
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSite-directed attachment of photoexcitable spin labels for light-induced pulsed dipolar spectroscopy†
Lara
Williams‡
 ,
Sonja
Tischlik‡
,
Andreas
Scherer
,
Jörg Wolfram Anselm
Fischer
and
Malte
Drescher
*
,
Sonja
Tischlik‡
,
Andreas
Scherer
,
Jörg Wolfram Anselm
Fischer
and
Malte
Drescher
*
Department of Chemistry and Konstanz Research School Chemical Biology (KoRS-CB), University of Konstanz, Universitätsstraße 10, 78457 Konstanz, Germany. E-mail: malte.drescher@uni-konstanz.de
First published on 7th November 2020
Abstract
Photoexcited triplet states are gaining popularity as spin labels in pulsed electron paramagnetic resonance (EPR) spectroscopy. Here, we demonstrate that the fluorophores Eosin Y, Rose Bengal and Atto Thio12 are suitable markers for distance determination by laser-induced magnetic dipole (LaserIMD) spectroscopy in proteins that lack an intrinsic photoexcitable center.
Pulsed electron paramagnetic resonance (EPR) spectroscopy has come into its own as a tool that provides insights into the structure and dynamics of biological macromolecules that elude investigation by other techniques such as NMR spectroscopy and X-ray crystallography, be it due to their size or difficult crystallization.1–3 The dipolar electron–electron interaction between two paramagnetic species, introduced at user-defined sites through site-directed spin labeling (SDSL),4,5 can be translated into a distance distribution which samples a range from 1.8 nm up to 16 nm6 and captures conformationally distinct states of the system under study. As the applications of pulsed dipolar spectroscopy (PDS) broaden, the repertory of spin labels now comprises not only the routinely used nitroxides7 but also other paramagnetic species.8–11 To accommodate the spectral and relaxational differences between these labels, other techniques than the established double electron–electron resonance (DEER) experiment,12 such as relaxation-induced dipolar modulation enhancement (RIDME)13,14 and double-quantum coherence (DQC),15 are stepping into the spotlight. Several examples have also illustrated the advantages of combining spectroscopically orthogonal spin labels, e.g., Cu(II) and nitroxides.16,17 Touching on this last point, 2014 marked the introduction of the photoactivated triplet state of porphyrin as a new type of selectively excitable paramagnetic label.18 The first experiments by Di Valentin et al. exploiting this approach employed light induced DEER (LiDEER) to determine the distance between a nitroxide and a porphyrin moiety attached to a rigid model peptide.18,19 A different take on this system was offered in 2016 by our group as the single frequency technique light-induced magnetic dipole (LaserIMD) spectroscopy.20 Since then, methodical work on light-induced PDS (LiPDS) has been ongoing, as demonstrated by the recently published refocussed (Re)LaserIMD pulse sequence and the first reported LiRIDME experiment.21 However, all measurements require a photoexcitable moiety that undergoes intersystem crossing to a paramagnetic triplet state. This makes the development of exogenously introducible labels a priority if LiPDS is to expand beyond the fraction of bio-macromolecules with a native photoexcitable center, such as a heme prosthetic group, that have been investigated so far.20–22 Serrer et al. proposed dyes that are used in photodynamic therapy or ground state depletion microscopy, as they reach high triplet quantum yields ΦT and, unlike other compounds with a photoexcited triplet state such as fullerenes,23 are water-soluble.24
In this work, we set out to expand the applicability of LiPDS and present a set of photoexcitable spin labels for site-directed attachment to proteins. Together with a characterization of their light-excited triplet state, we show successful distance determinations by LaserIMD spectroscopy between a nitroxide spin marker and these labels, site-selectively attached to the oxidoreductase thioredoxin (TRX). The latter is a well-established model system in SDSL owing to its rigid three-dimensional structure, the availability of X-ray structural data and the possibility to remove the native cysteines by site-directed mutagenesis without perturbing structural integrity.25
Based on the work of Serrer et al.,24 we selected Rose Bengal (RB),26 Eosin Y (EO)26 and the microscopy dye Atto Thio12 (AT12)27 as label candidates; all bear heavy atom substitutions promoting intersystem crossing to the triplet state through increased spin–orbit coupling.28 For the orthogonal, site-selective dual labeling of TRX, we opted for a combination of maleimide–cysteine conjugation and the copper-catalyzed azido-alkyne cycloaddition (CuAAC) reaction between azide-functionalized labels and an alkyne-bearing non-canonical amino acid (ncAA).29 A TRX mutant was used in which the residues C33 and C36 were replaced by serine, the residue D14 by the ncAA para-propargyloxy-L-phenylalanine (pPrF)25 and the residue R74 by cysteine. The labeling sites 14 and 74 are solvent-exposed and easily accessible. As has been shown previously, the strength of combining conventional cysteine chemistry with the orthogonal reactivity of genetically encoded ncAA is that it allows two non-identical spin labels to be introduced site-selectively to a monomeric biomacromolecule16,30 and does not rely on properties specific to the latter to achieve this. The photoexcitable spin labels were equipped with a linker containing either a maleimide (MA) or an azide (N3) moiety. While AT12-MA and EO-MA are commercially available, RB-MA was synthesized from RB disodium salt. For maleimide dyes, the second label, the nitroxide azido-proxyl (NO-N3), was attached by the CuAAC reaction.25 To explore the introduction of photoexcitable spin labels through bio-orthogonal chemistry, an azide-bearing EO derivative (EO-N3) was also prepared and used in combination with the thiol-reactive nitroxide 3-maleimido-proxyl (NO-MA) (Fig. 1). A detailed description of synthetic and protein labeling procedures is provided in the ESI.†
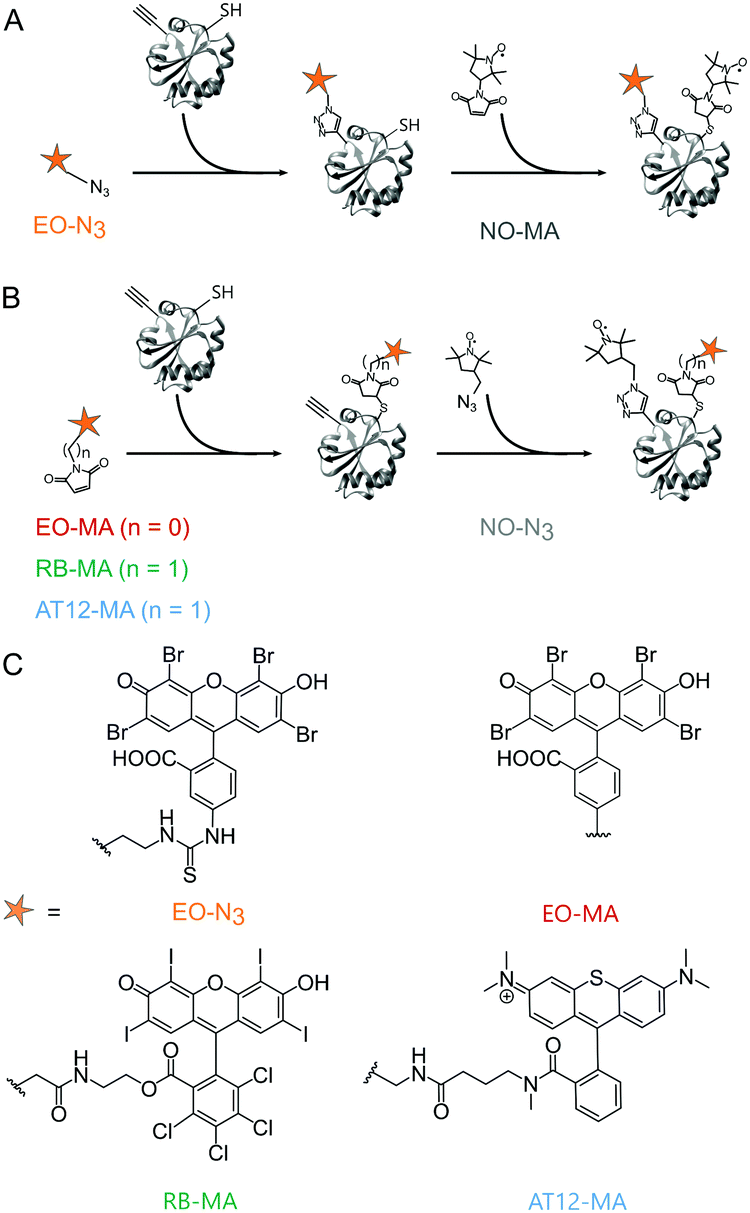 | ||
| Fig. 1 Orthogonal labeling of TRX (PDB ID: 2TRX) at the ncAA pPrF (residue 14) and at the cysteine (residue 74). (A) EO-N3 is attached via CuAAC, followed by NO-MA via cysteine–maleimide coupling. (B) EO-MA, RB-MA and AT12-MA are attached via cysteine–maleimide coupling, followed by NO-N3via CuAAC. (C) Chemical structure of the dyes. | ||
To assess the impact of introducing tethers on the photoexcited triplet state of the spin labels, time-resolved (tr) EPR spectroscopy measurements in X-band of the unbound compounds in frozen solution were performed, from which the zero-field splitting (ZFS) parameters D and E and sublevel populations px, py and pz (Fig. S9, ESI†) were derived. The values for RB-MA deviate slightly from those for unmodified RB24 (D and E decrease by ∼130 and 80 MHz, respectively), while the results for the EO derivatives and AT12-MA are in good agreement with EO disodium salt31 and with AT12 bearing a carboxylic acid residue in place of a maleimide.24 Consequently, the chemical modification appears to have a limited effect on the triplet parameters.
Echo-detected field sweep (EDFS) spectra in Q-band of the TRX proteins labeled with a nitroxide and one of the four photoexcitable spin labels (Fig. S10, ESI†) revealed a large separation between the global nitroxide maximum and maxima in the triplet spectrum (e.g., 230 MHz for TRX_EO-N3_NO-MA), and low triplet signal intensities within the microwave frequency offset from the nitroxide resonance allowed by our resonator profile (FWHM = 160 MHz, Fig. S11, ESI†). Therefore, we decided to perform LaserIMD experiments as a single-frequency technique observing on the nitroxide rather than LiDEER. So far, LaserIMD has only been shown for porphyrin as the photoexcitable center. As the ZFS parameters of RB, EO and AT12 are larger than for porphyrin, we wanted to confirm that LaserIMD distance determination can be done using these systems. To this end, we performed spin dynamic simulations of LaserIMD experiments between porphyrin, RB, EO and AT12 and a permanent organic radical for a fixed interspin distance of r = 2.5 nm and compared them with a simulation of porphyrin for the same distance (Fig. 2).
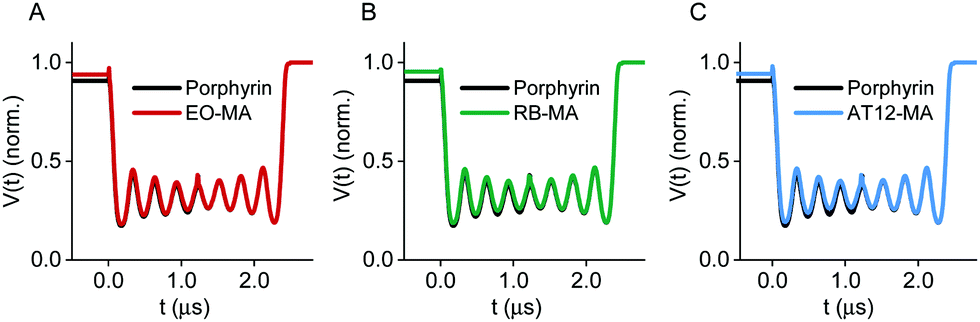 | ||
| Fig. 2 Simulation of LaserIMD traces in Q-band. The simulations with an exemplary interspin distance of r = 2.5 nm suggest that LaserIMD distance determination is feasible using the different photoexcitable spin labels by direct comparison with porphyrin (black). (A) EO-MA (red), (B) RB-MA (green), (C) AT12-MA (blue). The traces are normalized to the last data point. | ||
During the LaserIMD experiment the position of the laser flash at a time τ is swept through a Hahn-echo sequence, with the echo intensity being recorded as a position of the laser flash τ. Due to the acquired dipolar phase, the echo intensity is described as the sum of cosine oscillations of the form:
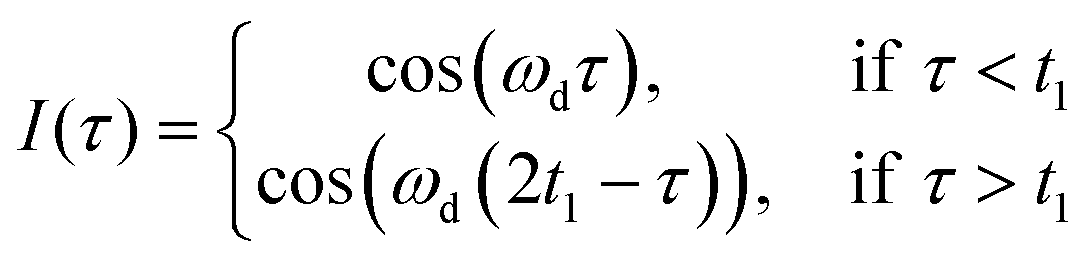 | (1) |
For LaserIMD, the efficiency of optical switching, i.e. of the “pump” pulse, relies solely on the triplet quantum yield ΦT and the populations p−1 and p+1 of the triplet sublevels.20 The theoretically achievable maximum modulation depth (MD) in LaserIMD is 2/3 (see ESI†) for a ΦT of 100%. Considering the reported ΦT for RB and EO, which are in the range between 64% and 98%34–36 for aqueous solvents, MDs from 43% to 66% should be possible. To avoid triplet-state saturation, the shot repetition time (SRT) of the LaserIMD experiments was set to exceed the triplet lifetimes determined by pulsed EPR spectroscopy37 (Fig. S12, ESI†).
Next, the LaserIMD sequence20 was experimentally applied to the labeled TRX samples. The LaserIMD datasets (Fig. 3) show modulation depths of around 20% for RB and EO and 30% for AT12, which are on a par with those of porphyrin–nitroxide pairs (typically between 15 and 34%). The mean distances lie between 3.2 and 3.8 nm and the corresponding distribution widths span a range from 0.7 to 1.1 nm, respectively (Table S2, ESI†). The bulk of the extended aromatic system of the photoexcitable labels, which might lead to steric clash with the protein backbone, as well as the flexibility of the appended linkers is expected to compromises the distance determination by broadening the distribution. Although this means our results cannot compare to the narrow inter-spin distance distributions obtained for biomolecules with an intrinsic, rigidly anchored chromophore, the achieved distance width is satisfactory for the expected Cα–Cα distance between the labeling sites of 2.4 nm, as derived from the TRX crystal structure.38 The contribution of the nitroxide label to the width of the distance distribution, roughly judged from the DEER data, is also noteworthy: the distribution for TRX doubly labeled with NO-MA (Fig. 3F) is narrower than for the NO-N3-tagged sample (Fig. 3E). Since the linkers for all photoexcitable spin labels are of similar length and flexibility, the difference in mobility between the two nitroxide labels might explain why the global maximum of the EO-N3/NO-MA distance distribution (Fig. 3A) is shifted to lower distances compared to all other combinations (Fig. 3B–E).
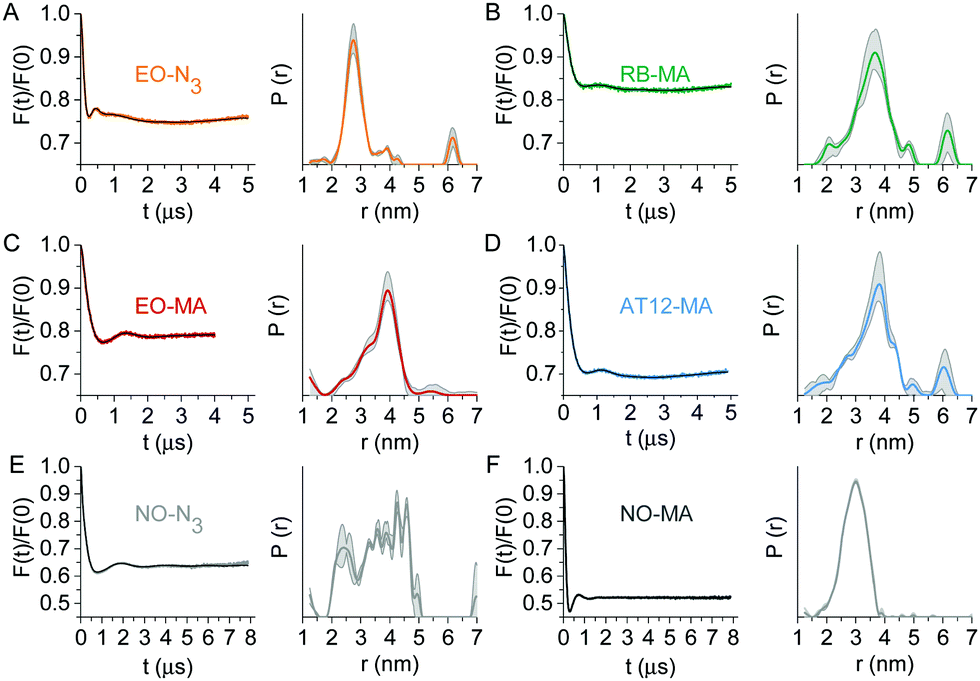 | ||
| Fig. 3 Q-Band dipolar spectroscopy on TRX. Form factors (left) after background correction with fits obtained by Tikhonov regularization (black) and corresponding distance distributions (right) with validation area (grey) of LaserIMD experiments at 10 K on TRX doubly labeled with (A) EO-N3/NO-MA, (B) RB-MA/NO-N3, (C) EO-MA/NO-N3 and (D) AT12-MA/NO-N3. (E and F) Form factors and distance distributions of a 4-pulse DEER measurement at 50 K on TRX labeled with NO-N3 or NO-MA at the same residues 14 and 74. More details are reported in the ESI.† | ||
Apart from their steric traits, high photostability and ΦT are crucial for photoexcitable spin labels. All LaserIMD measurements were performed as 2-dimensional experiments, i.e. each scan was saved separately, revealing a decrease in modulation depth over time (Table S3, ESI†) which we attributed to photobleaching. Consistent with reports on the excellent photostability of thiorhodamines,27 this effect is the least pronounced for AT12-MA, although we do not provide a quantitative comparison due to differences in excitation wavelength and laser power. Looking at the first scans only, i.e., before significant bleaching, the modulation depths for the EO- and RB-labeled TRX lie below our estimates. Incomplete excitation of the dye molecules in the sample was excluded, as the intensity of the triplet EPR signal did not increase any further at higher laser powers, as was excited state quenching by the nitroxide (details in ESI†); we rather believe that the dependence of ΦT on solvent and pH, and non-quantitative double labeling efficiencies in the range of 60–90% (Table S4, ESI†) are to blame. Overall, these effects do not detract from the successful implementation of our concept, which we perceive as a pioneering step towards opening up LiPDS and its promises of increased detection sensitivity, modulation-to-noise enhancement and access to shorter distances20 to virtually any bio-macromolecule of interest.
In summary, we have presented RB, EO and AT12 as photoexcitable spin labels for site-directed labeling by cysteine–maleimide conjugation or the bioorthogonal CuAAC reaction of proteins that lack an endogenous photoexcitable group. For the first time, LaserIMD distance determinations between a nitroxide and a photoexcitable spin label exogenously introduced at a user-defined protein site were performed, and modulation depths comparable to the established porphyrin–nitroxide pair were achieved. Compatibility with other spin labels and single-frequency techniques such as LiRIDME has yet to be investigated. For LaserIMD, AT12 outperforms RB and EO in terms of modulation depth and photostability, but the long lifetime of its triplet state requires longer measurement times. All four dyes incorporate a carboxyl group for functionalization, making it possible to compile a library with tethers of various lengths, flexibility and bioconjugation moieties. The design of these labels could be aided by an in silico rotamer library approach,39 in which the reference point for distance determination, in agreement with the literature,18,23 is located in the center of the molecule by a point-dipole approximation. In the bigger picture, the dual-addressable character of EO, RB and AT12 might provide a gateway to correlating the results from luminescence and EPR spectroscopy; and in turn, the field of bioimaging is expected to offer a steady flow of inspiration for the design of new labels for LiPDS.
This project has received funding from the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation program (Grant Agreement number: 772027—SPICE—ERC-2017-COG). We thank M. Adam and D. Brecht for experimental contributions, P. G. Schultz for the plasmid pEVOL_pCN-Phe and D. Bücker for helpful discussions.
Conflicts of interest
There are no conflicts to declare.References
- W. L. Hubbell, H. S. McHaourab, C. Altenbach and M. A. Lietzow, Structure, 1996, 4, 779–783 CrossRef CAS.
- G. Jeschke and Y. Polyhach, Phys. Chem. Chem. Phys., 2007, 9, 1895–1910 RSC.
- G. W. Reginsson and O. Schiemann, Biochem. J., 2011, 434, 353–363 CrossRef CAS.
- M. R. Fleissner, E. M. Brustad, T. Kálai, C. Altenbach, D. Cascio, F. B. Peters, K. Hideg, S. Peuker, P. G. Schultz and W. L. Hubbell, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 21637–21642 CrossRef CAS.
- I. M. S. de Vera, M. E. Blackburn, L. Galiano and G. E. Fanucci, Curr. Protoc. Protein Sci., 2013, 74 Search PubMed , 17.17.11-17.17.29.
- T. Schmidt, M. A. Wälti, J. L. Baber, E. J. Hustedt and G. M. Clore, Angew. Chem., Int. Ed., 2016, 55, 15905–15909 CrossRef CAS.
- G. I. Likhtenshtein, J. Yamauchi, S. i. Nakatsuji, A. Smirnov and R. Tamura, Nitroxides, Springer, 2008 Search PubMed.
- Y. Yang, B.-B. Pan, X. Tan, F. Yang, Y. Liu, X.-C. Su and D. Goldfarb, J. Phys. Chem. Lett., 2020, 11, 1141–1147 CrossRef CAS.
- D. Goldfarb, Phys. Chem. Chem. Phys., 2014, 16, 9685–9699 RSC.
- J. L. Wort, K. Ackermann, A. Giannoulis, A. J. Stewart, D. G. Norman and B. E. Bode, Angew. Chem., Int. Ed., 2019, 131, 11807–11811 CrossRef.
- K. Keller, M. Zalibera, M. Qi, V. Koch, J. Wegner, H. Hintz, A. Godt, G. Jeschke, A. Savitsky and M. Yulikov, Phys. Chem. Chem. Phys., 2016, 18, 25120–25135 RSC.
- G. Jeschke, Annu. Rev. Phys. Chem., 2012, 63, 419–446 CrossRef CAS.
- S. Milikisyants, F. Scarpelli, M. G. Finiguerra, M. Ubbink and M. Huber, J. Magn. Reson., 2009, 201, 48–56 CrossRef CAS.
- M. Azarkh, A. Bieber, M. Qi, J. W. A. Fischer, M. Yulikov, A. Godt and M. Drescher, J. Phys. Chem. Lett., 2019, 10, 1477–1481 CrossRef CAS.
- P. P. Borbat and J. H. Freed, Chem. Phys. Lett., 1999, 313, 145–154 CrossRef CAS.
- Z. Wu, A. Feintuch, A. Collauto, L. A. Adams, L. Aurelio, B. Graham, G. Otting and D. Goldfarb, J. Phys. Chem. Lett., 2017, 8, 5277–5282 CrossRef CAS.
- I. Ritsch, H. Hintz, G. Jeschke, A. Godt and M. Yulikov, Phys. Chem. Chem. Phys., 2019, 21, 9810–9830 RSC.
- M. Di Valentin, M. Albertini, E. Zurlo, M. Gobbo and D. Carbonera, J. Am. Chem. Soc., 2014, 136, 6582–6585 CrossRef CAS.
- M. Di Valentin, M. Albertini, M. G. Dal Farra, E. Zurlo, L. Orian, A. Polimeno, M. Gobbo and D. Carbonera, Chem. – Eur. J., 2016, 22, 17204–17214 CrossRef CAS.
- C. Hintze, D. Bücker, S. Domingo Köhler, G. Jeschke and M. Drescher, J. Phys. Chem. Lett., 2016, 7, 2204–2209 CrossRef CAS.
- M. G. Dal Farra, S. Richert, C. Martin, C. Larminie, M. Gobbo, E. Bergantino, C. R. Timmel, A. M. Bowen and M. Di Valentin, ChemPhysChem, 2019, 20, 931–935 CrossRef CAS.
- M. Di Valentin, M. G. Dal Farra, L. Galazzo, M. Albertini, T. Schulte, E. Hofmann and D. Carbonera, Biochim. Biophys. Acta, 2016, 1857, 1909–1916 CrossRef CAS.
- O. A. Krumkacheva, I. O. Timofeev, L. V. Politanskaya, Y. F. Polienko, E. V. Tretyakov, O. Y. Rogozhnikova, D. V. Trukhin, V. M. Tormyshev, A. S. Chubarov and E. G. Bagryanskaya, Angew. Chem., 2019, 131, 13405–13409 CrossRef.
- K. Serrer, C. Matt, M. Sokolov, S. Kacprzak, E. Schleicher and S. Weber, Mol. Phys., 2019, 117, 2688–2699 CrossRef CAS.
- P. Widder, F. Berner, D. Summerer and M. Drescher, ACS Chem. Biol., 2019, 14, 839–844 CrossRef CAS.
- W. J. Jones, A. Grofcsik, M. Kubinyi and D. Thomas, J. Mol. Struct., 2006, 792-793, 121–129 CrossRef CAS.
- A. Chmyrov, J. Arden-Jacob, A. Zilles, K.-H. Drexhage and J. Widengren, Photochem. Photobiol. Sci., 2008, 7, 1378–1385 RSC.
- K. N. Solov’ev and E. A. Borisevich, Phys.-Usp., 2005, 48, 231–253 CrossRef.
- S. I. Presolski, V. P. Hong and M. G. Finn, Curr. Protoc. Chem. Biol., 2011, 3, 153–162 CrossRef.
- L. Garbuio, E. Bordignon, E. K. Brooks, W. L. Hubbell, G. Jeschke and M. Yulikov, J. Phys. Chem. B, 2013, 117, 3145–3153 CrossRef CAS.
- A. Katsuki, S. Tero-Kubota and Y. Ikegami, Chem. Phys. Lett., 1993, 209, 258–262 CrossRef CAS.
- A. Bieber, D. Bücker and M. Drescher, J. Magn. Reson., 2018, 296, 29–35 CrossRef CAS.
- M. G. Dal Farra, S. Ciuti, M. Gobbo, D. Carbonera and M. Di Valentin, Mol. Phys., 2019, 117, 2673–2687 CrossRef CAS.
- M. Nemoto, H. Kokubun and M. Koizumi, Bull. Chem. Soc. Jpn., 1969, 42, 2464–2470 CrossRef CAS.
- A. Penzkofer, A. Beidoun and M. Daiber, J. Lumin., 1992, 51, 297–314 CrossRef CAS.
- O. Berndt, F. Bandt, I. Eichwurzel and H. Stiel, Acta Phys. Pol., A, 1999, 95, 207–220 CrossRef CAS.
- H. Seidel, M. Mehring and D. Stehlik, Chem. Phys. Lett., 1984, 104, 552–559 CrossRef CAS.
- S. K. Katti, D. M. LeMaster and H. Eklund, J. Mol. Biol., 1990, 212, 167–184 CrossRef CAS.
- M. Qi, A. Gross, G. Jeschke, A. Godt and M. Drescher, J. Am. Chem. Soc., 2014, 136, 15366–15378 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Theoretical description and simulations of LaserIMD, label synthesis and chemical characterization, cw and transient EPR, triplet relaxation, EDFS spectra, TRX protein expression, purification and labeling, LaserIMD and 4-pulse DEER distance measurements. See DOI: 10.1039/d0cc03101a |
| ‡ L. W. and S. T. contributed equally. |
| This journal is © The Royal Society of Chemistry 2020 |