excited electron at P700 on the luminal side transfers to the stromal side and is then used to oxidize the electron acceptor (in this case, oxygen), while the oxidized P700 receives an electron from the NiO surface (Fig. 1b). The charge on the NiO surface is transported to the FTO through the NiO/electrolyte interface,38 and consequently, a photocurrent is generated. Moreover, addition of the well-known electron acceptor methyl viologen (MV2+) increase the cathodic photocurrent of PSI to −1.10 ± 0.09 μA cm−2. This is because the reaction rate of MV2+ with PSI is higher than that with oxygen.39 For comparison, when using the n-type semiconductor TiO2, the resulting photocurrent is anodic.21,40 Overall, these results indicate that the PSI-NiO electrode acts as a photocathode. In addition, the photocurrent response at various potentials was also measured, and the results are shown Fig. S3.† At −200 mV vs. Ag/AgCl, a cathodic photocurrent also occurred, whereas at +300 mV vs. Ag/AgCl, an anodic photocurrent occurred. In this case, the electron flow begins with an excited electron on P700 transferring to NiO, after which the oxidized P700 receives an electron from the electron donor in the electrolyte. Therefore, the results indicate that PSI was adsorbed in different orientations on the NiO surface.
Photovoltaic characteristics
Biophotovoltaics based on p-type DSSCs were fabricated using the PSI-NiO photocathode. To investigate the photovoltaic performances of the p-type biophotovoltaics, a two-electrode sandwich cell was prepared with the PSI-NiO electrode, a Pt counter electrode, and a triiodide/iodide-based ionic liquid electrolyte according to a previous report.41 Table 1 and Fig. 3 show the photovoltaic performances under 1 sun conditions (AM 1.5G) and the IPCE spectrum, respectively. Our fabricated p-type biophotovoltaic exhibited a 0.16 ± 0.01 mA cm−2 short-circuit current density (JSC), a 0.10 ± 0.01 V open-circuit voltage (VOC), and a 0.0071 ± 0.0003% power conversion efficiency (PCE). The IPCE spectrum of the p-type biophotovoltaic is consistent with the distinct absorption peak at 680 nm, as shown Fig. 3. These results show that PSI acts as a photosensitizer in the biophotovoltaic based on p-type DSSCs.| JSC/mA cm−2 | VOC/V | FF | PCE/% | |
|---|---|---|---|---|
| PSI-NiO | 0.16 ± 0.01 | 0.10 ± 0.01 | 0.42 ± 0.01 | 0.0071 ± 0.0003 |
| Tandem cell | 0.16 ± 0.01 | 0.71 ± 0.01 | 0.50 ± 0.02 | 0.056 ± 0.001 |
| PSI-TiO2 | 0.15 ± 0.01 | 0.58 ± 0.02 | 0.59 ± 0.03 | 0.053 ± 0.008 |
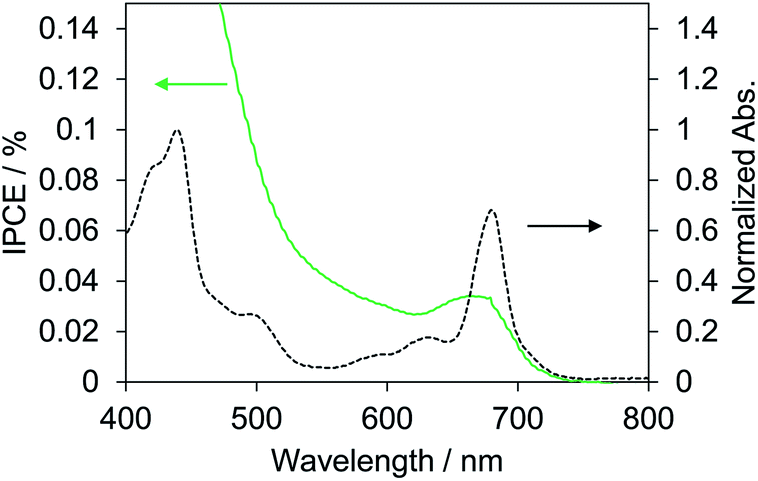 | ||
| Fig. 3 IPCE spectrum of the best biophotovoltaic cell using the PSI-NiO electrode (green solid) and normalized absorption spectrum of PSI in HEPES buffer (black dash). Absorbance is normalized against the maximum peak. | ||
To investigate whether the PSI-NiO photocathode could be used in a tandem cell, we demonstrated a PSI-based tandem cell consisting of a PSI-NiO photocathode and PSI-TiO2 photoanode in a simple sandwich configuration according to Fig. 4a. When light was incident to the tandem cell, the PSI adsorbed onto NiO reduces the redox agent, and an electron is received from NiO. On the other hand, the PSI adsorbed onto TiO2 receives an electron from the redox agent and injects it into TiO2, resulting in current flow. For comparison, PSI-sensitized solar cells each using a PSI-NiO electrode or PSI-TiO2 electrode were also demonstrated. The PSI-TiO2 electrode and photovoltaic were fabricated according to previously reported procedures.41 In the photovoltaic measurement, the light was illuminated on the TiO2 side, and the thickness of the top film was optimized to allow incident light on the bottom electrode (NiO film: 9 μm, TiO2 film: 3 μm). The amounts of PSI adsorbed onto the electrodes were calculated as 6.01 ± 0.30 μgChl cm−2 (NiO electrode) and 6.27 ± 0.15 μgChl cm−2 (TiO2 electrode). The series connection of the photoanode and photocathode in the tandem cell showed that the photocurrent is governed by the weakest photoelectrode, whereas the photovoltages are additive. Indeed, the photocurrent of the tandem cell was 0.16 ± 0.01 mA cm−2, which is similar to that of the PSI-TiO2 cell, whereas the VOC of the tandem cell matched the sum of the VOC values of the PSI-TiO2 and PSI-NiO cells, as shown in Fig. 4b. Consequently, the VOC was improved to 0.71 ± 0.01 V, and the PCE was 0.056 ± 0.001%. This VOC is comparable to that of other biophotovoltaics that have exhibited high photovoltages.30,31 However, the fill factor (FF) of the tandem cell was lower than that of PSI-TiO2. This is due to the low shunt resistance of the tandem cell, which might be attributable to shade from the top cell and/or increased recombination at the semiconductor/electrolyte interface.42,43 In summary, our fabricated PSI-NiO photocathode was utilized in a tandem cell that exhibited a high voltage when irradiated on the TiO2 side. However, the photocurrent was low because the same bio-sensitizer was used for the photocathode and photoanode. Thus the use of other biomaterials that match the solar spectrum will be required to further increase the photovoltaic performance.
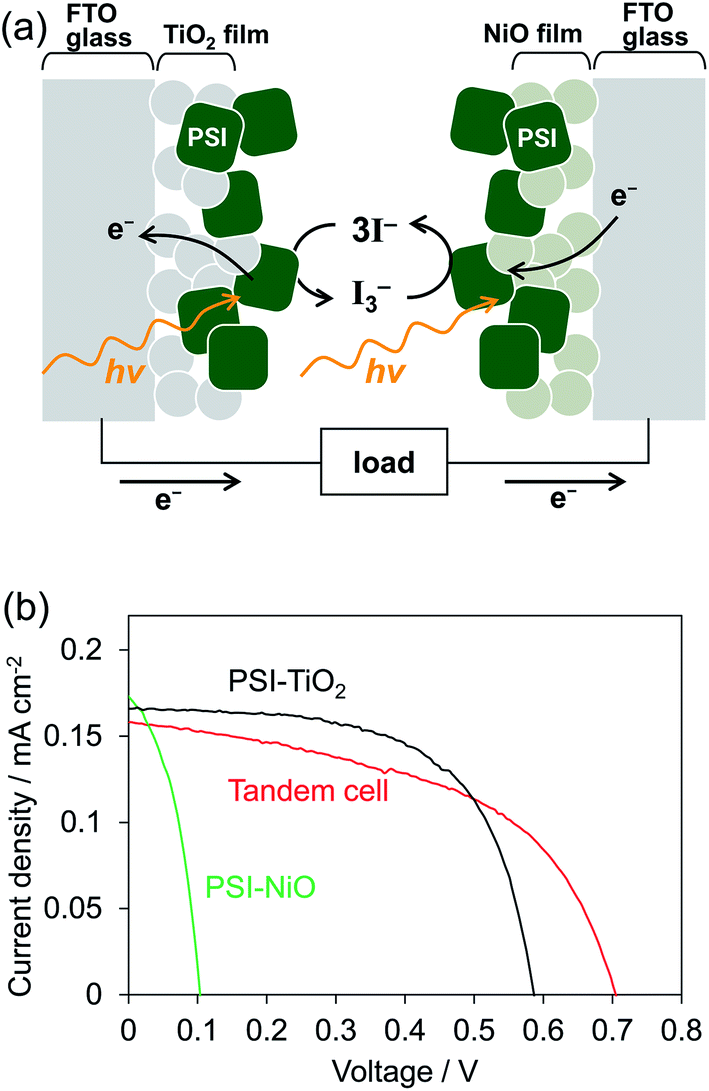 | ||
| Fig. 4 (a) Schematic representation of the tandem cell power generation mechanism. (b) J–V curves under AM 1.5G (100 mW cm−2) of the tandem cell and individual n- and p-biophotovoltaic cells. | ||
Conclusion
In summary, we fabricated a PSI-adsorbed NiO electrode for utilization as a component in a tandem cell and demonstrated p-type biophotovoltaics based on DSSCs using the PSI-NiO electrode. Under visible light illumination, the electrode showed a cathodic photocurrent, and the photovoltaic was powered by PSI, which indicates that the PSI-NiO electrode acts as a photocathode. Moreover, we demonstrated a tandem cell consisting of the PSI-NiO photocathode and a PSI-TiO2 photoanode. The tandem cell exhibited a high VOC of over 0.7 V under illumination on the TiO2 side.Tandem cells have the advantages of a simple fabrication method employed by changing the Pt counter electrode and a high VOC since it does not depend on the redox potential of the redox agents but instead on the conduction band minimum of the photoanode and valence band maximum of the photocathode. Therefore, selection of appropriate redox agents could lead to more efficient photovoltaic performances. Moreover, controlling the PSI orientation to match the current flow and using other photosynthetic proteins that can absorb a broad spectral region could also lead to further improvements to the efficiency of these biophotovoltaics.
Experiments
Materials
All chemicals were purchased from commercial suppliers and used without purification.Purification of PSI of T. vulcanus33–35,41
Harvested T. vulcanus cells were treated with lysozyme for 90 min at 38 °C in darkness, then centrifuged at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000g for 10 min. The obtained precipitants were suspended with a small amount of buffer containing 25% (w/v) glycerol, 20 mM HEPES–NaOH (pH 7.0), and 10 mM MgCl2. Subsequently, the samples were stored at −80 °C, and the thylakoid membranes were obtained by a freeze-thawing method.44 The thylakoid membranes were solubilized with 0.6% (w/v) n-dodecyl-β-D-maltoside (β-DDM), then centrifuged at 38900g for 90 min at 4 °C. Subsequently, the obtained supernatants were filtrated through a 0.45 μm filter and loaded onto an anion-exchange column (DEAE TOYOPEARL 650S) with 5% (w/v) glycerol, 30 mM Mes (pH 6.0), 3 mM CaCl2, and 0.03% (w/v) β-DDM. The column was washed with the same buffer containing 80 mM NaCl to remove the large amount of phycobilisome impurities. Elution of the PSI core complexes was carried out with a linear gradient from 80 to 140 mM NaCl in the same buffer.
000g for 10 min. The obtained precipitants were suspended with a small amount of buffer containing 25% (w/v) glycerol, 20 mM HEPES–NaOH (pH 7.0), and 10 mM MgCl2. Subsequently, the samples were stored at −80 °C, and the thylakoid membranes were obtained by a freeze-thawing method.44 The thylakoid membranes were solubilized with 0.6% (w/v) n-dodecyl-β-D-maltoside (β-DDM), then centrifuged at 38900g for 90 min at 4 °C. Subsequently, the obtained supernatants were filtrated through a 0.45 μm filter and loaded onto an anion-exchange column (DEAE TOYOPEARL 650S) with 5% (w/v) glycerol, 30 mM Mes (pH 6.0), 3 mM CaCl2, and 0.03% (w/v) β-DDM. The column was washed with the same buffer containing 80 mM NaCl to remove the large amount of phycobilisome impurities. Elution of the PSI core complexes was carried out with a linear gradient from 80 to 140 mM NaCl in the same buffer.
Fabrication of electrodes for photoelectrochemical measurements
The NiO pastes consisted of 18 wt% NiO nanoparticles, 9 wt% ethyl cellulose (EC), and 73 wt% terpineol. Two kinds of EC powders, EC (Kanto Kagaku, 10cP) and EC (Kanto Kagaku, 45cP) (65/35, v/v), were dissolved in ethanol. The ethanolic mixtures were added to a round-bottom flask containing NiO nanopowder (Sigma-Aldrich, <50 nm particle size) and terpineol. This mixture was then ultrasonicated for 48 h, and the ethanol was removed by rotary-evaporator. Finally, the pastes were made with a three-roll mill (EXAKT, Nagase Screen Printing Research). The nanocrystalline NiO paste was coated onto fluorine-doped SnO2 (FTO) conductive glass plates (Nippon Sheet Glass) by squeegee for photoelectrochemical measurements, and the electrodes were sintered at 450 °C for 30 min and then 550 °C for 15 min in air. PSI was deposited onto the NiO electrode according to the vacuum-assisted drop-casting method reported Faulkner et al.;36 an aqueous solution of PSI (ca. 0.4 mgChl mL−1) containing 0.1 M HEPES buffer was dropped onto the NiO electrode, and the electrode was placed under vacuum for 120 min. Subsequently, the electrode was rinsed with water and dried with N2 gas.Electrochemical and photoelectrochemical measurements
All measurements were performed using a potentiostat (HSV-110, Hokuto Denko) with a Pt counter electrode and Ag/AgCl (saturated KCl) reference electrode in an aqueous solution containing phosphate buffer (0.1 M, pH 7) and 0.1 M NaClO4. For the cyclic voltammetry measurements, the potential of the working electrode was scanned from −0.3 to 0.8 V vs. Ag/AgCl at a rate of 50 mV s−1. Photochronoamperometric measurements were performed with a Xe lamp solar simulator (XB-500E, Peccell Technologies) and a 420 nm long-pass filter with using an illumination time of 60 s (ca. 85 mW cm−2). The light intensity was measured by Si solar cell (BS-520, Bunkokeiki) placed in front of the electrochemical cell. MV2+ was employed as an electron donor at 3 mM.Fabrication of biophotovoltaics
The FTO glass was coated with a 0.05 M nickel acetate ethanol solution by dip-coating and subsequently dried at room temperature before screen printing. The NiO photocathode was screen-printed using the above-mentioned NiO paste, and the electrodes were sintered at 450 °C for 30 min and then at 550 °C for 15 min in air. PSI was adsorbed onto the NiO electrode by immersion in a PSI buffer solution (ca. 0.4 mgChl mL−1). The photovoltaic device was fabricated by sandwiching a 30 μm Surlyn spacer between the stained NiO photocathode and a Pt counter electrode. The electrolyte solution was introduced into the cell via a predrilled hole in the Pt counter electrode. The electrolyte consisted of 0.2 M I2 and 0.1 M guanidine thiocyanate in 1-ethyl-3-methylimidazolium tricyanomethanide and 1-methyl-3-propylimidazolium iodide (13:7 v/v). The tandem cell was fabricated in the same manner but with the PSI-TiO2 photoanode instead of the Pt counter electrode. The TiO2 photoanode was fabricated by screen-printing, and the thickness was optimized to allow light incident to the bottom NiO electrode.
Characterization of bio-photovoltaic devices
The photovoltaic performances of the fabricated solar cells were evaluated by the incident photon-to-current conversion efficiency (IPCE) spectra and photocurrent density–voltage (J–V) measurements. The IPCE spectra were measured with a Peccell Technologies S10AC system and the J–V curves were measured under AM 1.5G one-sun conditions (100 mW cm−2) with a Yamashita Denso YSS-150A. The light intensity was calibrated with reference to a Si solar cell (BS-520, Bunkokeiki)Conflicts of interest
There are no conflicts to declare.Acknowledgements
Y. Takekuma and M. Nagata would like to thank Mr Masaki Saito (Department of Industrial Chemistry, Tokyo University of Science, Japan) for measuring a scanning electron microscope image. M. Nango thanks for the support of the Grant, AOARD (Grant No. FA2386-19-1-4033).Notes and references
- S. K. Ravi, V. S. Udayagiri, L. Suresh and S. C. Tan, Adv. Funct. Mater., 2018, 28, 1705305 CrossRef
.
- N. Kornienko, J. Z. Zhang, K. K. Sakimoto, P. Yang and E. Reisner, Nat. Nanotechnol., 2018, 13, 890–899 CrossRef CAS PubMed
- A. J. McCormick, P. Bombelli, R. W. Bradley, R. Thorne, T. Wenzel and C. J. Howe, Energy Environ. Sci., 2015, 8, 1092–1109 RSC
- A. Antonacci and V. Scognamiglio, TrAC Trends Anal. Chem., 2019, 115, 100–109 CrossRef CAS
- L. Liu and S. Choi, Biosens. Bioelectron., 2019, 140, 111354 CrossRef CAS PubMed
- M. Rosenbaum, Z. He and L. T. Angenent, Curr. Opin. Biotechnol., 2010, 21, 259–264 CrossRef CAS PubMed
- M. Sawa, A. Fantuzzi, P. Bombelli, C. J. Howe, K. Hellgardt and P. J. Nixon, Nat. Commun., 2017, 8, 1327 CrossRef PubMed
- K. Nguyen and B. D. Bruce, Biochim. Biophys. Acta, 2014, 1837, 1553–1566 CrossRef CAS PubMed
- T. Kothe, N. Plumeré, A. Badura, M. M. Nowaczyk, D. A. Guschin, M. Rögner and W. Schuhmann, Angew. Chem., Int. Ed., 2013, 52, 14233–14236 CrossRef CAS PubMed
- V. Hartmann, T. Kothe, S. Pöller, E. El-Mohsnawy, M. M. Nowaczyk, N. Plumeré, W. Schuhmann and M. Rögner, Phys. Chem. Chem. Phys., 2014, 16, 11936–11941 RSC
- M. Rasmussen, A. Shrier and S. D. Minteer, Phys. Chem. Chem. Phys., 2013, 15, 9062–9065 RSC
- K. P. Sokol, W. E. Robinson, A. R. Oliveira, J. Warnan, M. M. Nowaczyk, A. Ruff, I. A. C. Pereira and E. Reisner, J. Am. Chem. Soc., 2018, 140, 16418–16422 CrossRef CAS PubMed
- F. Zhao, P. Wang, A. Ruff, V. Hartmann, S. Zacarias, I. A. C. Pereira, M. M. Nowaczyk, M. Rögner, F. Conzuelo and W. Schuhmann, Energy Environ. Sci., 2019, 12, 3133–3143 RSC
- E. Musazade, R. Voloshin, N. Brady, J. Mondal, S. Atashova, S. K. Zharmukhamedov, I. Huseynova, S. Ramakrishna, M. M. Najafpour, J.-R. Shen, B. D. Bruce and S. I. Allakhverdiev, J. Photochem. Photobiol., C, 2018, 35, 134–156 CrossRef CAS
- B. O'Regan and M. Gratzel, Nature, 1991, 353, 737–740 CrossRef
- A. Mershin, K. Matsumoto, L. Kaiser, D. Yu, M. Vaughn, M. K. Nazeeruddin, B. D. Bruce, M. Graetzel and S. Zhang, Sci. Rep., 2012, 2, 234 CrossRef PubMed
- M. Kondo, M. Amano, T. Joke, S. Ishigure, T. Noji, T. Dewa, Y. Amao and M. Nango, Res. Chem. Intermed., 2014, 40, 3287–3293 CrossRef CAS
- D. Yu, M. Wang, G. Zhu, B. Ge, S. Liu and F. Huang, Sci. Rep., 2015, 5, 9375 CrossRef CAS PubMed
- E. A. Gizzie, J. Scott Niezgoda, M. T. Robinson, A. G. Harris, G. Kane Jennings, S. J. Rosenthal and D. E. Cliffel, Energy Environ. Sci., 2015, 8, 3572–3576 RSC
- K. Ocakoglu, T. Krupnik, B. van den Bosch, E. Harputlu, M. P. Gullo, J. D. J. Olmos, S. Yildirimcan, R. K. Gupta, F. Yakuphanoglu, A. Barbieri, J. N. H. Reek and J. Kargul, Adv. Funct. Mater., 2014, 24, 7467–7477 CrossRef CAS
- V. B. Shah, W. R. Henson, T. S. Chadha, G. Lakin, H. Liu, R. E. Blankenship and P. Biswas, Langmuir, 2015, 31, 1675–1682 CrossRef CAS PubMed
- M. Nagata, M. Amano, T. Joke, K. Fujii, A. Okuda, M. Kondo, S. Ishigure, T. Dewa, K. Iida, F. Secundo, Y. Amao, H. Hashimoto and M. Nango, ACS Macro Lett., 2012, 1, 296–299 CrossRef CAS
- J. He, H. Lindström, A. Hagfeldt and S.-E. Lindquist, Sol. Energy Mater. Sol. Cells, 2000, 62, 265–273 CrossRef CAS
- A. Nattestad, A. J. Mozer, M. K. R. Fischer, Y. B. Cheng, A. Mishra, P. Bäuerle and U. Bach, Nat. Mater., 2010, 9, 31–35 CrossRef CAS PubMed
- S. K. Ravi, Z. Yu, D. J. K. Swainsbury, J. Ouyang, M. R. Jones and S. C. Tan, Adv. Energy Mater., 2017, 7, 1601821 CrossRef
- I. R. Perera, T. Daeneke, S. Makuta, Z. Yu, Y. Tachibana, A. Mishra, P. Bäuerle, C. A. Ohlin, U. Bach and L. Spiccia, Angew. Chem., Int. Ed., 2015, 54, 3758–3762 CrossRef CAS PubMed
- F. Odobel, Y. Pellegrin, E. A. Gibson, A. Hagfeldt, A. L. Smeigh and L. Hammarström, Coord. Chem. Rev., 2012, 256, 2414–2423 CrossRef CAS
- N. Nelson and C. F. Yocum, Annu. Rev. Plant Biol., 2006, 57, 521–565 CrossRef CAS PubMed
- S. W. Hogewoning, E. Wientjes, P. Douwstra, G. Trouwborst, W. van Ieperen, R. Croce and J. Harbinson, Plant Cell, 2012, 24, 1921–1935 CrossRef CAS PubMed
- P. I. Gordiichuk, G.-J. A. H. Wetzelaer, D. Rimmerman, A. Gruszka, J. W. de Vries, M. Saller, D. A. Gautier, S. Catarci, D. Pesce, S. Richter, P. W. M. Blom and A. Herrmann, Adv. Mater., 2014, 26, 4863–4869 CrossRef CAS PubMed
- V. K. Singh, S. K. Ravi, J. W. Ho, J. K. C. Wong, M. R. Jones and S. C. Tan, Adv. Funct. Mater., 2018, 28, 1703689 CrossRef
- S. Ito, T. N. Murakami, P. Comte, P. Liska, C. Grätzel, M. K. Nazeeruddin and M. Grätzel, Thin Solid Films, 2008, 516, 4613–4619 CrossRef CAS
- J.-R. Shen and N. Kamiya, Biochemistry, 2000, 39, 14739–14744 CrossRef CAS PubMed
- K. Kawakami, M. Iwai, M. Ikeuchi, N. Kamiya and J.-R. Shen, FEBS Lett., 2007, 581, 4983–4987 CrossRef CAS PubMed
- J.-R. Shen, K. Kawakami and H. Koike, in Photosynthesis Research Protocols, ed. R. Carpentier, Humana Press, Totowa, NJ, 2011, pp. 41–51, DOI:10.1007/978-1-60761-925-3_5
- C. J. Faulkner, S. Lees, P. N. Ciesielski, D. E. Cliffel and G. K. Jennings, Langmuir, 2008, 24, 8409–8412 CrossRef CAS PubMed
- G. Boschloo and A. Hagfeldt, J. Phys. Chem. B, 2001, 105, 3039–3044 CrossRef CAS
- H. Zhu, A. Hagfeldt and G. Boschloo, J. Phys. Chem. C, 2007, 111, 17455–17458 CrossRef CAS
- G. E. Milanovsky, A. A. Petrova, D. A. Cherepanov and A. Y. Semenov, Photosynth. Res., 2017, 133, 185–199 CrossRef CAS PubMed
- V. V. Nikandrov, Y. V. Borisova, E. A. Bocharov, M. A. Usachev, G. V. Nizova, V. A. Nadtochenko, E. P. Lukashev, B. V. Trubitsin, A. N. Tikhonov, V. N. Kurashov, M. D. Mamedov and A. Y. Semenov, High Energy Chem., 2012, 46, 200–205 CrossRef CAS
- Y. Takekuma, H. Nagakawa, T. Noji, K. Kawakami, R. Furukawa, M. Nango, N. Kamiya and M. Nagata, ACS Appl. Energy Mater., 2019, 2, 3986–3990 CrossRef CAS
- S. Chen, J. Weng, Y. Huang, C. Zhang, L. Hu, F. Kong, L. Wang and S. Dai, J. Phys. D: Appl. Phys., 2010, 43, 305102 CrossRef
- H. Choi, T. Hwang, S. Lee, S. Nam, J. Kang, B. Lee and B. Park, J. Power Sources, 2015, 274, 937–942 CrossRef CAS
- R. Nagao, A. Ishii, O. Tada, T. Suzuki, N. Dohmae, A. Okumura, M. Iwai, T. Takahashi, Y. Kashino and I. Enami, Biochim. Biophys. Acta, Bioenerg., 2007, 1767, 1353–1362 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra01793k |
| This journal is © The Royal Society of Chemistry 2020 |