
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Insights from semi-oriented EPR spectroscopy studies into the interaction of lytic polysaccharide monooxygenases with cellulose†
Luisa
Ciano‡
 ab,
Alessandro
Paradisi
a,
Glyn R.
Hemsworth
c,
Morten
Tovborg
d,
Gideon J.
Davies
a and
Paul H.
Walton
*a
ab,
Alessandro
Paradisi
a,
Glyn R.
Hemsworth
c,
Morten
Tovborg
d,
Gideon J.
Davies
a and
Paul H.
Walton
*a
aDepartment of Chemistry, University of York, Heslington, York, YO10 5DD, UK. E-mail: paul.walton@york.ac.uk
bSchool of Chemistry and Photon Science Institute, University of Manchester, Oxford Road, Manchester, M13 9PL, UK
cAstbury Centre for Structural Molecular Biology and School of Molecular and Cellular Biology, Faculty of Biological Sciences, University of Leeds, LS2 9JT, UK
dNovozymes A/S, Krogshoejvej 36, 2880 Bagsvaerd, Denmark
First published on 3rd March 2020
Abstract
Probing the detailed interaction between lytic polysaccharide monooxygenases (LPMOs) and their polysaccharide substrates is key to revealing further insights into the mechanism of action of this class of enzymes on recalcitrant biomass. This investigation is somewhat hindered, however, by the insoluble nature of the substrates, which precludes the use of most optical spectroscopic techniques. Herein, we report a new semi-oriented EPR method which evaluates directly the binding of cellulose-active LPMOs to crystalline cellulose. We make use of the intrinsic order of cellulose fibres in Apium graveolens (celery) to orient the LPMO with respect to the magnetic field of an EPR spectrometer. The subsequent angle-dependent changes observed in the EPR spectra can then be related to the orientation of the g matrix principal directions with respect to the magnetic field of the spectrometer and, hence, to the binding of the enzyme onto the cellulose fibres. This method, which does not require specific modification of standard CW-EPR equipment, can be used as a general procedure to investigate LPMO–cellulose interactions.
Introduction
Lytic polysaccharide monooxygenases (LPMOs, also known as PMOs) are copper metalloenzymes which activate O2 for the oxidative cleavage of glycosidic bonds in polysaccharides.1–3 The active site of LPMOs contains a mononuclear copper ion that is coordinated by an N-terminal histidine along with the side chain of another histidine, in a T-shaped configuration known as the histidine brace (Fig. 1).2,4 LPMOs have attracted attention not only because of the mechanistic questions they pose about the oxidation of a recalcitrant substrate with strong C–H bonds (ca. 100 kcal mol−1), but also for the potential that these enzymes offer the second-generation biofuel industry through improving the accessibility of abundant biomass, most notably cellulose.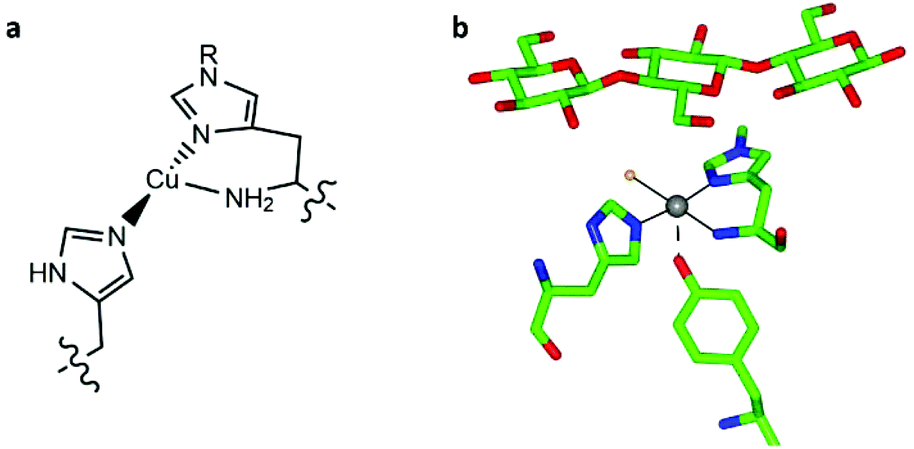 | ||
| Fig. 1 (a) Scheme of the copper histidine brace (R = Me or H). (b) Active site of LsAA9A bound to cellotriose (PDB: 5ACF), showing the position of the oligosaccharide with respect to the histidine brace (copper ion in grey, coordinated chloride ion in orange). | ||
LPMOs are widespread throughout life, with many bacteria, fungi, insects and viruses holding LPMO sequences within their genomes. Indeed, as a measure of the prevalence of LPMOs, the Carbohydrate Active enZyme (CAZy) database currently lists over 5700 known LPMO sequences spanning seven different sequence-distinct classes, which are named “Auxiliary Activity” enzymes (abbreviation AA) followed by an identifying number, i.e. AA9–AA11, AA13–AA16.6–12 Despite their importance, detailed molecular insight into the mechanism of action of LPMOs is hampered by the heterogeneous and insoluble nature of the polysaccharide substrate, precluding the use of traditional single crystal X-ray diffraction (XRD) studies and most optical spectroscopic methods. To circumvent this issue, some studies have been performed on the handful of LPMOs which are known to be active on soluble oligosaccharides, from which XRD and NMR structural studies have provided detailed information on the nature of oligosaccharide binding to the enzyme, particularly the interaction of the substrate with the copper active site.5,13–15 These studies have offered a key starting point for theoretical studies from which insights into the catalytic mechanism of LPMOs have been obtained.16–18 Notwithstanding these advances, however, the detailed interaction of LPMOs with their natural, insoluble, polysaccharide substrates is an area which presents significant experimental challenges. While computational methods have been employed by some laboratories,19,20 no general experimental method for examining the detailed molecular interaction between LPMO and solid polysaccharides is currently available, with the only exception of a handful of Electron Paramagnetic Resonance (EPR) studies.2,5,7,9,11,13,21 Since EPR spectroscopy can be performed on powders, frozen solutions and slurries, this particular technique has been able to give valuable information about the electronic structure of the copper active site of LPMOs. For instance, EPR spectroscopy has been pivotal in the characterisation of LPMOs at the early stages of their discovery and, more recently, in the determination of the structure of the copper coordination sphere within LPMO-substrate complexes.5 Perhaps most conspicuously, EPR revealed that some LPMOs (within CAZy classifications AA9 and AA10) show significant perturbation of the spin Hamiltonian parameters of the copper ion upon substrate addition,5,13,20,21 whereas some LPMOs show no perturbation to the spectrum on substrate addition.2,10
It is in this context that we sought to exploit EPR spectroscopy to give new insights into the binding of LPMOs to cellulose fibrils. Accordingly, we report here a new CW-EPR investigation of two AA9 LPMOs interacting with a natural semi-crystalline form of cellulose which is readily available from the stalks of Apium graveolens (celery), following the work of Jarvis et al. on this form of cellulose.22,23 We take advantage of the fact that the centimetre-wavelength microwaves used in X-band EPR are not significantly scattered by polysaccharide in this form and thus can be used to study LPMOs in the presence of natural substrates. We show that simple CW-EPR can be used to gain dimensional information about the LPMO–substrate interaction when the sample is (partially) orientated, akin to EPR studies on single crystals of transition metal complexes. In other words, rather than using crystallization to orientate the LPMO, we have made use of a naturally semi-orientated form of cellulose to bind and thus orientate an LPMO within the magnetic field of an EPR spectrometer. From the resulting dependence of the EPR spectrum on the orientation of the LPMO-loaded fibres within the  field of the spectrometer, the direction cosines of the principal components of the g matrix with respect to the
field of the spectrometer, the direction cosines of the principal components of the g matrix with respect to the  field can be evaluated, from which it is possible to obtain dimensional information about the LPMO–substrate interaction. The substrate is readily available and the method is experimentally straightforward with standard CW-EPR equipment; it is therefore applicable to any cellulose-active LPMO. We thus propose that the method can be used as a general approach for evaluation of all LPMO–cellulose interactions.
field can be evaluated, from which it is possible to obtain dimensional information about the LPMO–substrate interaction. The substrate is readily available and the method is experimentally straightforward with standard CW-EPR equipment; it is therefore applicable to any cellulose-active LPMO. We thus propose that the method can be used as a general approach for evaluation of all LPMO–cellulose interactions.
Results and discussion
Cellulose fibre preparation and activity of LPMOs on fibres
The stalks of Apium graveolens (celery) offer a readily available source of oriented, semi-crystalline cellulose, ideal for the proposed EPR experiments. Therefore, cellulose fibres were prepared following reported literature procedures,22,23 which—in brief—is as follows: the fibres were manually extracted from the fluted ridges of celery stalks using a scalpel and then washed to remove extraneous proteins, giving fibres of ∼10 cm length. Fibres grow in one direction with the reducing end of the individual cellulose fibrils laying at the anterior end of the fibre. As such, all collected fibres were marked at the anterior end as to permit subsequent identification of the direction of cellulose chains within the fibres when used in later EPR studies. The orientation of the fibres is visible to the naked eye, a feature which was used to orientate the fibres on a glass slide for subsequent EPR studies (see below). The macroscopic orientation of the cellulose fibrils within the fibres was further evident upon inspection with an optical microscope under polarised light (Fig. S1 and Movie S1†). The intensity of polarised light passing through the fibres was much reduced when the long axis of the fibre is placed perpendicular to the plane of polarisation, as expected for a uniaxially-orientated sample.Two LPMOs were used in this study, one from Lentinus similis (LsAA9A), for which a handful of oligosaccharide–enzyme structures are available,5,13 and one from Thermoascus aurantiacus (TaAA9A). Both enzymes are known to be active on PASC (phosphoric-acid swollen cellulose) and Avicel, but their activity on more natural forms of cellulose has not yet been reported, with the only exception of one study by Harris et al. in which TaAA9A was used with corn stover.1 Accordingly, LsAA9A and TaAA9A were tested for their activity on celery fibres by incubation of a solution of the enzyme and ascorbic acid with the fibres. Any resulting soluble reaction products were then analysed by MALDI-TOF mass spectrometry, the results of which clearly show that the action of the LPMO on the cellulose fibres afforded oxidised oligosaccharides (Fig. S2†); this is a hallmark of LPMO action.
Unlike the action of LPMOs on non-natural sources of cellulose (e.g. Avicel, PASC) however, action on the cellulose fibres also gave mixed glucose–pentose oligosaccharide products, along with oxidised cello-oligosaccharides. These products were identified in the MALDI-TOF spectra (Fig. S3†) and are in accord with reports in the literature of presence of xyloglucan on the cellulose fibres,23 which are only removed by prolonged treatment with a concentrated alkali solution.24 (The activity of LsAA9A and TaAA9A on xyloglucan has previously been investigated using tamarind xyloglucan,13,25 and the assays reported herein confirm that these LPMOs are active on both the cellulosic and the hemicellulosic components of the substrate.) All peaks in the mass spectra could be assigned to either mixed hexose–pentose oligomers or native glucose oligosaccharides. In each case the parent ion peak lays at −2 Da with respect to the native oligosaccharide mass, consistent with oxidation at position C4 of the glycosidic bond. It was notable that the activity of TaAA9A on the native cellulose fibres afforded C4 oxidation products whereas in previous studies2,25 the action of the same enzyme on PASC gave C1 (or mixed C1/C4) oxidation—the reason for the difference is not clear but is likely related to the macroscopic structure of the crystalline cellulose fibrils guiding the positioning of the enzyme on the substrate surface (see later). This observation highlights the differences that can emerge when studying LPMOs on non-natural substrates as compared to the natural ones. For the purposes of the study herein, the observed activity of LPMOs on celery-derived cellulose fibres, especially the site-selective C4 oxidation, demonstrates that celery fibres are a substrate for both enzymes employed in this study, and that a substrate–LPMO interaction must form upon the addition of the enzyme to the fibres. Taking advantage of this feature we then performed angle-dependent studies of the enzyme-soaked fibres within an X-band EPR spectrometer. The objective of this study was to establish the orientation of the g matrix principal directions with respect to the main axis of the cellulose fibrils, from which a determination of the orientation of the enzyme on the surface of the fibril might be made.
Semi-orientated CW-EPR spectroscopy
CW-EPR experiments using the celery fibres were performed to evaluate how the crystallinity of the cellulose induced orientation of the LPMOs within the field of the spectrometer. Such a (partial) orientation could then be detected by collecting the EPR spectra at different angles of the fibres with respect to the magnetic field of the spectrometer (
field of the spectrometer. Such a (partial) orientation could then be detected by collecting the EPR spectra at different angles of the fibres with respect to the magnetic field of the spectrometer ( ). In performing this experiment, it is important to know the orientation of the cellulose fibrils with respect to the celery fibre axis. In this regard, it is known from previous studies that the polysaccharide chains are largely orientated along a single axis which is near-coincident with the long dimension of mature celery fibres (mean angular orientation of cellulose microfibrils with respect to fibre axis is ∼5°).23,26 Furthermore, cellulose within the fibril is present in the cellulose I crystalline form which contains parallel adjacent chains, where the reducing ends of the individual chains lie at one end of the fibre. There are, however, known areas of disorder which complicate or potentially preclude any orientation-dependent EPR studies of LPMO-binding to celery cellulose fibres. Firstly, the fibrils are twisted. This twist necessarily introduces complete disorder in the plane which lies normal to the main axis of the fibre. Moreover, some fibrils in the part of the fibre near the plasma membrane are arranged in a somewhat disordered cross-lamellate structure (akin to a 60–120° herringbone pattern with its 2-fold symmetry axis parallel to the fibre axis) that essentially introduces a second disorder axis to the fibril orientation, which—if the LPMO bound to these fibrils arranged in this way—would essentially create a near-completely disordered system from which little or no orientational information could be extracted.26 Thus, on the assumption that LPMO binding occurs to some extent on the sites of uniaxially ordered cellulose fibrils in the celery fibre, it is expected that any EPR orientation dependence observed experimentally reports only on this aspect of LPMO binding to the fibre. LPMO-binding to the disordered or cross-lamellate regions would give rise to a disordered background to the orientation-dependent part of the EPR signal.
). In performing this experiment, it is important to know the orientation of the cellulose fibrils with respect to the celery fibre axis. In this regard, it is known from previous studies that the polysaccharide chains are largely orientated along a single axis which is near-coincident with the long dimension of mature celery fibres (mean angular orientation of cellulose microfibrils with respect to fibre axis is ∼5°).23,26 Furthermore, cellulose within the fibril is present in the cellulose I crystalline form which contains parallel adjacent chains, where the reducing ends of the individual chains lie at one end of the fibre. There are, however, known areas of disorder which complicate or potentially preclude any orientation-dependent EPR studies of LPMO-binding to celery cellulose fibres. Firstly, the fibrils are twisted. This twist necessarily introduces complete disorder in the plane which lies normal to the main axis of the fibre. Moreover, some fibrils in the part of the fibre near the plasma membrane are arranged in a somewhat disordered cross-lamellate structure (akin to a 60–120° herringbone pattern with its 2-fold symmetry axis parallel to the fibre axis) that essentially introduces a second disorder axis to the fibril orientation, which—if the LPMO bound to these fibrils arranged in this way—would essentially create a near-completely disordered system from which little or no orientational information could be extracted.26 Thus, on the assumption that LPMO binding occurs to some extent on the sites of uniaxially ordered cellulose fibrils in the celery fibre, it is expected that any EPR orientation dependence observed experimentally reports only on this aspect of LPMO binding to the fibre. LPMO-binding to the disordered or cross-lamellate regions would give rise to a disordered background to the orientation-dependent part of the EPR signal.
Thus, on the basis that any orientation dependence of the EPR spectrum arises from LPMO-binding to orientated fibrils laying along the axis of the fibres, bunches of individual celery fibres which had previously been incubated with solutions of LsAA9A were carefully arranged by hand onto three different custom-made quartz supports, each with a different orientation with respect to the long axis of the EPR sample holder (Fig. 2). In each case, the previously identified anterior end of each fibre was positioned on one side of the support—thus maintaining a common cellulose chain direction across all fibres placed on the slide or cell. It is estimated that the angular spread of the orientated fibres was ca. 10° (note that this is the orientation of the fibres, not the cellulose fibrils). The quartz supports with the three different orientations of fibres were then placed within the sample cavity of an X-band EPR spectrometer and individual spectra were collected between 0° and 180° in 10° steps with respect to the rotation around the long axis of the sample holder and the  field of the spectrometer.
field of the spectrometer.
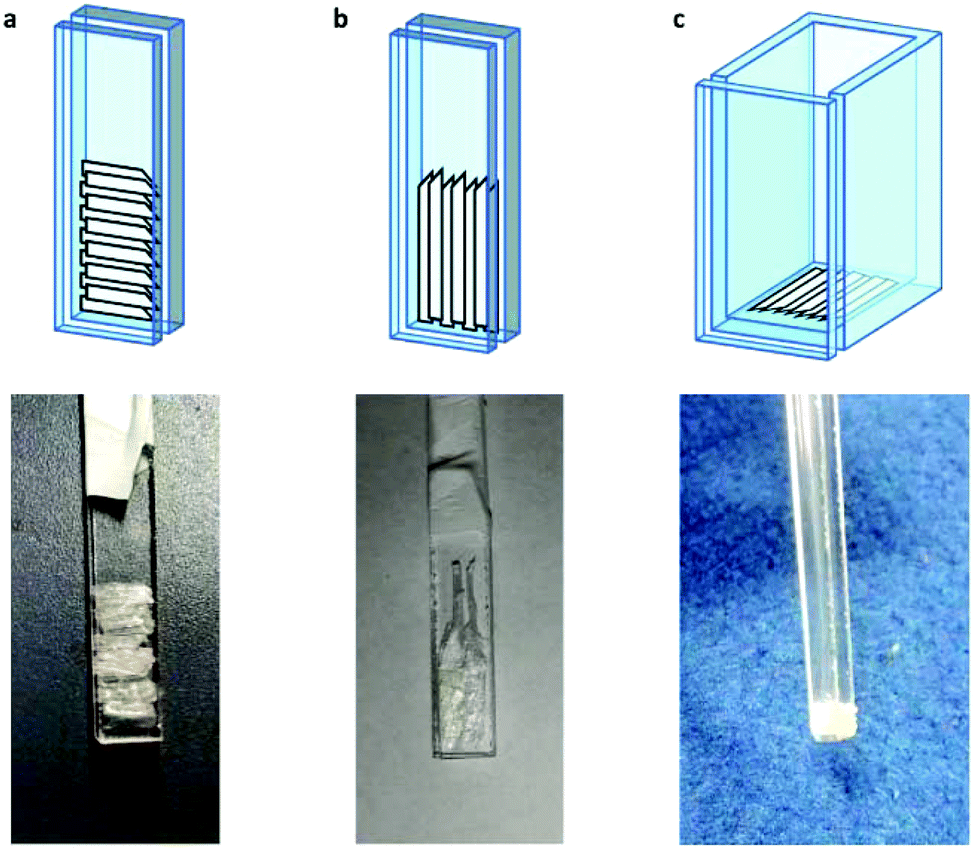 | ||
| Fig. 2 Schemes and corresponding pictures of the cellulose fibres in the custom made quartz supports. | ||
For samples arranged in the configuration depicted in Fig. 2b no angle dependent changes were observed in the Cu EPR spectrum collected at 165 K (Fig. S4a†). For the samples with the quartz slides arranged as shown in Fig. 2a and c, EPR spectra collected from 0° to 180° in 10° intervals showed clear angle-dependent changes, with an increase in intensity of the z region of the spectrum and concomitant decrease of the y region over the 0° to 90° rotation (Fig. 3 and Fig. S4b, respectively, and Movie S2†). The observed changes in intensity resemble the EPR spectra of previously reported uniaxially oriented samples,27 confirming the presence of a preferential axis of orientation. To further confirm that the observed spectra arise from LsAA9A directly interacting with the cellulose, the spin Hamiltonian parameters derived from fitting the spectra were found to match closely those of the substrate-bound species, which has been previously shown to differ markedly from the substrate-free resting state of LsAA9A.5 Notwithstanding the clear orientation-dependence, the EPR spectra also indicated that some fraction of the protein was randomly oriented with respect to the fibres. As the cellulose fibres had been washed with buffer after incubation with LsAA9A to remove possible enzyme bound to soluble oligosaccharides and unbound protein, the portion of non-oriented sample was attributed to some intrinsic disorder within the fibres and their unstructured hemicellulose content.
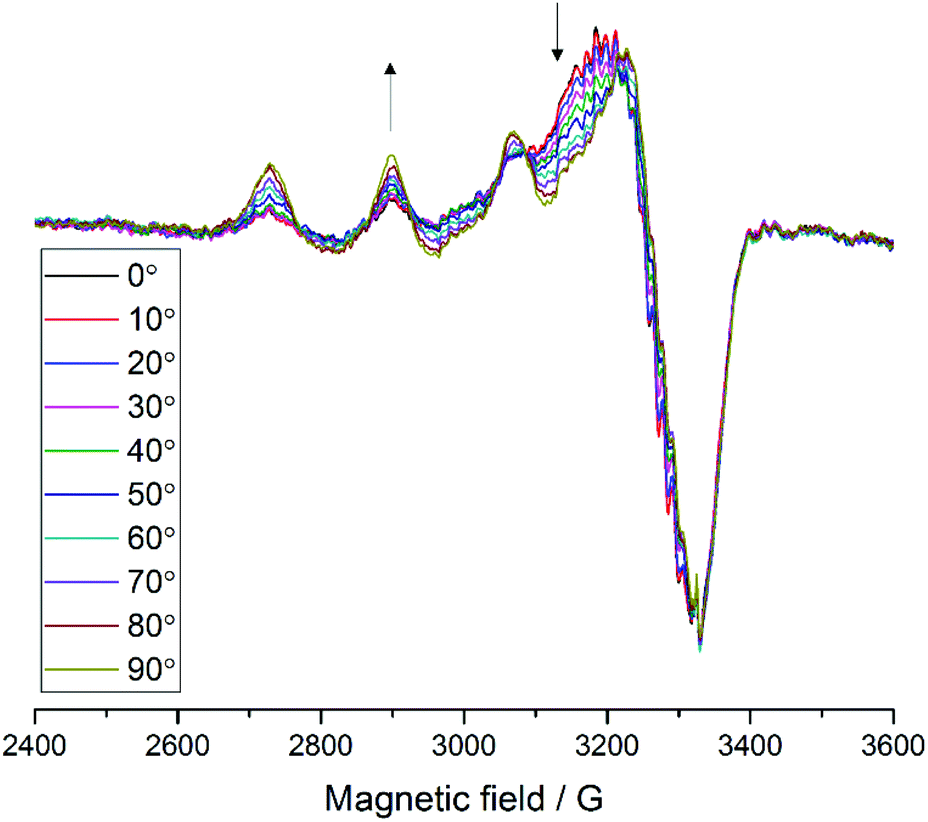 | ||
Fig. 3 EPR spectra of LsAA9A on celery fibres in the arrangement shown in Fig. 2a, collected between 0° and 90° with respect to the static magnetic field, where 0° represent the position with the long axis of the fibres parallel to  . The arrows indicate the direction of the change of signal intensity over the rotation. . The arrows indicate the direction of the change of signal intensity over the rotation. | ||
As a control experiment, no angle-dependent changes were observed when a solution of CuCl2 was used with the support, thereby excluding any changes that may have arisen from changes in cavity filling factor upon rotation (Fig. S6†). Additionally, the spectra with the LsAA9A-loaded fibres were recorded with several different spectrometer settings to exclude distortions induced by the presence of the fibres. In a final negative control experiment, an LPMO active on xylan but not cellulose, PcAA14B, was loaded onto the fibres, from which no copper signal in the EPR could be detected even after very long incubation times, showing that the spectra obtained for LsAA9A were not due to physisorption of the LPMO onto the fibres.
In relating the changes in the EPR spectrum of LsAA9A with the molecular details of the LPMO/cellulose interaction, the relative orientations of four sets of axes (‘frames’) need to be ascertained. These frames are the molecular frame, the g matrix frame, the “crystal” frame (where the crystal is considered to be the celery cellulose fibril) and the laboratory frame. Determination of the relative orientation of the molecular frame with the crystal frame is the experimental objective, from which information about the LPMO–cellulose interaction can be determined.
The relative orientation of the crystal and laboratory frames is straightforward and related to the rotation of the support with respect to 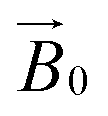 during the experiment. The relative orientation of the g and the crystal frames, instead, is more complex due to the rotational disorder of the cellulose strands along the long axis. As such, any g matrix positioned on the cellulose fibres is fully disordered in two axes. This disorder has to be factored into the subsequent EPR simulations, discussed below, which require the calculation of the effective g value over a restricted set of orientations of the g matrix with respect to the
during the experiment. The relative orientation of the g and the crystal frames, instead, is more complex due to the rotational disorder of the cellulose strands along the long axis. As such, any g matrix positioned on the cellulose fibres is fully disordered in two axes. This disorder has to be factored into the subsequent EPR simulations, discussed below, which require the calculation of the effective g value over a restricted set of orientations of the g matrix with respect to the 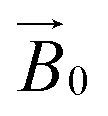 field of the spectrometer. While somewhat complex, a pictorial description of the orientation of the g matrix on the fibre is helpful. The pictorial description is as follows. If the principal directions of the g matrix are positioned where the gz component is perpendicular to the fibre axis, then the disorder induced by the twisting of the fibre is such that the gz component describes a circle (Fig. 4) of orientations. The fibre axis is a normal to the plane of the circle. Depending on which orientation of sample holder is employed, the relative orientation of the plane of this circle with respect to the
field of the spectrometer. While somewhat complex, a pictorial description of the orientation of the g matrix on the fibre is helpful. The pictorial description is as follows. If the principal directions of the g matrix are positioned where the gz component is perpendicular to the fibre axis, then the disorder induced by the twisting of the fibre is such that the gz component describes a circle (Fig. 4) of orientations. The fibre axis is a normal to the plane of the circle. Depending on which orientation of sample holder is employed, the relative orientation of the plane of this circle with respect to the 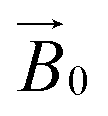 -field of the spectrometer varies upon orientation of the sample holder within the spectrometer (Fig. 4). Therefore, in the “vertical” arrangement of the fibres (Fig. 2b and 4b), the gz component lies along the
-field of the spectrometer varies upon orientation of the sample holder within the spectrometer (Fig. 4). Therefore, in the “vertical” arrangement of the fibres (Fig. 2b and 4b), the gz component lies along the 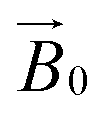 direction and does not vary during the 0° to 90° rotation of the sample holder, thus the contribution of gz to geff for any single orientation of the LPMO does not change. For a uniaxially orientated sample this lack of change in contribution manifests itself as no change in intensity of the EPR spectrum in the parallel region upon rotation. When instead the fibres are arranged as in Fig. 2a and c, the gz contribution to geff changes depending on the angle that the fibres make with the magnetic field over the 0° to 90° rotation of the fibres (Fig. 4a). Pictorially, the circle generated by the presence of the disorder axis goes from being almost normal to
direction and does not vary during the 0° to 90° rotation of the sample holder, thus the contribution of gz to geff for any single orientation of the LPMO does not change. For a uniaxially orientated sample this lack of change in contribution manifests itself as no change in intensity of the EPR spectrum in the parallel region upon rotation. When instead the fibres are arranged as in Fig. 2a and c, the gz contribution to geff changes depending on the angle that the fibres make with the magnetic field over the 0° to 90° rotation of the fibres (Fig. 4a). Pictorially, the circle generated by the presence of the disorder axis goes from being almost normal to  at 0° (and hence giving the lowest intensity of the parallel region) to being collinear to it at 90°, producing the highest relative intensity of the parallel region of the spectrum. Such qualitative behaviour is indeed observed in the variation of the EPR spectral intensity for a sample holder in this configuration.
at 0° (and hence giving the lowest intensity of the parallel region) to being collinear to it at 90°, producing the highest relative intensity of the parallel region of the spectrum. Such qualitative behaviour is indeed observed in the variation of the EPR spectral intensity for a sample holder in this configuration.
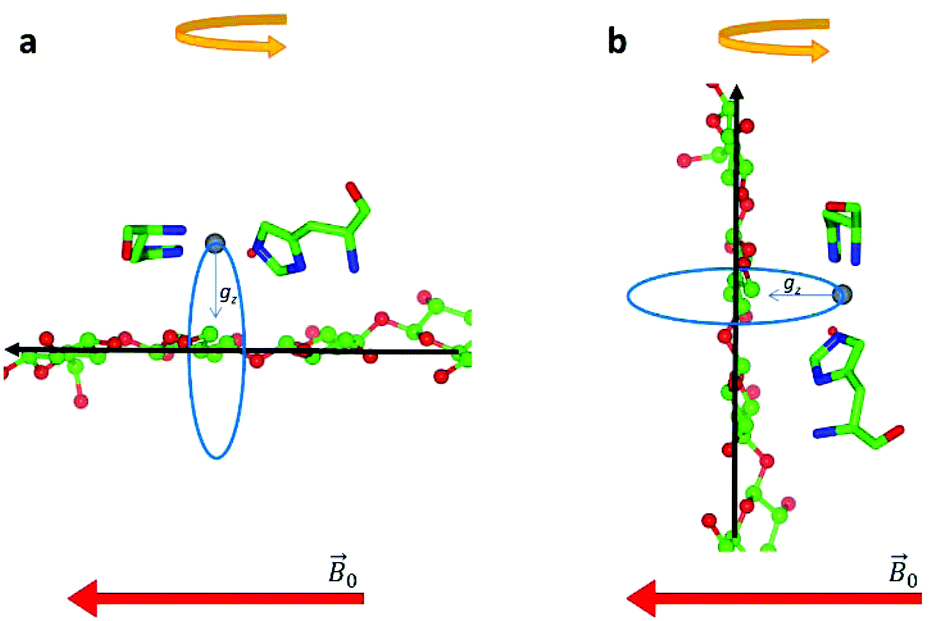 | ||
| Fig. 4 Schematic representation of the disorder within the cellulose fibres. The orientation of the Cu-His brace with respect to a cello-oligosaccharide chain is taken from reported crystal structures (PDB: 5ACI).5 The disorder axis is shown in black, the direction of the static magnetic field in red and the 0°–90° rotation of the fibres during the experiment in yellow. The direction of the gz axis of the Cu g matrix is shown in blue, while the blue circle represents the array of possible orientations of gz due to the disorder axis. (a) Fibre arrangement as in Fig. 2a; (b) fibre arrangement as in Fig. 2b. | ||
Orientation of g matrix and molecular structure
To associate the g matrix orientation with the LPMO structure, it was necessary to align the principal directions of the matrix to the copper active site coordination geometry. Here, a common qualitative strategy is to assume that there is little ligand-field induced mixing between d orbitals and that the coordinating atoms of the histidine brace along with the exogenous ligand (water molecule or chloride ion) define the xy plane of the g matrix. A more robust method is to use Density Functional Theory (DFT) calculations on the active site structure. These were duly performed on a small cluster model of the active site to evaluate the relative orientation of the g matrix and ACu tensor principal components, and with respect to the molecular frame (Fig. 5). The model of the active site was built using the X-ray structure of LsAA9A bound to cellotriose (PDB: 5ACF), with appropriate truncations and constraints (see Experimental for details about methodology). The molecular frame was defined with the origin on the Cu ion, the X axis along the Cu–Nδ (imidazole ring, His1) bond, the Y axis along the Cu–N (amino terminus) bond and Z axis normal to these two vectors.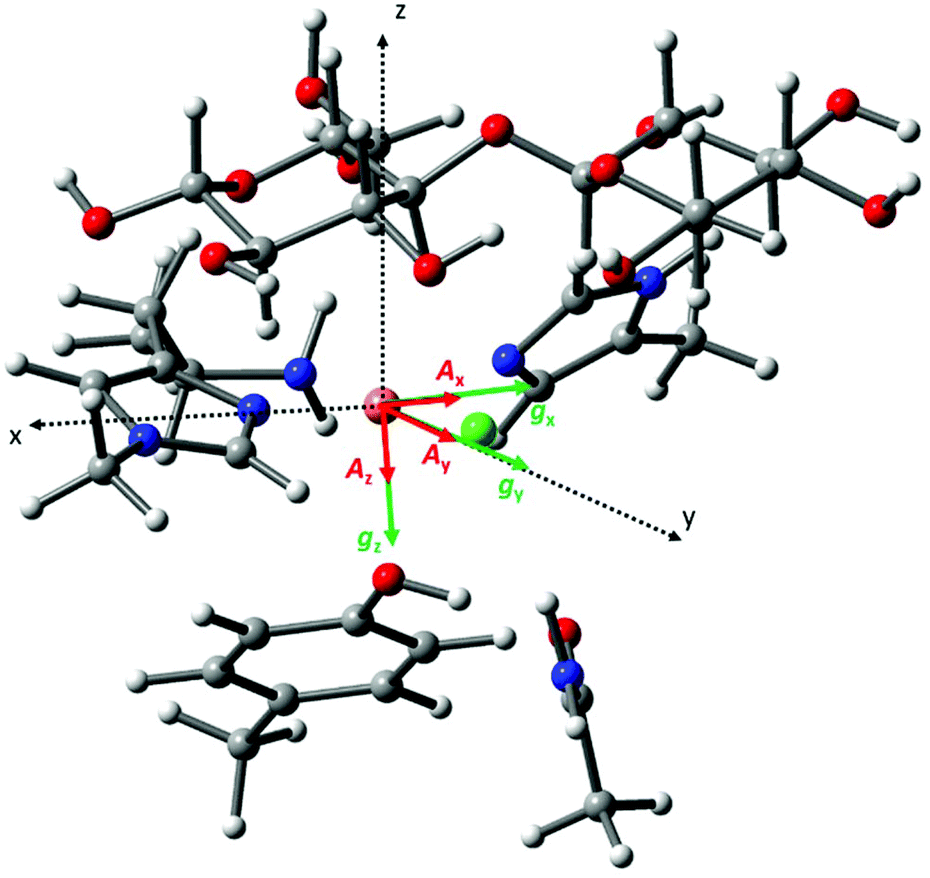 | ||
| Fig. 5 Cartoon representation of the calculated relative orientation of the g (green) and ACu (red) principal components with respect to the molecular frame (black dotted vectors). | ||
The calculated g matrix principal components are close to those expected from the qualitative analysis described above apart from a small rotation (∼7°) along the Y axis with respect to the molecular frame. The calculated ACu hyperfine tensor is essentially co-linear with the g principal components, with a 2.5° rotation along the gz direction. Table S1† reports the eigenvector matrices for the g and ACu principal components. The calculated spin Hamiltonian parameters are in reasonable agreement with the experimental ones, although there is some deviation from the experimental gz and Az, a well know situation in DFT studies of Cu(II) complexes (Table S1†).28
Simulation of spectral data
Methods for simulating the EPR spectra of partially oriented paramagnetic centres have been described previously,27,29 focussing on determining the orientation distribution functions and hence the degree of order in the sample. These methods allow the user to determine the probability of finding a particular molecular axis aligned with the orientation axis in the partially ordered sample. Despite their use however, these methods have often been optimised for a specific system or rely on in-house developed software and so are not broadly available. As such, in our work we chose to use EasySpin30 to simulate our partially oriented data.The objective of the simulations was the determination of the Euler angles that define the relative orientation of the molecular and crystal frames. This was achieved by simulating the spectra collected at 0° and 90° over a range of Euler angles until a satisfactory fit could be obtained to data at both angles. A disorder axis was added in the simulations to account for the rotational disorder along the long axis of the celery cellulose, described above. Using these angles, simulations were then carried out for all orientations of the LPMO-loaded fibres with respect to the  field of the spectrometer by addition of a linear combination of the simulation of the 0° and 90° spectra (40% of the total) to the frozen solution spectrum (for the remaining 60% of the total to account for the non-oriented enzyme). Under these constraints the spectral data and their variance with experimental orientation could be excellently simulated (Fig. S7 and Movie S3†, Table 1). From these simulations, the resulting Euler angles of the relative orientations of the molecular frame with the g matrix frame (Euler angles 1) and the molecular frame with the crystal frame (Euler angles 2) define the orientation of the LPMO protein with the crystal fibril (see below).
field of the spectrometer by addition of a linear combination of the simulation of the 0° and 90° spectra (40% of the total) to the frozen solution spectrum (for the remaining 60% of the total to account for the non-oriented enzyme). Under these constraints the spectral data and their variance with experimental orientation could be excellently simulated (Fig. S7 and Movie S3†, Table 1). From these simulations, the resulting Euler angles of the relative orientations of the molecular frame with the g matrix frame (Euler angles 1) and the molecular frame with the crystal frame (Euler angles 2) define the orientation of the LPMO protein with the crystal fibril (see below).
| LsAA9A | TaAA9A | ||
|---|---|---|---|
| a Due to the disorder axis in the fibres, the first Euler angle (α) had very little effect on the simulations, therefore it could not be determined accurately. The value of −15° was taken from the docking model (vide infra). | |||
| g values | g x | 2.04 | 2.05 |
| g y | 2.06 | 2.07 | |
| g z | 2.23 | 2.27 | |
| A Cu (MHz) | |Ax| | 10 | 75 |
| |Ay| | 77 | 75 | |
| |Az| | 517 | 460 | |
| SHF AN principal values (MHz) | 36, 31, 19 | 38, 36 | |
| SHF ACl principal value (MHz) | 40 | — | |
| A Cu strains (MHz) | 15, 35, 70 | 95, 95, 120 | |
| Euler angles 1 (°) | 0, 7, 0 | 0, 10, 0 | |
| Euler angles 2 (°)a | −15, 90, 45 | −15, 90, 40 | |
| Linewidths (mT) | 0.4, 0.4 | 0.4, 0.4 | |
| Frequency (GHz) | 9.33 | 9.33 | |
The same analysis was carried out for the interaction of TaAA9A with celery fibres. TaAA9A has clear activity on cellulose, but—in contrast to LsAA9A—standard CW-EPR spectra of this enzyme do not show any perturbation at the copper active site upon addition of substrate. When incubated with the celery cellulose fibres in the arrangement depicted in Fig. 2a, the EPR of TaAA9A shows some angle dependent changes (Fig. S8†), although not as prominent as those observed for LsAA9A. The EPR spectra of TaAA9A show the presence of free copper (ca. 20% with respect to the protein concentration, estimated from the first Cu hyperfine peak), the origin of which is unknown, but possibly due to partial protein deterioration caused by long incubation times. Following simulation as described above, the angle-dependent changes observed in the spectra of TaAA9A bound to the celery fibres could be well reproduced using the parameters reported in Table 1 (Fig. S9†). Similar to LsAA9A, the simulations were obtained by summing the frozen solution spectrum of TaAA9A (60%), a linear combination of the simulations at 0° and 90° (for a total fraction of 40%) and the free copper spectrum. It should be noted that the uncertainty in the Euler angles determined for TaAA9A is higher than for LsAA9A due to the overall weaker signal and the presence of free copper. Nevertheless, the second Euler angle in the molecular frame rotation is diagnostic, and only ±10° deviation from the reported angle of 90° could be tolerated in the simulations.
Binding of LPMOs to cellulose fibrils
Analysis of the experimental data shows that the simulations are particularly sensitive to the value of the middle Euler angle (β) of the molecular frame rotation. This angle relates to the tilt (θ, where θ = 90° − β) of the gz axis with respect to a plane normal to the flat surface of the cellulose fibrils and the direction of the glycosidic bonds (Fig. S10†); in other words, the β angle defines the amount of gz component along the long axis (the disorder axis) of the cellulose fibres. When θ = 0° (i.e. β = 90°), gz lies completely on the plane defined above, while a value of 90° (i.e. β = 0°) indicates that gz is normal to such a plane and is parallel to the flat surface of the fibres. For the LPMOs studied herein, only small deviations of β (±10°) from the value of 90° could be tolerated in the simulations. Indeed, this is the essential finding from these experiments, showing that the N–Cu–N direction in the His brace lies parallel to the plane of the sugar rings in the fibre.Under this constraint we therefore examined how the protein could form a complementary fit with the known structure of a cellulose fibril. Accordingly, a 24-chain rectangular model of cellulose Iβ, which has been reported to be the most likely form of crystalline cellulose in celery,23 was generated using the cellulose builder toolkit31 and the crystal faces identified as shown in Fig. S11a.† Docking of the LPMOs was subsequently carried out, guided by the known structure of LsAA9A bound to cellohexaose (5ACI)5 (although the overlay of the cellulose to the cello-oligo showed that the latter presents a twist of the saccharide chain which is not compatible with the extended cellulose crystal, Fig. S11b†). The subsequent 3D models show that both LsAA9A and TaAA9A can only be accommodated at the edge between two crystal faces of the cellulose fibre, rather than on the flat hydrophobic surface (Fig. 6). In particular, the contour of the protein determines the orientation of the His brace with respect to the cellulose chain. For LsAA9A, the gx axis, defined by the N–Cu–N direction, can be either parallel to the sugar rings, if the binding occurs at the edge between the (200)–(010) or the (100)–(020) faces (Fig. S11c and d†), or tilted by about 35° with respect to the sugar rings for binding at the edge between the (200)–(020) or the (100)–(010) faces (Fig. S11e and f†). The former binding positions are in excellent agreement with the enzyme orientation determined by EPR simulations, hence supporting the hypothesis of binding at the (200)–(010) or (100)–(020) edge of the cellulose fibrils (Fig. 6, Fig. S11c, d, Table 1 and Movie S4†). Furthermore, binding on these edges positions any reactive oxygen species, which during the catalytic cycle occupies the site held by a water molecule in the crystal structures (red sphere in Fig. S11†), towards the glycosidic bond. Conversely, the exogenous ligand is directed away from the polysaccharide chain if binding occurs on the (200)–(020) or the (100)–(010) edges. Similar considerations can be made for TaAA9A, where good agreement between the EPR parameters and the structural docking also suggests binding to the same edges as for LsAA9A. We note from this analysis that the plane of the ring of the conserved distal Tyr residue (Tyr203 for LsAA9A and Tyr212 for TaAA9A) is rotated by ca. 30° with respect to the sugar rings in the suggested binding position. This rotation is in contrast with previous findings, which showed the involvement of Tyr203 in the binding of LsAA9A to oligosaccharides.5 Although the discrepancy cannot be rationalised on the basis of the current data, it could be hypothesised that binding of the LPMO disrupts the cellulose network, partially “lifting” the edge chain from the surface of the fibril.
 | ||
| Fig. 6 Representation of binding of LsAA9A on crystalline cellulose derived from the oriented EPR study. The cellulose fibre was generated using the cellulose builder toolkit30 and the image produced in CCP4MG. | ||
Conclusions
Effective utilisation of carbohydrate active enzymes in biomass conversion requires understanding of their detailed interaction with complex, extended polysaccharide surfaces. We have thus developed an experimentally straightforward semi-oriented EPR method to assess the binding of LPMOs to crystalline cellulose using fibres extracted from celery stalks. In exemplifying this method, we have used two cellulose-active LPMOs, LsAA9A and TaAA9A. Both enzymes showed clear angle-dependent changes in their EPR spectra, demonstrating partial orientation of the proteins on the substrate. Furthermore, the data provided the first experimental evidence of cellulose binding for an LPMO that does not show electronic structure changes at the active site upon addition of substrate. Simulation of the EPR spectra and subsequent analysis of the orientation frames guided the 3D modelling of the enzyme binding onto an extended cellulose surface. The orientation of the frames and this the positioning of the LPMO on the cellulose fibril was guided by potential fits determined from modelling. Very good fits with the experimentally derived orientations could only be obtained for binding to the (200)–(010) or (100)–(020) edge of the cellulose fibrils for both enzymes used in this study, thus providing an experimental verification of the interaction of the LPMO with the fibril. This methodology has therefore allowed us to expand the current knowledge of LPMO action on recalcitrant substrates and it can be used as a general procedure to assess the binding of these enzymes to crystalline cellulose.Experimental
General
Ultrapure water (resistivity >18.0 MΩ cm) produced by a Merck Simplicity® water purification system was used in the preparation of all buffers and solutions. Acetone, 37% HCl solution, glacial acetic acid, NaOH and Tween 20 were purchased from Sigma-Aldrich or Fisher Scientific. The pH of the solutions was measured with a Radiometer Analytical ION450® pH-meter calibrated using standard buffer solutions at pH 4.01, 7.00 and 10.01.LPMO preparation
Lentinus similis AA9 (LsAA9A) was prepared as described by Frandsen et al.5 Briefly, the gene encoding LsAA9A was PCR-amplified from genomic DNA of Lentinus similis and cloned in E. coli as described by the patent number reported in ref. 32 using the following forward primer F-P247JK: 5′-ACACAACTGGGGATCCACCATGAAGTACTCCATCCTCGGGCT-3′ and reverse primer R-P247JK: 5′-CCCTCTAGATCTCGAGCCTTGTCGAGCGACTCT ATCCA-3′, containing insertion sites for the vector pDau109 used for cloning.LsAA9A was expressed in Aspergillus oryzae MT3568 as also described by the patent number reported in ref. 32. A transformant producing the recombinant LsAA9A was inoculated in 2 L of Dap-4C medium and incubated at 30 °C for 4 days. Mycelia were removed by filtration and the broth collected for purification. Ammonium sulfate was added to the sterile filtered broth to a conductivity of 200 mSi cm−1 and the pH adjusted to 7.5. The broth was applied to a 50/15 Butyl Toyopearl column (Tosoh Biosciences, Stuttgart, Germany) equilibrated with 25 mM Tris, 1.5 M ammonium sulfate, pH 7.5. The column was washed in the same buffer and eluted with a gradient to 25 mM Tris pH 7.5. Fractions containing LsAA9A were combined and washed with milliQ water by ultrafiltration (10 kDa MWCO, PES filter, Sartorius, Goettingen, Germany) to a conductivity of 1.2 mSi cm−1. The pH was adjusted to 8.0 and applied to a 50/40 Q Sepharose FF column (GE Healthcare, Pittsburgh, USA) equilibrated with 20 mM Tris pH 8.0. The column was washed in the same buffer and the enzyme eluted with a gradient from 0 to 0.5 M sodium chloride. Fractions containing LsAA9A were combined and concentrated by ultrafiltration using VIVASPIN 20 (10 kDa MWCO) spin concentrators. The Apo-protein was incubated with excess CuCl2 and was then buffer exchanged on a HiLoad 16/600 Superdex 75 column (GE Healthcare) into 20 mM MES pH 6, 200 mM NaCl to remove any excess copper. For chloride free samples LsAA9A was copper loaded with an equimolar amount of CuSO4 before being buffer exchanged into 20 mM sodium phosphate buffer pH 6 on a HiLoad 16/600 Superdex 75 column (GE Healthcare).
Thermoascus aurantiacus AA9 (TaAA9A) was expressed and purified as previously reported.1
Celery cellulose fibres preparation
Cellulose fibres were extracted from Apium graveolens (celery) stems following reported literature procedures.22,23 Celery stems were purchased from Waitrose and only the most mature, outer stems were used for cellulose extraction. The celery stems were cut to eliminate the bottom 5 cm and the top 2 cm. With a scalpel, the petioles were scored along the length of the stem at either side of the cellulose strands, so that the collenchyma could be removed from the petiole. The soft tissue around the fibres was peeled off by gently pulling between the finger and thumbnail and the fibres were scored with the scalpel at the top to keep track of the direction of growth of the stems. The strands were kept in water until the process was finished and then dried at room temperature for 20 h. The cellulose strands were placed in acetone for 5 min, rinsed with water for 5 min and immersed in a 1 g L−1 solution of Tween 20 in water for 5 min to dissolve the plasma membrane. The treatment with surfactant was followed by a thorough rinse with water and another acetone wash for 5 min. The strands were dried at room temperature for 16 h, then heated to reflux for 1 h in ca. 100 mL of a 1 M HCl solution, thoroughly washed with water and dried for 16 h. The fibres were soaked in 150 mL of a 0.1 M NaOH solution for 30 min and the pH of the solution was then adjusted to ∼7 using a 20% v/v acetic acid solution. The strands were carefully washed with water, placed individually in straight lines on a large glass watch, dried for 16 h and stored.Activity assays
Celery cellulose fibres prepared as described above were used in activity assays without further purification. Reactions were set up in 1 mL total volume with 2 mg of solid substrate in 10 mM ammonium acetate, pH 6.0, 1 mM ascorbic acid and 1 μM Cu(II)-LsAA9A or Cu(II)-TaAA9A and were incubated at 30 °C rotating overnight. Remaining substrate was removed by centrifugation at 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000g for 5 min and the supernatant used for the analysis. 1 μL of sample was mixed with 2 μL of 10 mg mL−1 2,5-dihydroxybenzoic acid in 50% v/v acetonitrile, 0.1% v/v trifluoroacetic acid in water, on a SCOUT-MTP 384 target plate (Bruker). The spotted samples were then dried under a lamp in air, before being analysed by mass spectrometry on an Ultraflex III matrix-assisted laser desorption ionization–time of flight (MALDI-TOF) instrument (Bruker), as described by Vaaje-Kolstad et al.3
000g for 5 min and the supernatant used for the analysis. 1 μL of sample was mixed with 2 μL of 10 mg mL−1 2,5-dihydroxybenzoic acid in 50% v/v acetonitrile, 0.1% v/v trifluoroacetic acid in water, on a SCOUT-MTP 384 target plate (Bruker). The spotted samples were then dried under a lamp in air, before being analysed by mass spectrometry on an Ultraflex III matrix-assisted laser desorption ionization–time of flight (MALDI-TOF) instrument (Bruker), as described by Vaaje-Kolstad et al.3
MALDI-TOF data are available on request through the Research Data York (DOI: 10.15124/e6bd5772-738f-4108-88c3-9d5f9e65e00e).
Electron paramagnetic resonance (EPR) spectroscopy
Continuous wave (CW) X-band frozen solution EPR spectra were acquired on a Bruker micro EMX spectrometer operating at ∼9.30 GHz, with modulation amplitude of 4 G, modulation frequency 100 kHz and microwave power of 10.02 mW at 165 K. EPR spectra of a 0.2 mM solution of LsAA9A, prepared and copper loaded as described above, were collected in the presence of excess cellohexaose and 200 mM NaCl in 20 mM sodium phosphate buffer pH 6.0 (Fig. S5†).Experiments with cellulose from celery fibres were carried out at X band frequency with the same settings as reported above. The celery fibres were cut into pieces of ca. 5 mm in length for the arrangements shown in Fig. 2a and c, ca. 1 cm for the arrangement in Fig. 2b. The fibres were then incubated with 70 μL of a 0.6 mM solution of LsAA9A in 20 mM MES buffer pH 6, 200 mM NaCl or 20 mM sodium phosphate buffer pH 6, 200 mM NaCl for 4 h. Alternatively, fibres were incubated with 60 μL of a 0.5 mM solution of TaAA9A in 20 mM sodium phosphate pH 6 for 18 h or with 60 μL of 0.45 mM solution of PcAA14B in 50 mM sodium acetate buffer pH 5 for 18 h. After the incubation, the supernatant solution of protein was removed and 60 μL of the appropriate buffer added to the vial containing the fibres. The cellulose fibres were carefully arranged on the custom made quartz support making sure that the long axes of the pieces were aligned. For reference, in the arrangement shown in Fig. 2a, ca. 40 pieces of cellulose fibres were aligned over 3 to 4 layers. The fibres were then carefully covered with a quartz slide, ensuring that the orientation was maintained in the process. The supports with the fibres were then inserted in the EPR cavity and the spectra recorded at 165 K in 5° or 10° intervals, measuring the angles with a protractor.
To make sure that the changes were not due to the shape of the flat cell, controls were performed adding a few microliters of a 5 mM solution of CuCl2 in water with 10% v/v glycerol between the quartz slides. No angle dependency was detected in the spectra (Fig. S6†). Furthermore, the spectra with LsAA9A and the celery fibres were recorded using different power settings (2, 5 or 10 mW power) and different modulation amplitude (2 or 6G), showing no distortion (data not shown, but available on request).
Spectral simulations were carried out using EasySpin 5.2.1630 integrated into MATLAB R2017a software on a desktop PC. Simulation parameters are given in Table 1. gz and |Az| values were determined accurately from the absorptions at low field. It was assumed that g and A tensors were axially coincident. The superhyperfine coupling values for the nitrogen atoms could not be determined accurately, although it was noted that satisfactory simulation could only be achieved with the addition of two (for TaAA9A) or three (for LsAA9A) nitrogen atoms with coupling values as reported in Table 1. Analysis of the celery cellulose experiments was carried out using the EasySpin frame reference system, as reported in the EasySpin documentation. Briefly, the lab frame z axis (Lz) was set parallel to the static magnetic field 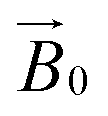 as for convention, while the long axis of the celery fibres was labelled as the crystal z (Cz). The axis laying along the flat surface of the fibres and normal to Cz was considered the crystal y axis (Cy). The molecular frame of the His brace was set in accordance to EPR experiments and the DFT calculations, with the x axis (Mx) along the His-Cu-His direction, the y axis (My) along the NH2−Cu–Cl direction and the z axis (Mz) orthogonal to these two. Spectra and simulations are shown in Fig. S7 and Movie S3.†
as for convention, while the long axis of the celery fibres was labelled as the crystal z (Cz). The axis laying along the flat surface of the fibres and normal to Cz was considered the crystal y axis (Cy). The molecular frame of the His brace was set in accordance to EPR experiments and the DFT calculations, with the x axis (Mx) along the His-Cu-His direction, the y axis (My) along the NH2−Cu–Cl direction and the z axis (Mz) orthogonal to these two. Spectra and simulations are shown in Fig. S7 and Movie S3.†
Raw EPR data are available on request through the Research Data York (DOI: 10.15124/e6bd5772-738f-4108-88c3-9d5f9e65e00e).
DFT methods
Geometry optimization was performed using the Gaussian 09 software package.33 The starting point of the geometry optimized structure was obtained from the coordinates of the substrate-bound LsAA9A crystal structure (PDB: 5ACF),5 and included four amino acid residues (His1, His78, Gln162, and Tyr164) with the following modifications: carbonyl of His1 is replaced by a methyl group, His78 and Tyr164 are truncated by methyl substitution of the Cβ, Gln162 is truncated by methyl substitution of the Cγ. The equatorial Cl− ligand was retained, together with two glucose units of the cellotriose substrate, and the ‘pocket’ water molecule that is hydrogen bonded to the amino terminus and the substrate. Asterisks in Fig. S12† indicate atoms kept frozen during the optimization. The structure was optimized at the density functional theory level of theory, using the uB3LYP hybrid functional and the Ahlrichs's Def2-TZVP on the Cu(II), the C and N atoms of the two coordinating imidazole rings, the N atom of the amino terminus, the O atom of the axial Tyr and on the halide ion. Ahlrichs's Def-2 SVP was used on all the remaining atoms.34,35 Dispersion correction were included using Grimme's D3 method (GD3),36 as implemented in Gaussian 09. Solvation effects were included with the polarized continuum model as implemented in Gaussian 09 with a dielectric constant of 4.0. Selected distances and angles for the Cu coordination sphere in the optimized geometry are reported in Table S2.† The atomic coordinates list of the geometry optimized model is reported in the appendix (ESI†).All EPR properties calculations were done using the ORCA 4.0 program37 at the DFT level of theory. The integration grid was kept large thorough all the calculations (AngularGrid = 7 for all the atoms and IntAcc = 7 on the Cu(II) ion) to ensure that the core density was correctly described. The B3LYP functional with 38% of Hartree–Fock exchange was used, which, for this model, led to somewhat better EPR property predictions, with respect to the standard B3LYP. The CP(PPP)38 basis set was used for the Cu ion, and the IGLO-III basis set39 for the coordination N of the amino terminus, the C and N atoms of the two imidazole rings and the halide ion. All the remaining atoms were described with the Def2-SVP basis set. Solvation effects were included with the conductor-like polarized continuum model (C-PCM) as implemented in ORCA, with a dielectric constant of 4.0.
The g tensor was calculated through the solution of the coupled perturbed Kohn–Sham equations, as implemented in ORCA. The origin was chosen as the centre of the electronic charge. The calculations include the relativistic mass correction, diamagnetic spin–orbit, and paramagnetic spin orbit terms. The hyperfine coupling calculations include the Fermi-contact term, the spin-dipolar contribution and the spin–orbit coupling correction (SOC) for the Cu(II) and the halides ion. The calculation of the nitrogen hyperfine tensors only included the first order terms, since SOC corrections are small for light ligand nuclei.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank Dr Michael Jarvis for helpful discussions, Mrs Abigail Mortimer and Mr Stuart Murray for making the EPR cells and Dr Jean-Guy Berrin for kindly providing PcAA14B for control experiments. LC, GJD and PHW gratefully acknowledge the support of the UK's Biotechnology and Biological Sciences Research Council (BBSRC) through grants BB/L001926/1 and BB/L021633/1. LC thanks the EPSRC National EPR Facility and Service (NS/A000055/1) for funding. GJD is supported by the Royal Society “Ken Murray” Research professorship. AP and PHW gratefully thank the Lesley Wild Scholarship for funding. We thank an anonymous reviewer for her/his comments on the structure of celery fibrils, which added considerably to our analysis. We dedicate the work described in this manuscript to Professor Robin Perutz: mentor, colleague, scientist and friend.Notes and references
- P. V. Harris, D. Welner, K. C. McFarland, E. Re, J.-C. Navarro Poulsen, K. Brown, R. Salbo, H. Ding, E. Vlasenko, S. Merino, F. Xu, J. Cherry, S. Larsen and L. Lo Leggio, Biochemistry, 2010, 49, 3305–3316 CrossRef CAS PubMed
.
- R. J. Quinlan, M. D. Sweeney, L. Lo Leggio, H. Otten, J.-C. N. Poulsen, K. S. Johansen, K. B. R. M. Krogh, C. I. Jørgensen, M. Tovborg, A. Anthonsen, T. Tryfona, C. P. Walter, P. Dupree, F. Xu, G. J. Davies and P. H. Walton, Proc. Nat. Acad. Sci. U. S. A., 2011, 108, 15079–15084 CrossRef CAS PubMed
- G. Vaaje-Kolstad, B. Westereng, S. J. Horn, Z. Liu, H. Zhai, M. Sørlie and V. G. H. Eijsink, Science, 2010, 330, 219–222 CrossRef CAS PubMed
- L. Ciano, G. J. Davies, W. B. Tolman and P. H. Walton, Nat. Catal., 2018, 1, 571–577 CrossRef CAS
- K. E. H. Frandsen, T. J. Simmons, P. Dupree, J.-C. N. Poulsen, G. R. Hemsworth, L. Ciano, E. M. Johnston, M. Tovborg, K. S. Johansen, P. von Freiesleben, L. Marmuse, S. Fort, S. Cottaz, H. Driguez, B. Henrissat, N. Lenfant, F. Tuna, A. Baldansuren, G. J. Davies, L. Lo Leggio and P. H. Walton, Nat. Chem. Biol., 2016, 12, 298 CrossRef CAS PubMed
- A. Levasseur, E. Drula, V. Lombard, P. M. Coutinho and B. Henrissat, Biotechnol. Biofuels, 2013, 6, 41 CrossRef CAS PubMed
- G. R. Hemsworth, B. Henrissat, G. J. Davies and P. H. Walton, Nat. Chem. Biol., 2014, 10, 122–126 CrossRef CAS PubMed
- V. V. Vu, W. T. Beeson, E. A. Span, E. R. Farquhar and M. A. Marletta, Proc. Nat. Acad. Sci. U. S. A., 2014, 111, 13822–13827 CrossRef CAS PubMed
- L. Lo Leggio, T. J. Simmons, J. C. Poulsen, K. E. Frandsen, G. R. Hemsworth, M. A. Stringer, P. von Freiesleben, M. Tovborg, K. S. Johansen, L. De Maria, P. V. Harris, C. L. Soong, P. Dupree, T. Tryfona, N. Lenfant, B. Henrissat, G. J. Davies and P. H. Walton, Nat. Commun., 2015, 6, 5961 CrossRef CAS PubMed
- M. Couturier, S. Ladevèze, G. Sulzenbacher, L. Ciano, M. Fanuel, C. Moreau, A. Villares, B. Cathala, F. Chaspoul, K. E. Frandsen, A. Labourel, I. Herpoël-Gimbert, S. Grisel, M. Haon, N. Lenfant, H. Rogniaux, D. Ropartz, G. J. Davies, M.-N. Rosso, P. H. Walton, B. Henrissat and J.-G. Berrin, Nat. Chem. Biol., 2018, 14, 306 CrossRef CAS PubMed
- F. Sabbadin, G. R. Hemsworth, L. Ciano, B. Henrissat, P. Dupree, T. Tryfona, R. D. S. Marques, S. T. Sweeney, K. Besser, L. Elias, G. Pesante, Y. Li, A. A. Dowle, R. Bates, L. D. Gomez, R. Simister, G. J. Davies, P. H. Walton, N. C. Bruce and S. J. McQueen-Mason, Nat. Commun., 2018, 9, 756 CrossRef PubMed
- C. Filiatrault-Chastel, D. Navarro, M. Haon, S. Grisel, I. Herpoël-Gimbert, D. Chevret, M. Fanuel, B. Henrissat, S. Heiss-Blanquet, A. Margeot and J.-G. Berrin, Biotechnol. Biofuels, 2019, 12, 55 CrossRef PubMed
- T. J. Simmons, K. E. H. Frandsen, L. Ciano, T. Tryfona, N. Lenfant, J. C. Poulsen, L. F. L. Wilson, T. Tandrup, M. Tovborg, K. Schnorr, K. S. Johansen, B. Henrissat, P. H. Walton, L. Lo Leggio and P. Dupree, Nat. Commun., 2017, 8, 1064 CrossRef CAS PubMed
- G. Courtade, Z. Forsberg, E. B. Heggset, V. G. H. Eijsink and F. L. Aachmann, J. Biol. Chem., 2018, 293, 13006–13015 CrossRef CAS PubMed
- F. L. Aachmann, M. Sørlie, G. Skjåk-Bræk, V. G. H. Eijsink and G. Vaaje-Kolstad, Proc. Nat. Acad. Sci. U. S. A., 2012, 109, 18779–18784 CrossRef CAS PubMed
- B. Wang, E. M. Johnston, P. Li, S. Shaik, G. J. Davies, P. H. Walton and C. Rovira, ACS Catal., 2018, 8, 1346–1351 CrossRef CAS
- B. Wang, P. H. Walton and C. Rovira, ACS Catal., 2019, 9, 4958–4969 CrossRef CAS PubMed
- L. Bertini, R. Breglia, M. Lambrughi, P. Fantucci, L. De Gioia, M. Borsari, M. Sola, C. A. Bortolotti and M. Bruschi, Inorg. Chem., 2018, 57, 86–97 CrossRef CAS PubMed
- V. V. Vu, J. A. Hangasky, T. C. Detomasi, S. J. W. Henry, S. T. Ngo, E. A. Span and M. A. Marletta, J. Biol. Chem., 2019, 294, 12157–12166 CrossRef CAS PubMed
- B. Bissaro, I. Isaksen, G. Vaaje-Kolstad, V. G. H. Eijsink and Å. K. Røhr, Biochemistry, 2018, 57, 1893–1906 CrossRef CAS PubMed
- A. S. Borisova, T. Isaksen, M. Dimarogona, A. A. Kognole, G. Mathiesen, A. Várnai, Å. K. Røhr, C. M. Payne, M. Sørlie, M. Sandgren and V. G. H. Eijsink, J. Biol. Chem., 2015, 290, 22955–22969 CrossRef CAS PubMed
- A. Šturcová, I. His, D. C. Apperley, J. Sugiyama and M. C. Jarvis, Biomacromolecules, 2004, 5, 1333–1339 CrossRef PubMed
- L. H. Thomas, V. T. Forsyth, A. Šturcová, C. J. Kennedy, R. P. May, C. M. Altaner, D. C. Apperley, T. J. Wess and M. C. Jarvis, Plant Physiol., 2013, 161, 465–476 CrossRef CAS PubMed
- M. Pauly, P. Albersheim, A. Darvill and W. S. York, Plant J., 1999, 20, 629–639 CrossRef CAS PubMed
- D. M. Petrović, B. Bissaro, P. Chylenski, M. Skaugen, M. Sørlie, M. S. Jensen, F. L. Aachmann, G. Courtade, A. Várnai and V. G. H. Eijsink, Protein Sci., 2018, 27, 1636–1650 CrossRef PubMed
- D. Chen, L. D. Melton, D. J. McGillivray, T. M. Ryan and P. J. Harris, Planta, 2019, 250, 1819–1832 CrossRef CAS PubMed
- J. C. Swartz, B. M. Hoffman, R. J. Krizek and D. K. Atmatzidis, J. Magn. Reson., 1979, 36, 259–268 CAS
-
F. Neese, A critical evaluation of DFT, including time-dependent DFT, applied to bioinorganic chemistry, 2006 Search PubMed
- A. K. Vorobiev and N. A. Chumakova, J. Magn. Reson., 2005, 175, 146–157 CrossRef CAS PubMed
- S. Stoll and A. Schweiger, J. Magn. Reson., 2006, 178, 42–55 CrossRef CAS PubMed
- T. C. F. Gomes and M. S. Skaf, J. Comput. Chem., 2012, 33, 1338–1346 CrossRef CAS PubMed
-
Novozymes A/S, WO2014066141A2, 2014
-
M. J. T. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Revision D.01, 2016 Search PubMed
- A. Schäfer, C. Huber and R. Ahlrichs, J. Chem. Phys., 1994, 100, 5829–5835 CrossRef
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed
- F. Neese, WIREs Comput. Mol. Sci., 2018, 8, e1327 Search PubMed
- F. Neese, Inorg. Chim. Acta, 2002, 337, 181–192 CrossRef
-
W. Kutzelnigg, U. Fleischer and M. Schindler, The IGLO-Method: Ab-initio Calculation and Interpretation of NMR Chemical Shifts and Magnetic Susceptibilities, in Deuterium and Shift Calculation, NMR basic Principles and Progress, ed. P. Diehl, Springer, Berlin, Heidelberg, 1991, vol. 23, pp. 165–262 Search PubMed
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9dt04065j |
| ‡ Current address: School of Chemistry, University of Nottingham, University Park, Nottingham, NG7 2RD, UK. |
| This journal is © The Royal Society of Chemistry 2020 |