
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
From plant to probe: semi-synthesis of labelled undecaprenol analogues allows rapid access to probes for antibiotic targets†
Rachel V. K.
Cochrane
 a,
Francesca M.
Alexander
a,
Coilín
Boland
b,
Susan K.
Fetics
b,
Martin
Caffrey
b and
Stephen A.
Cochrane
*a
a,
Francesca M.
Alexander
a,
Coilín
Boland
b,
Susan K.
Fetics
b,
Martin
Caffrey
b and
Stephen A.
Cochrane
*a
aSchool of Chemistry and Chemical Engineering, Queen's University Belfast, David Keir Building, Stranmillis Road, Belfast, UK BT9 5AG. E-mail: s.cochrane@qub.ac.uk
bSchool of Medicine and School of Biochemistry and Immunology, Trinity Biomedical Sciences Institute, Trinity College Dublin, 152-160 Pearse Street, Dublin 2, D02 R590, Ireland
First published on 29th June 2020
Abstract
Undecaprenol-containing glycolipids (UCGs) are essential precursors of bacterial glycopolymers and glycoproteins. We report a novel semi-synthetic strategy to prepare labelled UCGs directly from undecaprenol. This one-size-fits-all approach offers a concise and efficient method for obtaining labelled-UCGs, which will allow new mechanistic studies and inhibitor screens to be performed on novel antibiotic targets.
The rapid emergence of multidrug resistant (MDR) bacteria is one of the biggest challenges facing our generation. By 2050 it could result in 300 million deaths and cost the global economy over $100 trillion.1 Undecaprenol-containing glycolipids (UCGs) are found in bacteria, where they are used as substrates to synthesize glycopolymers, such as peptidoglycan (PG) and lipopolysaccharide (LPS),2,3 and glycoproteins (Fig. 1).4,5 Therefore, UCGs and the enzymes that process them are excellent antibiotic targets.6 UCGs are synthesized on the cytoplasmic side of the membrane by phosphotransferases and glycosyltransferases,2,4,7 before being flipped8–11 across the membrane for further processing. Many different UCGs are found in bacteria, and labelled UCGs are highly valuable probes for studying enzyme mechanism and screening antimicrobial compounds.9,11–13 Common to every UCG is the undecaprenol motif. This is an excellent modification position to introduce chemical labels, as a single labelled undecaprenol analogue can be used to synthesize any UCG by chemical or enzymatic synthesis. Previous efforts towards labelled undecaprenol derivatives have relied on either total chemical synthesis or chemoenzymatic synthesis (Fig. 1). Wong and co-workers previously described the total synthesis of a labelled hexaprenol analogue.12 This approach is laborious, requiring >25 synthetic steps, utilizes air- and moisture-sensitive reactions and gives low overall yields. Troutman and co-workers have devised a chemoenzymatic approach.14–17 Although shorter than total chemical synthesis, labelled intermediates must still be synthesized (usually 6–8 steps) and the use of an enzyme may limit substrate scope and reaction scale. Thus, alternative methods for the preparation of labelled-undecaprenol analogues are still needed. We envisioned an improved method to prepare labelled undecaprenol analogues could involve semi-synthesis, starting from isolated undecaprenol. Bacterial undecaprenol, which has the (Z8, E2, ω)-configuration, is present in low amounts and is not easily isolated but (Z7, E3, ω)-undecaprenol can be extracted from various plant sources and is the version used in most UCG studies given its easier accessibility.18 Herein, we describe the large-scale extraction of undecaprenol from bay leaves and its derivatization into novel labelled analogues.
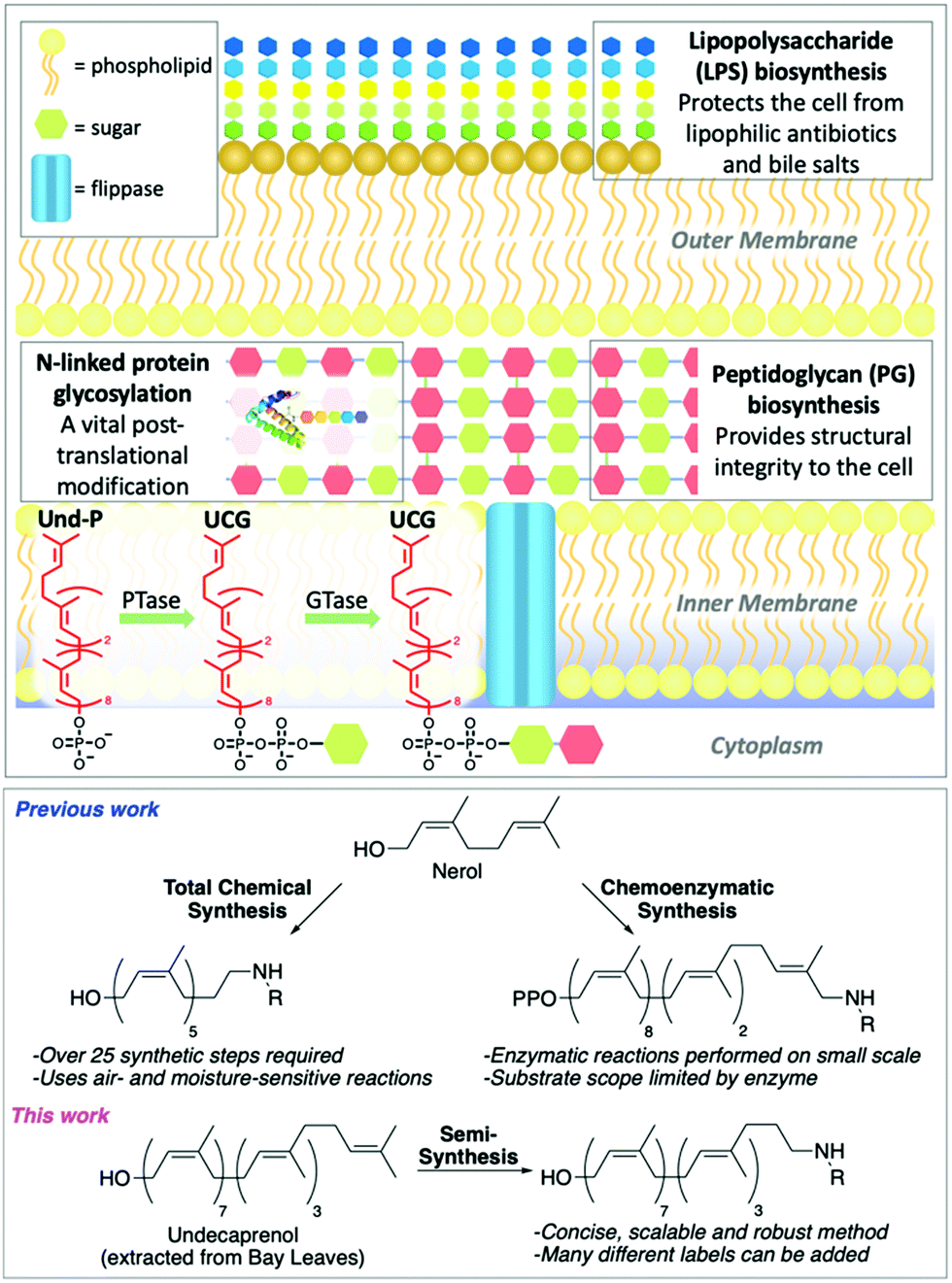 | ||
| Fig. 1 Top: Cross section of Gram-negative bacterial cell showing key glyco-polymers and -proteins assembled using UCGs. Conversion of undecaprenyl phosphate (Und-P) to different UCGs by phosphotransferases (PTases) and glycosyltransferases (GTases) is shown, with the common undecaprenyl tail in red. Bottom: Previous methods to prepare labelled polyprenols and our novel semi-synthetic approach. | ||
The semi-synthesis of labelled undecaprenol analogues requires access to multigram quantities of undecaprenol. Although plant undecaprenol (1) is commercially available, it is very expensive (∼$100 per mg). Previous reports have described the isolation of undecaprenol on a small scale.19 In our optimized approach, we start by sequentially extracting two portions (900 g each) of powdered bay leaves by Soxhlet extraction (Fig. S1, ESI†), each for 48 h. Bay leaf powder is cheap (<$15 for 1 kg) and widely available. A methanolysis step is then performed, which is reported to improve polyprenol yields and remove phenolic impurities (Scheme 1).20 A key modification that allows this process to be performed on a larger scale is to only partially purify isolated undecaprenol. Subsequent acetylation of this enriched undecaprenol with Ac2O in pyridine, followed by column chromatography, then yields pure undecaprenyl acetate (Und-OAc) (2) (Fig. S2, ESI†). This material can be deacetylated back to undecaprenol or used as a starting material to prepare labelled analogues. Using this optimized procedure, we routinely obtained >10 g of Und-OAc per extraction.
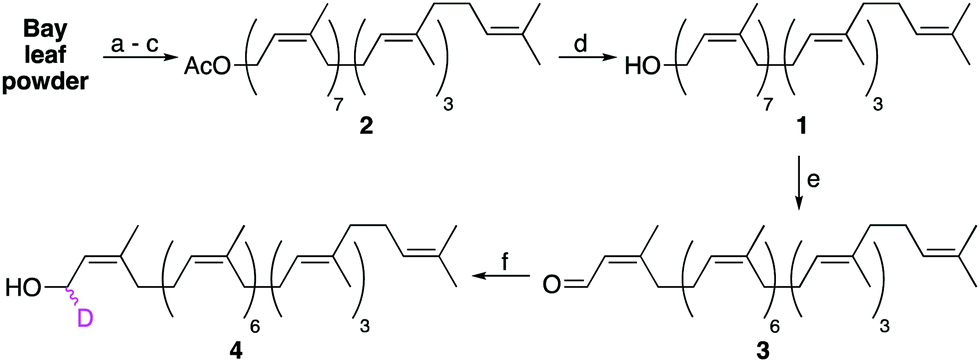 | ||
| Scheme 1 Extraction of undecaprenol (1) from bay leaf powder and isotopic labelling of the α-isoprene unit. Conditions: (a) Soxhlet extraction, petroleum ether (PE) 48 h per batch; (b) K2CO3, PE/MeOH, 72 h, RT; (c) Ac2O, pyridine, 18 h, RT; (d) K2CO3, THF/MeOH, 18 h, RT, 96%; (e) MnO2, CH2Cl2, 24 h, RT, 80%; (f) NaBD4, CeCl3.7H2O, MeOH, 5 min, 0 °C, 88%. | ||
Upon considering the structure of undecaprenol, we identified the α-isoprene unit as the most accessible position to install a chemical label, owing to the directing effect of the hydroxyl group. However, we were aware that labelling here could prove problematic for later enzymatic processing, as this is where the carbohydrate head-group is attached during UCG synthesis. Therefore, only isotopic labelling at this position was considered. If a deuterium label could be incorporated at the α-position, the resulting d1-undecaprenol (4) could be used to synthesize deuterated Und-P/PP or UCGs. Such compounds would then enable mechanistic studies on a host of enzymes using kinetic isotope effects or deuterium NMR. To prepare d1-undecaprenol (4), allylic oxidation of undecaprenol (1) with MnO2 was first performed to yield aldehyde 3. A subsequent Luche Reduction then afforded d1-undecaprenol (4) in good yields.
We next turned our attention to alternative labelling sites. An excellent labelling position would be the ω-isoprene unit, which is unlikely to interfere with later enzymatic processing. A terminal epoxidation of short chain polyprenols using N-bromosuccinimide (NBS) and K2CO3/MeOH was previously reported,21,22 therefore we investigated whether this approach could be translated to the much larger undecaprenol scaffold (Scheme 2). Remarkably, treatment of Und-OAc (2) with NBS in THF/H2O at 0 °C, followed by elimination and re-acetylation yielded ω-epoxide 5 in 42% yield over 3 steps (Scheme 2). ω-Epoxidation was confirmed by 2D-NMR, with clear 1H–13C HMBC correlations visible between the epoxide CH (δC 64.3 ppm) and ω-methyl groups (δH 1.29 & 1.25 ppm) (Fig. S3, ESI†).
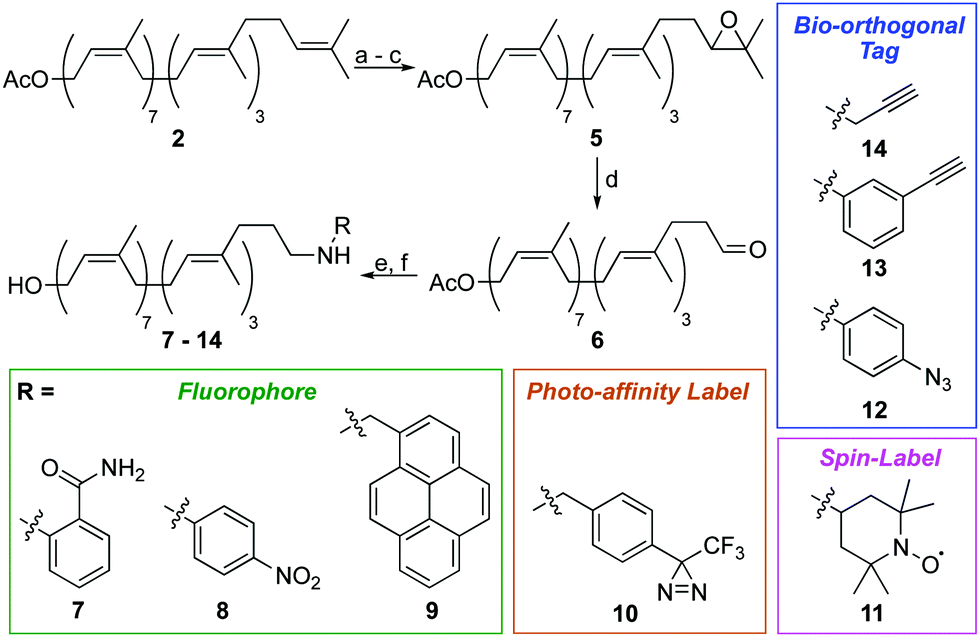 | ||
| Scheme 2 Synthesis of ω-labelled undecaprenol using reductive amination. Conditions: (a) NBS, THF/H2O, 6 h, 0 °C; (b) K2CO3, THF/MeOH, 18 h, RT; (c) Ac2O, DMAP, DIPEA, 16 h, RT, 42% over 3 steps; (d) HIO4, THF/H2O, 4 h, 0 °C to RT, 94%; (e) R-NH2, NaBH(OAc)3, AcOH, CH2Cl2 or ClCH2CH2Cl, 18 h, RT; (f) K2CO3, THF/MeOH, 18 h, RT, yields from 5: 46% (7), 68% (8), 39% (9), 38% (10), 42% (11), 24% (12), 35% (13) and 41% (14). | ||
Epoxide 5 was then converted to ω-aldehyde 6 in excellent yield by treatment with HIO4 in THF/H2O. From aldehyde 6, we envisaged the use of Wittig reactions to label the ω-position but despite our best efforts these attempts were low yielding and gave complex mixtures, making purification difficult. We therefore investigated reductive amination as an alternative labelling strategy. Using this approach, we successfully installed a variety of chemical labels on undecaprenol. Fluorophores, including 2-aminobenzamide (7), 4-nitroaniline (8) and pyrene (9) were added. Fluorescent probes can be used in cell-imaging, tracking biomolecule metabolism and for monitoring enzymatic reactions. Fluorescent undecaprenol derivatives have previously been used to study enzymes that process UCGs.7,12,23 Diazirine-tagged undecaprenol derivative 10 was also prepared. The 3-trifluoromethyl-3-phenyldiazirinyl group is an excellent photo-affinity label.24 Photo-affinity probes are often used to identify enzymes that process them, as well as where they make contact with those enzymes.24 Very little is known about the location of the undecaprenyl chain during UCG-processing, therefore diazirine-labelled derivative 10 is an excellent probe for this purpose. Spin-labelled undecaprenol 11 could also be used to study where the undecaprenyl tail interacts with UCG-processing enzymes. Radicals quench the fluorescence of tryptophan in proteins and electron-spin resonance (ESR) has been used extensively to study lipid–protein interactions.25 Bio-orthogonal tags, including azides (12) and alkynes (13–14) were also introduced. Such tags will allow the in vivo labelling of UCGs, as well as the attachment of UCGs to solid-supports for applications such as solid-supported synthesis, determining enzyme-UCG binding affinities and/or high-throughput screening assays.
Other labelling-methods that relied on the SN2 displacement of an ω-alcohol were also explored (Scheme 3). Starting from ω-epoxide 15, sequential oxidative cleavage of the epoxide, followed by alcohol protection as a THP ether and reduction of the ω-aldehyde, yielded ω-alcohol 16 in 51% yield over 3 steps. Mesylation of the ω-alcohol and displacement with either KSAc or NaN3, followed by removal of the THP protecting group, yielded ω-thioacetate 17 and ω-azide 18 in moderate yields. Although successful, this is a less robust method than reductive amination as it requires extra steps and utilizes moisture-sensitive reactions. In contrast, the reductive amination strategy requires just six steps and two purifications by column chromatography.
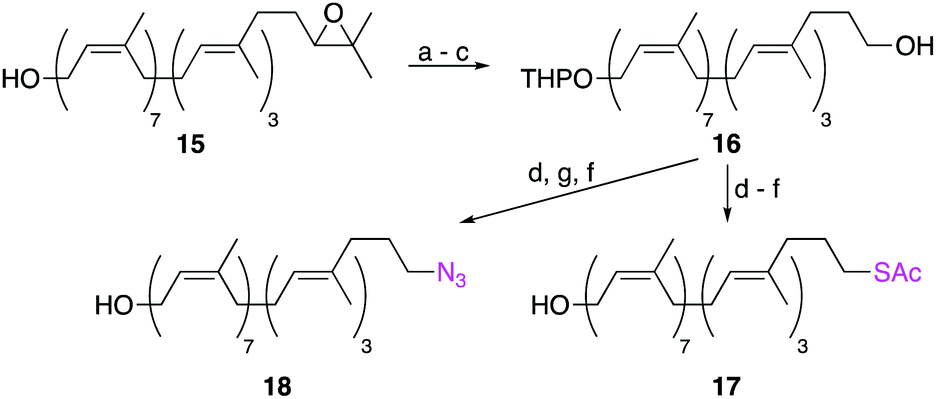 | ||
| Scheme 3 Synthesis of ω-labelled undecaprenol analogues using SN2 displacement reactions. Conditions: (a) HIO4, THF/H2O, 3 h, 0 °C to RT; (b) DHP, PPTS, CH2Cl2, 8 h, RT; (c) NaBH4, MeOH, Et2O, 2 h, RT, 51% over 3 steps; (d) MsCl, NEt3, CH2Cl2, 2 h, 0 °C; (e) KSAc, DMF, 2 h, 60 °C; (f) p-TsOH, THF/MeOH, 18 h, RT; (g) NaN3, DMF, 2 h, 60 °C, yields from 16: 50% (17) and 53% (18). DHP = dihydropyran, PPTS = pyridinium para-toluenesulfonate. | ||
We next proceeded to test if these synthetic modifications were compatible with enzyme function (Table 1). Undecaprenol kinase (UdpK) is an important enzyme in Gram-positive bacteria that converts undecaprenol to Und-P.26 Until now, the simple structure of undecaprenol had hindered the development of substrate-based probes for UdpK. Using a previously described kinase assay,27 the activity of UdpK towards synthetic undecaprenol derivatives was assessed (Table 1). UdpK processed all synthetic analogues with comparable activity to its natural substrate undecaprenol (1). These results corroborate our hypothesis that modification of the ω-isoprene unit is ideal for introducing chemical labels as it is far removed from the UCG head-group attachment point. Interestingly, bishomoallylic ω-alcohol 16, in which the order of the E- and Z-alkenes is reversed, is processed by UdpK. The extra flexibility next to the alcohol likely allows it to adopt the correct conformation in the active site. This result also suggests that internal alkene configuration may not be important for enzymatic processing.
| Analogue | Modification | Activitya [μmol min−1 mg−1] |
|---|---|---|
| a UdpK activity determined colorimetrically using a pyruvate kinase/lactate dehydrogenase coupled assay. See ESI for details. | ||
| 1 | Unmodified | 8.5 ± 0.7 |
| 4 | α-d1 | 9.4 ± 0.5 |
| 7 | ω-(2-Aminobenzamide) | 6.9 ± 0.8 |
| 8 | ω-(4-Nitroaniline) | 9.0 ± 0.9 |
| 9 | ω-(1-Pyrenemethylamine) | 6.2 ± 0.1 |
| 10 | ω-Diazirine | 5.1 ± 0.3 |
| 11 | ω-(4-Amino-TEMPO) | 5.1 ± 0.3 |
| 12 | ω-(4-Azidoaniline) | 9.4 ± 0.2 |
| 13 | ω-(3-Ethynylaniline) | 9.0 ± 0.5 |
| 14 | ω-Propargylamine | 7.1 ± 0.5 |
| 15 | ω-Epoxide | 8.1 ± 0.7 |
| 16 | α-THP ether, ω-alcohol | 3.4 ± 0.3 |
| 17 | ω-Thioacetate | 11.8 ± 0.1 |
| 18 | ω-Azide | 10.8 ± 0.6 |
Finally, to show that synthetic undecaprenol derivatives can be easily converted into labelled UCGs, we prepared a novel deuterated analogue of the peptidoglycan intermediate lipid II, d1-lipid II (19) from d1-undecaprenol (4) (Scheme 4). Lipid II intermediate 21 (Fig. S4, ESI†) was previously prepared for use in other studies.28,29 d1-Undecaprenol was conveniently converted to d1-Und-P (20) in 58% yield over 3 steps. d1-Und-P (20) was then coupled to activated phosphate 21, followed by global deprotection and HPLC purification, to yield d1-lipid II (19). It is important to emphasize that the labelled undecaprenyl chain is being added in the penultimate step, before global deprotection. Therefore, a single labelled undecaprenol analogue can be used to make many different labelled-UCGs, all of which are processed by potential antibiotic targets.
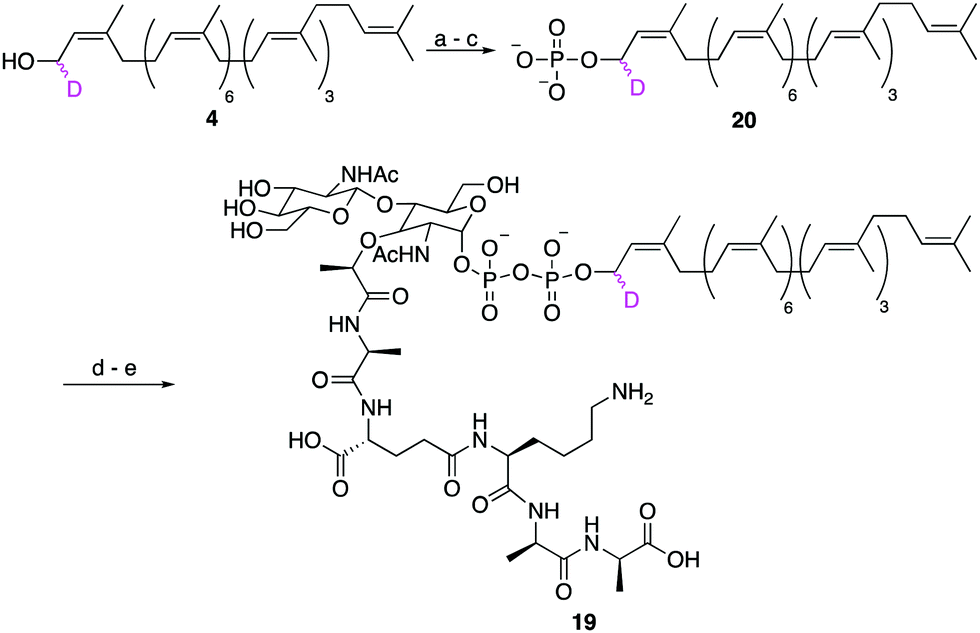 | ||
| Scheme 4 Synthesis of d1-lipid II. Conditions: (a) 5-ethylthio-1H-tetrazole, (iPr)2NP(OCH2CH2CN)2, CH2Cl2, 3 h, RT; (b) H2O2, THF, 3 h, −78 °C to RT; (c) NaOMe, MeOH, 18 h, RT, 53% over 3 steps; (d) 21, DMF, 4 d, RT; (e) NaOH, 1,4-dioxane/H2O, 22% over 4 steps. | ||
We have developed a novel strategy to prepare labelled analogues of the important bacterial lipid undecaprenol, using a semi-synthetic approach on undecaprenol extracted from bay leaves. This method is concise, highly robust, gives improved yields with less steps, and allows numerous flavours of label to be incorporated, including fluorophores, spin labels, photo-affinity labels and bio-orthogonal tags. Labelled undecaprenol analogues can be chemically or enzymatically phosphorylated and utilized in the synthesis of labelled undecaprenol-containing glycolipids, which are valuable probes used to study old and new antimicrobial targets. This work will enable new mechanistic studies and inhibitor screens to be performed on UCG-processing enzymes, which before now were limited by the availability of suitable chemical probes.
We would like to thank Professor Eric Anslyn, Dr Chris Lohans and Dr Shaun McKinnie for advice during the preparation of this manuscript. We acknowledge financial support from the EPSRC (EP/S015892/1, S. A. C.) and Science Foundation Ireland (16/IA/4435, M. C).
Conflicts of interest
There are no conflicts to declare.Notes and references
- J. O‘Neil, Review on Antimicrobial Resistance, 2014 Search PubMed.
- A. Typas, M. Banzhaf, C. A. Gross and W. Vollmer, Nat. Rev. Microbiol., 2011, 10, 123–136 CrossRef PubMed.
- C. R. Raetz and C. Whitfield, Annu. Rev. Biochem., 2002, 71, 635–700 CrossRef CAS PubMed.
- H. L. Tytgat and S. Lebeer, Microbiol. Mol. Biol. Rev., 2014, 78, 372–417 CrossRef CAS PubMed.
- H. Nothaft and C. M. Szymanski, Nat. Rev. Microbiol., 2010, 8, 765–778 CrossRef CAS PubMed.
- A. Müller, A. Klöckner and T. Schneider, Nat. Prod. Rep., 2017, 34, 909–932 RSC.
- R. Woodward, W. Yi, L. Li, G. Zhao, H. Eguchi, P. R. Sridhar, H. Guo, J. K. Song, E. Motari, L. Cai, P. Kelleher, X. Liu, W. Han, W. Zhang, Y. Ding, M. Li and P. G. Wang, Nat. Chem. Biol., 2010, 6, 418–423 CrossRef CAS PubMed.
- L. T. Sham, E. K. Butler, M. D. Lebar, D. Kahne, T. G. Bernhardt and N. Ruiz, Science, 2014, 345, 220–222 CrossRef CAS PubMed.
- T. Mohammadi, V. van Dam, R. Sijbrandi, T. Vernet, A. Zapun, A. Bouhss, M. Diepeveen-de Bruin, M. Nguyen-Distèche, B. de Kruijff and E. Breukink, EMBO J., 2011, 30, 1425–1432 CrossRef CAS PubMed.
- S. T. Islam and J. S. Lam, Environ. Microbiol., 2013, 15, 1001–1015 CrossRef CAS PubMed.
- C. Perez, S. Gerber, J. Boilevin, M. Bucher, T. Darbre, M. Aebi, J. L. Reymond and K. P. Locher, Nature, 2015, 524, 433–438 CrossRef CAS PubMed.
- S. H. Huang, W. S. Wu, L. Y. Huang, W. F. Huang, W. C. Fu, P. T. Chen, J. M. Fang, W. C. Cheng, T. J. Cheng and C. H. Wong, J. Am. Chem. Soc., 2013, 135, 17078–17089 CrossRef CAS PubMed.
- E. Breukink, H. E. van Heusden, P. J. Vollmerhaus, E. Swiezewska, L. Brunner, S. Walker, A. J. Heck and B. de Kruijff, J. Biol. Chem., 2003, 278, 19898–19903 CrossRef CAS PubMed.
- D. K. Lujan, J. A. Stanziale, A. Z. Mostafavi, S. Sharma and J. M. Troutman, Carbohydr. Res., 2012, 359, 44–53 CrossRef CAS PubMed.
- A. Z. Mostafavi, D. K. Lujan, K. M. Erickson, C. D. Martinez and J. M. Troutman, Bioorg. Med. Chem., 2013, 21, 5428–5435 CrossRef CAS PubMed.
- S. Dodbele, C. D. Martinez and J. M. Troutman, Biochemistry, 2014, 53, 5042–5050 CrossRef CAS PubMed.
- J. M. Troutman, K. M. Erickson, P. M. Scott, J. M. Hazel, C. D. Martinez and S. Dodbele, Biochemistry, 2015, 54, 2817–2827 CrossRef CAS PubMed.
- M. Lee, D. Hesek, J. Zajíček, J. F. Fisher and S. Mobashery, Chem. Commun., 2017, 53, 12774–12777 RSC.
- R. T. Gale, E. W. Sewell, T. A. Garrett and E. D. Brown, Chem. Sci., 2014, 5, 3823–3830 RSC.
- R. Bizzarri, B. Cerbai, F. Signori, R. Solaro, E. Bergamini, I. Tamburini and E. Chiellini, Biogerontology, 2003, 4, 353–363 CrossRef CAS PubMed.
- R. P. Hanzlik, Org. Synth., 1988, 560–564 Search PubMed.
- S. Handa, P. S. Nair and G. Pattenden, Helv. Chim. Acta, 2000, 83, 2629–2643 CrossRef CAS.
- S. Sharma, K. M. Erickson and J. M. Troutman, ACS Chem. Biol., 2016, 12, 92–101 CrossRef PubMed.
- E. Smith and I. Collins, Future Med. Chem., 2015, 7, 159–183 CrossRef CAS PubMed.
- D. Marsh, Eur. Biophys. J., 2010, 39, 513–525 CrossRef CAS PubMed.
- L. Y. Huang, S. C. Wang, T. R. Cheng and C. H. Wong, Biochemistry, 2017, 56, 5417–5427 CrossRef CAS PubMed.
- L. Y. Huang, S. H. Huang, Y. C. Chang, W. C. Cheng, T. J. Cheng and C. H. Wong, Angew. Chem., Int. Ed., 2014, 53, 8060–8065 CrossRef CAS PubMed.
- Y. Y. Dong, H. Wang, A. C. W. Pike, S. A. Cochrane, S. Hamedzadeh, F. J. Wyszyński, S. R. Bushell, S. F. Royer, D. A. Widdick, A. Sajid, H. I. Boshoff, Y. Park, R. Lucas, W. M. Liu, S. S. Lee, T. Machida, L. Minall, S. Mehmood, K. Belaya, W. W. Liu, A. Chu, L. Shrestha, S. M. M. Mukhopadhyay, C. Strain-Damerell, R. Chalk, N. A. Burgess-Brown, M. J. Bibb, C. E. Barry Iii, C. V. Robinson, D. Beeson, B. G. Davis and E. P. Carpenter, Cell, 2018, 175, 1045–1058 CrossRef CAS PubMed . e16.
- S. J. Bann, R. D. Ballantine, C. E. McCallion, P. Y. Qian, Y. Li and S. A. Cochrane, J. Med. Chem., 2019, 62, 10466–10472 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cc03388j |
| This journal is © The Royal Society of Chemistry 2020 |