
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAcetylcysteine in paracetamol poisoning: a perspective of 45 years of use
D. Nicholas
Bateman
 * and
James W.
Dear
* and
James W.
Dear
Pharmacology, Toxicology and Therapeutics, University of Edinburgh, Edinburgh, UK. E-mail: drnickbateman@gmail.com
First published on 29th April 2019
Abstract
Paracetamol poisoning was first reported in 1966. The development of antidotes followed within 10 years, and by 1980 acetylcysteine (NAC) was acknowledged as the optimal therapy available. This article examines the history of the development of NAC and recent developments in its use. We offer suggestions for improvements in the way NAC may be administered and outline new developments that should have major impacts on the way we manage paracetamol poisoning in the near future.
Introduction
Paracetamol was introduced into clinical medicine in the 1950s as an antipyretic analgesic.1,2 At that time presentations with poisoning, either accidental or from self-harm, were unusual in Europe. The profile of the epidemiology of poisoning changed over the next 40 years with poisoning becoming a common reason for emergency admission to hospital.3 The precise psychosocial factors behind this increase are unclear, but by the early 1970s admissions to hospital from patients deliberately ingesting aspirin and paracetamol had become a regular, problematic medical emergency. The first documented cases of organ toxicity from paracetamol were in Scotland in 1966.4,5 At that time the biochemical mechanisms underlying paracetamol toxicity were unknown, and thus treatment was entirely symptomatic. The major clinical presentation was of hepatic failure due to acute liver necrosis.The breakthrough came in the early 1970s with the development of the concept that free radical generation during drug metabolism was a potential cause of toxicity. Animal experiments clearly demonstrated that the hepatic toxicity of some molecules was by metabolism which could be both induced and inhibited, and that it involved production of reactive intermediates in metabolism that were normally neutralised by glutathione.6–8 The important finding was that toxicity was glutathione dependant.9 This elegant chemistry explains why at standard doses paracetamol remains one of the safest drugs used in clinical practice, but that with increasing dose hepatic toxicity becomes more likely. The problem then, and now, is to determine the dose threshold for toxicity in order to tailor therapy optimally.
In the early 1970s the development of an assay for paracetamol, and presence of a body of patients who had ingested paracetamol in overdose in Edinburgh, lead to the development of nomograms in man that illustrated the dose–response relationship of paracetamol toxicity (Fig. 1).10,11 This approach thus facilitated the development of antidotes, as it allowed comparison of outcomes in patients in pre-defined risk categories.
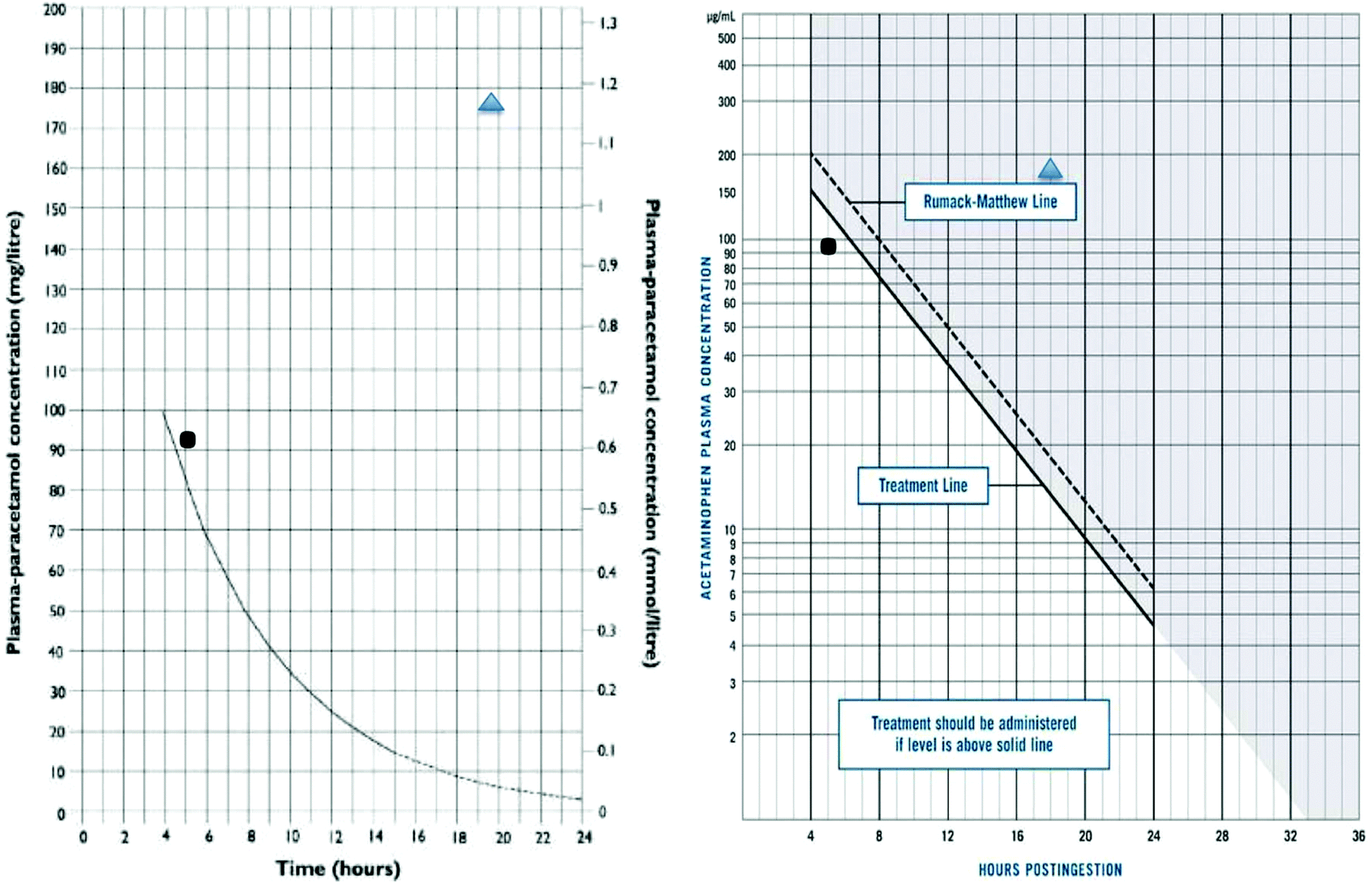 | ||
Fig. 1 Examples of nomograms for decisions on use of acetylcysteine in clinical use, showing paracetamol concentration and time after ingestion. Left panel is the present UK nomogram (‘100 mg’ line) on a linear scale. On the right is shown the nomogram originally determined by Rumack and Matthew (‘150 mg’ line) and the original UK (‘200 mg’ Prescott line) both on log scales. The markers on the graphs show timing and sampling of 2 examplar patients, one ( ) who developed hepatotoxicity at a concentration above UK but below other current nomograms, and one ( ) who developed hepatotoxicity at a concentration above UK but below other current nomograms, and one ( ) who had no hepatotoxicity, despite having a concentration well above all current treatment lines. ) who had no hepatotoxicity, despite having a concentration well above all current treatment lines. | ||
In recent years paracetamol has become an important laboratory tool for investigation of drug-induced liver injury. However, it still represents a major drug toxicity seen in overdose and is the most common cause of drug-induced liver failure in many countries.12
This article is intended to give both clinicians and non-clinical toxicologists an overview of the development of treatments for paracetamol poisoning and an outline of future therapeutic directions. We review aspects of the clinical development of the use of NAC since the 1970s. It is based on a literature review using search terms including paracetamol, NAC and poisoning. We have selected relevant references that illustrate the problems in the use of NAC, and approaches to address these, together with the challenges in the indications for NAC use in paracetamol overdose.
Paracetamol toxicity in medicine
Paracetamol toxicity presents in a number of scenarios. These are principally in two types of presentation: firstly following a single excessive ingestion, as an intentional act of self-harm, following accidental ingestion (usually in a child), or rarely deliberately by a third party; secondly after repetitive excess ingestion, sometimes deliberate repeated self-harm attempts, or in a therapeutic attempt to control a problem such as dental pain.Paracetamol is dosed over a 24 h period by patient weight. The dose of paracetamol that causes liver injury varies from patient to patient, and this is a major issue for decisions on treatment.13 Rarely, use of intravenous or oral paracetamol may also be associated with excess dosing due to miscalculation of the dose in children, or thin and/or malnourished adults. While antidote therapy is appropriate in all these cases, it is likely to be most effective when given early in acute poisoning as repeated overdose is a risk for organ toxicity.14
NAC as an antidote
The development of NAC as an antidote for paracetamol has been described in detail elsewhere, but is relevant in relation to current use of antidotes in paracetamol poisoning.15 The basic understanding of toxic mechanisms of paracetamol, in particular the potential protective role of endogenous glutathione, facilitated trials in animal models of sulfhydryl donors as potential antidotes,15,16 and demonstrated that they were effective particularly if given in the early stages of toxicity. Three antidotes were tested in animals, cysteamine, methionine and NAC.17 In the 1970s, of these agents, only NAC was licensed in a liquid formulation for human use in the UK, and that was as an inhalational mucolytic agent. The practical problems of drug licensing and development now encountered were not present in the 1970s, and this facilitated the rapid introduction and use of these untested antidotes in man.The first antidote successfully tested in man was cysteamine,18 and although this was used in patients, when given to volunteers it caused them to feel so ill that the lead investigator, Laurie Prescott, was actually admitted to his own unit overnight! Investigations then focused on the other two antidotes that had been successful in animals, the orally administered methionine and the liquid formulation of NAC, which was administered intravenously (IV) through a microfilter in Edinburgh, since it was not yet formulated for IV administration.19 The microfilter was because the investigators were concerned that, although it was sterile, the NAC might contain micro-particulates. At that time gastric lavage was standard therapy for overdose, and patients were often left nauseated and vomiting after procedure. Use of an oral antidote therefore was problematic, and this meant that although methionine was tested, IV NAC soon became the lead agent in Europe.20–22 In the USA the FDA had required a different approach and for many years a 3 day oral regimen of NAC was used, before being generally replaced by an IV regimen in the 1990s.15,23
In the 1970s patients who had ingested paracetamol seemed to develop hepatic toxicity with monotonous regularity, and there was a desire to reduce this risk by rapid administration of the antidote. An empirical regimen was developed which involved the use of a weight-related dose of NAC of 300 mg per kilogram bodyweight, given in three divided doses, 150 mg per kilogram over 15 minutes, 50 mg per kilogram over 4 hours and 100 mg per kilogram over 16 hours.21 This regimen, often described as the Prescott regimen, was used in the UK until 2012, when the initial infusion was changed to 1 hour.24 It was also widely used elsewhere in the world, although in some countries the initial dose was given over one hour rather than 15 minutes (Table 1).
| Total duration (prior to check blood tests) | Initial dose and duration | Second dose and duration | Third dose and duration (if part of initial regimen) | Origin and exemplar reference |
|---|---|---|---|---|
| 20.25 h | 150 mg kg−1 over 15 min | 50 mg kg−1 over 4 h | 100 mg kg−1 over 16 h | Prescott regimen21 |
| 48 h | 140 mg kg−1 over 1 h | 70 mg kg−1 over 1 h given 4 h × 12 doses | North America60 | |
| 21 h | 150 mg kg−1 over 1 h | 50 mg kg−1 over 4 h | 100 mg kg−1 over 16 h | North America and Australia62 |
| 12 h | 100 mg kg−1 over 2 h | 200 mg kg−1 over 10 h | Subsequent doses dependent on blood tests at 10–12 h post IV commencement | SNAP study37 |
| 20 h | 200 mg kg−1 over 4 h | 100 mg kg−1 over 16 h | Modified Australian study70 |
That this NAC regimen was being used by 1975, less than 10 years from the first description of paracetamol toxicity, and less than five years from the identification of the toxic mechanism, was an amazing achievement which no doubt saved many lives.
Progress in indication for antidote use
The only data published on the prognosis of untreated patients who ingest different doses of paracetamol are those of Prescott (Table 2).25 This categorises 83 untreated patients seen in Edinburgh in the early 1970s by the estimated dose of paracetamol based on blood concentration at presentation as related to a paracetamol risk nomogram. These data are illuminating, as they show the individual variability in hepatic toxicity at apparently similar ingested doses of paracetamol. Factors that may impact this could be variability in rates of paracetamol metabolism to the reactive toxic intermediate, or stores of endogenous glutathione.26 Measuring these in vivo in man has not proved possible in real time, although there are recent data showing that rates of production of paracetamol sulfhydryl metabolites are greater in those who develop liver injury, thus supporting the hypothesis that rates of paracetamol metabolism are important in humans.27| Paracetamol line (mg L−1) | Number of patients | Liver injury N, (%) | Renal failure N, (%) | Death N, (%) |
|---|---|---|---|---|
| <100 | 9 | 0 (0) | 0 | 0 |
| 100–200 | 22 | 5 (23) | 0 | 0 |
| 200–300 | 25 | 6 (24) | 1 (4) | 0 |
| >300 | 27 | 25 (93) | 5 (20) | 3 (12) |
Kinetic considerations
There are few studies on the pharmacokinetics of acetylcysteine, and the pharmacokinetics of paracetamol are dose dependant and altered by liver injury. The absorption of paracetamol after oral overdose is generally considered complete within 4 h of ingestion. While this may not always be the case, due to interactions of drugs that delay intestinal absorption such as anticholinergics and opiates, for the vast majority of cases it has proved a useful landmark for determining the timing of paracetamol concentrations for risk assessment.28 Clearance of paracetamol is normally first order, but the rate of clearance varies depending on the individual, but also early onset liver injury. After therapeutic doses the half-life is 90 min to 2 h, but in liver injury it is much longer, often over 10 h.10 In contrast, the half-life of paracetamol on the nomograms used for treatment decisions is 4 h (Fig. 1).The major metabolites of paracetamol are sulphate and glucuronide conjugates, with small amounts of the drug being metabolised via the reactive N-acetyl-p-benzoquinone imine (NAPQI) intermediary that is neutralised by SH groups, principally glutathione, to cysteine, mercapturic and methoxy metabolites.29 As the dose of paracetamol increases the simple conjugation pathways are saturated and more passes down the reactive, potentially toxic route. This pathway can be both induced and inhibited in animal models, and probably therefore man, and may also be altered by pharmacogenetic polymorphisms.30,31 However, the relevance of pharmacogenetics on treatment decision is unclear, and at present phenotyping has no practical utility in patient care. These effects may explain some of the differences in individual susceptibility but testing this is challenging. Importantly to support this hypothesis regarding human mechanisms of toxicity, the amounts of SH-metabolites derived from NAPQI are higher in blood in those who develop liver injury following paracetamol overdose.32 Starvation is often used in standard animal models of hepatotoxicity studies using paracetamol, but evidence that this is important in man is unclear, though some clinical guidelines have suggested starvation may increase the risk of hepatotoxicity in man and that a lower threshold for treatment with NAC should be used.24,26
There is interaction between NAC and the metabolism of paracetamol at moderate increase in paracetamol dose, up to 3 g. Increased sulphation and glucuronidation was seen in volunteers who were given NAC at these doses of higher doses of paracetamol, indicating glutathione supply is affected at even small excesses of paracetamol.29
The mechanisms of toxicity have been recently reviewed and are now recognised to more complex than originally believed.33 As liver injury occurs the rate of paracetamol clearance falls and the half-life lengthens, of itself a marker of developing liver injury. The aim of NAC therapy is to neutralise this process by providing excess glutathione. While the kinetics of acetylcysteine itself are understood and terminal half-life values of between 2.7 and 5.7 h are reported,34,35 the rates of conversion to glutathione are poorly studied in man.36 The original dose estimates for NAC were based on extrapolation from animal models, with the concept that rapid dosing was clinically important.15 Models for administering NAC by novel regimens have been used to suggest a modified 12 h infusion would give serum concentrations of NAC adequate for therapy.37 Comparative clinical trials have not been adequate to determine the optimum NAC dose, but there is some evidence on case series that patients ingesting very large, ‘massive’, doses of paracetamol are more likely to develop liver injury despite prompt treatment with NAC.13,38 This has led some to suggest higher doses of antidote should be given to patients with this history.39
From this it is obvious that the clearance of paracetamol is a marker of hepatic injury, but in clinical practice it has been extremely uncommon to use this routinely in risk assessment, other than a measurement at the end of the NAC infusion to confirm the absence of toxicity, or suggest need for additional antidote. The data collected on untreated patients in Edinburgh in the early 70s still forms the basis for the nomograms used internationally (Table 2).25 In the UK Prescott advocated a treatment cut-off line of 200 mg L−1 at four hours, with a half-life of 4 h declining from that point. In the USA, however, the cut-off for treatment was set out a concentration of 150 mg L−1 at four hours post ingestion, as this was 50% of the concentration cut off for death in the untreated case series.11,23 As illustrated in Fig. 1 some patients will be below the cut off line and still suffer hepatotoxicity, and others be well above them and not.
Uncertainty about ingested dose in many cases means that treatment is based on rapid estimation of paracetamol concentration in a blood sample taken at least 4 h after overdose. This is now the standard of care for all patients admitted within 12 hours of paracetamol ingestion. Since the onset of hepatotoxicity occurs relatively quickly following paracetamol ingestion the need to provide antidote therapy in a timely manner results in many patients who present from 10 to 12 hours after ingestion being treated on the basis of the history of ingested dose.
There are, however, a number of problems associated with this rather simplistic approach to therapy. The nomograms were based on death and occurrence of liver injury, as defined in the 1970s as the rise in the ALT above 1000 IU L−1. This cut-off was only chosen because it represented the enzyme activity in blood which was the maximal measurable without sample dilution with the assay in use at the time. Thus, although potentially informative, it certainly does not invariably indicate the onset of liver failure as it is based on a small Scottish population.25
It is now appreciated that a more reliable biomarker is the rate of increase of the ALT, which has been used either alone or as a multiplier of paracetamol.40–42 The other important marker traditionally used has been prothrombin time, usually reported as INR. Changes in clotting requires exhaustion of circulating vitamin K dependent clotting factors, which have variable half-lives. This means it is generally 24 hours before clotting becomes disturbed. Clinical experience has also shown that some individual patients will develop features of liver damage, including hepatic failure, when presenting with an apparently verifiable history but concentrations of paracetamol in blood below the nomogram interventions. This was a problem identified with the UK treatment line (200 mg L−1 at 4 h) in the 1980s and resulted in a change in policy by the UK poison services and a recommendation to treat patients considered at a ‘higher risk’ at an intervention line of 100 mg L−1 at 4 h.26 These problems do not appear to have been so prominently reported in North America where the intervention level was always set at 150 mg L−1 at 4 h. The Australian intervention concentration has also been set at the ‘150’ line.43 A further change in treatment intervention paracetamol concentration occurred in the UK in 2012 following a single case in which death was apparently caused by paracetamol as the patients risk factors for toxicity were not identified at presentation. The UK now operates a nomogram with treatment indicated in patients above 100 mg L−1 at 4 h hours post overdose (Fig. 1).24
The major problem for clinicians is that the standard markers of hepatotoxicity (ALT and INR) do not usually begin to change until sometime after ingestion at least 10 hours in the case of ALT, and 24 hours for INR. A further complication is that NAC and paracetamol both affect INR, resulting in a moderate increase in some patients usually in the range of 1.2 to 1.7.44–46
The effect of these problems has been that patients are treated because they may be at risk rather than that they are at risk. It is difficult to estimate how many patients are treated who would not get liver damage if left untreated. So few patients were in the original untreated cohort that confidence intervals to estimate this accurately are not really possible, although the data shown in Table 2 suggests that many patients would presumably not develop major clinical problems, even with paracetamol concentrations in the range in which some patients do. It is worth bearing in mind that tens of thousands of patients are admitted every year to UK hospitals for NAC treatment, and likely hundreds of thousands worldwide.47
This problem has led to the consideration of other approaches to risk assessment in early presentations with paracetamol overdose. Over the past few years it has become evident that when injured, liver cells release a range of molecules into the circulation. A full-length version of the protein keratin-18 (K18) is released by necrotic cell death. A shorter, caspase cleaved form of K18 (ccK18) is released following cell apoptosis (programmed cell death). Both forms of K18, when measured in the first serum sample at presentation at the hospital after paracetamol overdose, correlate with peak ALT activity during the hospital stay.48,49 K18 is more sensitive than ALT – it distinguishes patients with and without acute liver injury at hospital presentation when ALT activity is still in the normal range. K18 is supported for exploratory use in assessing drug-induced liver injury in clinical trials, both by the EMA and the FDA.50,51 MicroRNA-122 (miR-122) is a microRNA biomarker specific for liver injury that is fully conserved (translational) across in vitro models, in vivo models and humans. Similar to K18, miR-122 is an early marker for acute liver injury that predicts a rise in ALT activity following paracetamol overdose. When miR-122 was measured at hospital presentation after a paracetamol overdose in patients requiring subsequent NAC therapy the circulating miR-122 concentration correlated significantly with peak hospital stay ALT activity and was significantly higher in those patients who developed subsequent acute liver injury.48,49 This is consistent with miR-122 having enhanced sensitivity and specificity in this context-of-use.
The uncertainty at this point is when it would be safe to discharge patients without rises in the novel markers. At present, all patients in these cohorts studied have received NAC and the abnormalities in ALT and other biomarkers seen are all in patients who received the antidote. Developing studies that allow us to understand whether an initial normal biomarker indicator at a given time interval will safely allow discharge without therapy or will permit administration of perhaps a single dose of antidote prior to discharge is one of the next big challenges.
Such information is necessary because of the other problems associated with NAC, in particular its propensity to cause adverse effects and need for hospital admission.
Adverse effects of NAC
As intravenous NAC became more widely used problems associated with its administration became clearer. The marketed formulation of the drug required calculation of the numbers of ampoules required to provide the correct dose, and in the early days of its use by staff unfamiliar with the mode of administration dosing errors occurred, resulting in rare deaths. Flushing had been recognised by the early investigators in Edinburgh in some patients who had received NAC, but as it came to be used more widely more serious adverse effects were reported, in particular anaphylactoid reactions, including in severe cases major bronchospasm and hypotension. Interestingly, the first case reported was in a patient who had low concentrations paracetamol at the time of treatment, and almost certainly therefore did not require therapy.52 This low concentration of paracetamol was not recognised at the time as a likely key factor in the adverse event. It was not until the early years of the 20th century that retrospective studies, using prescription of antihistamines and bronchodilators after use of NAC as a surrogate for anaphylaxis, showed a clear relationship between the concentration of paracetamol and risk of reaction. Rarely NAC has caused serious reactions and death, but these seem likely related to inadvertent excess dosing.53,54Earlier studies had suggested individual susceptibility to the reactions based on skin testing using the antidote solution intradermally.55 As physicians became familiar with this problem standard approaches were developed to treatment. Importantly this reaction does not involve an immunological mechanism but is almost certainly related to histamine release from basophils exposed to high concentrations of NAC. Cohort studies comparing concentrations of paracetamol in patients receiving NAC showed a clear concentration effect relationship of paracetamol as protective of the anaphylactoid response.46,56,57 From a practical clinical perspective this means that patients who are treated with NAC at lower concentrations of paracetamol, either because of the lower ingestion, or a later presentation to hospital, are at increased risk of adverse effects. Unfortunately, even though the adverse reaction is not immunological, previous history of such reactions sometimes deterred both patients and doctors from use of the antidote. Furthermore, in the patients who developed adverse reactions the antidote regimen had to be discontinued to alleviate the symptoms, thus potentially delaying therapy and prolonging hospital stay.
The frequency of detection of adverse reactions is well known to be dependent upon the methods chosen to ascertain them. The case of NAC illustrates this very well. Many clinicians will say they have never seen an adverse reaction, but of course most doctors will not be present at the time this occurs since adverse effects normally begin about an hour after the infusion of the antidote has commenced, when the physician has left to see other patients.
The incidence of reactions will be best tested under conditions of a formal clinical trial, and only one placebo-controlled trial has been conducted in which therapies were used to mitigate the risk of anaphylactoid responses.37 This was a study conducted using an antiemetic as the prophylactic, since vomiting is also a recognised adverse effect of intravenous NAC. In this study anaphylactoid responses requiring intervention or therapy were detected in 30% of the cases receiving the standard NAC regimen.47 This is much higher than has been previously documented, where the techniques used to detect reactions have tended to involve monitoring of specific therapies such as antihistamines and bronchodilators using prescription records. Using this methodology ADR rates generally range between about 5 and 10%.58
The precise mechanisms underlying the adverse reaction have been examined in a study in patients receiving NAC for paracetamol overdose. This indicated that peak concentrations of NAC were related to the onset of the adverse effect, but interestingly there was no obvious difference in that concentration in patients who developed or did not develop an anaphylactoid response. The key difference was in the concentration of circulating histamine, which was much higher in those with the reactions.46 The origins of this histamine release seem most likely to be basophils, since the normally accepted marker of mast cell degranulation, tryptase, was unchanged. These findings therefore suggest, as previously demonstrated in volunteers using intradermal injections,55 that some patients are indeed predisposed to have an anaphylactoid response. Remarkably though the concentration of paracetamol in blood also affects the frequency of these reactions, although the mechanism of this effect has not been determined. This is been shown in a number of studies in which patients who received NAC were grouped by paracetamol concentration at time of admission. The incidence of reactions is inversely related to paracetamol concentration above be 100 mg L−1 treatment line.59 The net effect of these findings is that reducing the intervention concentration for paracetamol, in an attempt to completely prevent risk of hepatic toxicity, will inevitably increase the incidence of adverse effects to the antidote.
Implications for dosing of NAC: newer regimens
The dose of NAC used in the initial studies intravenously was empirical and based on theoretical calculations. In North America the oral regimen adopted, principally because of the lack of licensed liquid preparation in the USA at that time, involved 140 mg kg−1 as a first loading dose followed by maintenance dose of 70 mg kg−1 4 hours after the loading dose, repeated every 4 hours for a total of 17 maintenance doses.23 This regimen, therefore, took approximately three days to administer, whereas in the UK the majority of patients had been discharged within 24 hours. This mismatch has been resolved over the years as US physicians using an oral regimen have examined the efficacy of shorter durations of oral therapy, but more importantly with the adoption of the intravenous approach across the USA, albeit initially over 48 h.60There has been debate about which is the most effective route for NAC, since the oral one should deliver more efficiently to the liver during first pass, but no direct comparative trials.61 Retrospective comparison of 2 cohorts of patients treated in North America, intravenous in Canada and orally in the USA, showed very little difference in efficacy measured as incidence of liver injury. Death rates were too low to allow any demonstration of the effect on mortality. Thus there seemed little evidence of difference in benefit to the majority of patients of giving the potentially larger doses provided orally.62
One key difference has been the far fewer anaphylactoid ADRs reported in the literature with oral NAC. There has also been some uncertainty as to the need to give such large initial doses of NAC IV as the oral regimen seems as effective.15 The clinical trial conducted in the UK of a simpler 12 h infusion regimen, using 200 mg per kg body weight over 4 h followed by 100 mg kg−1 over 8 h was not powered to demonstrate equivalent efficacy, though there was no signal of a greater risk of hepatic injury using the shorter, simpler regimen using conventional or novel biomarkers.47 Since this study others have confirmed that a slower initial infusion results in fewer ADRs. The 12 h NAC regimen is now routine clinical practice in a number of UK hospitals.63 In an audit of over 3300 patients there was no difference in liver injury or liver synthetic dysfunction between regimens (unpublished). This multi-centre study confirmed that the 12 h regimen was associated with fewer anaphylactoid reactions as measured by rate of anti-histamine prescription (11% with the 21 h regimen, 2% with the 12 h regimen). We believe that the 12 h regimen should be the standard of care given its enhanced safety profile, equivalent efficacy and shorter duration. Furthermore, patients with liver injury receive NAC at a quicker rate (compared to the 21 h regimen). For example, in the first 21 h, patients with the new regimen receive 500 mg kg−1 NAC if they have injury (such as an increase in ALT) compared with 300 mg kg−1 of NAC with the standard regimen (Table 1). An alternative approach to a shorter IV treatment time has been studied in a study in which a lower (250 mg kg−1 NAC over 12 h) was compared to a standard 20 h 300 mg regimen, with the decision to stop therapy being made on blood tests at 12 h.64 Whist this study confirms that blood testing at 12 h into an infusion is useful, the logic of the reduced initial dose of NAC in the 12 h group seems questionable to us, and this seems to offer no advance on SNAP.
Future developments
Predicting the future is always a challenge, however, there are some indicators that recent developments may allow the development of a new approach to managing paracetamol overdose, including a better targeting of the use of NAC.The first change possible should be the better identification of patients at risk of paracetamol induced liver injury. Based on the studies done to date looking at new microRNA and protein biomarkers it is clear that changes in these markers occur earlier than the traditional measures currently used. This indicates that liver injury has started before NAC is administered in virtually all patients who develop a subsequent ALT increase. Importantly the newer biomarkers provide better differentiation than that provided by the current treatment nomograms. One caveat is that all patients in whom biomarkers have been evaluated to date have received NAC. Further work will have to be done, firstly to provide biomarker analysis that is reliable and quick, and current developments suggest it was will be possible, and secondly to ensure that a normal biomarker concentration at a given time point after overdose is a reliable indicator of good outcome without NAC treatment.65 It is clear, however, that new biomarkers can accurately identify those patients who will get liver injury despite NAC treatment, which provides a new mechanism for identifying those patients who should be treated with different therapeutic approaches (see below).
Secondly, from our experience we would urge all clinicians now treating paracetamol poisoning with slower initial NAC infusions to also consider a shorter overall course in line with that we have published.47 This has 3 advantages, simplicity in use, fewer ADRs and shorter time on an infusion for most patients. As described above, it also allows additional NAC to be administered earlier in those who are identified at risk of developing hepatotoxicity at an earlier time of 12 h into therapy.
Any pharmacologist or toxicologist who looks at NAC dosing regimens will be surprised that the same dose of antidote is used initially in all patients irrespective of the dose of paracetamol. Since NAC is theoretically given to replace endogenous glutathione consumed by the production of the reactive paracetamol metabolite the logic of giving the same dose of antidote to everyone seems clearly flawed. Indeed, studies in the 1980s show that in patients who developed liver failure benefited from continuing doses of NAC after the end of the 21 hours initial infusion.66 This has led to the use of extended NAC infusion in patients to have any evidence of significant liver damage, now usually assessed by changes in the concentrations of ALT or in the INR.
Establishing whether higher doses of antidote are necessary in patients taking large overdoses is a clinical challenge. There is evidence that patients taking doses of paracetamol causing concentrations in blood above a concentration nomogram commencing at 300 mg L−1 four hours post overdose have larger rises in hepatic enzymes and a greater frequency of significant liver injury when compared to cohorts treated in the same hospitals with lower concentrations of paracetamol.38,67 Some clinicians already advise using higher doses of NAC in such patients, but clinical trial evidence supporting this approach is lacking. Because the new 12 h regimen is not dose limited by toxicity (such as anaphylactoid reactions), there is the potential to give higher doses to patients with high paracetamol body loads without inducing unacceptable side effects.
The final challenge is to consider what other treatments might be possible in patients who are either at a significant risk of, or are developing, liver injury with its potentially fatal outcome. Studies have shown that a number of agents offer potential for alleviating or preventing liver injury in animal models.68 The difficulty has always been that these drugs are potentially toxic and expensive. Determining which patients to enter into a clinical trial early enough for them to be effective in man is also problematic. A new approach may now be possible, however, because of the availability of biomarkers that are differentiating early in the course of liver injury. Newer, potentially safer, hepato-protective agents are in development, and one agent, calmangafodipir,69 has completed phase 1 studies in patients (clinicaltrials.gov: NCT03177395). Is to be hoped within the next 10 years that such agents, in combination with NAC, will eventually fully mitigate the effects of paracetamol overdose.
Conclusion
We are now in a position to significantly refine the treatment of paracetamol overdose. To date we have used a sub-optimal ‘one size fits all’ dosing regimen that was not based on robust dose finding studies. In the near future a precision medicine approach will be used in clinical practice with patients identified as higher or lower risk at first presentation to hospital. This will allow robust clinical trials testing the clinical and cost effectiveness of new treatment pathways. It is the authors’ opinion that a 12 h NAC regimen should be the standard of care because it is safer in use, is as effective, and shorter in duration compared with historic protocols.Conflicts of interest
There are no conflicts to declare.References
- B. B. Brodie and J. Axelrod, The fate of acetanilide in man, J. Pharmacol. Exp. Ther., 1948, 94(1), 29–38 CAS.
- D. N. N. Bateman and J. Dear, Medicine, poison, and mystic potion: a personal perspective on paracetamol Louis Roche lecture, Stockholm, 2009, Clin. Toxicol., 2010, 48(2), 97–103 CrossRef CAS PubMed.
- D. R. Camidge, R. J. Wood and D. N. Bateman, The epidemiology of self-poisoning in the UK, Br. J. Clin. Pharmacol., 2003, 56, 613–619 CrossRef CAS PubMed.
- D. G. Davidson and W. N. Eastham, Acute liver necrosis following overdose of paracetamol, Br. Med. J., 1966, 2(5512), 497–499 CrossRef CAS PubMed.
- J. S. Thomson and L. F. Prescott, Liver damage and impaired glucose tolerance after paracetamol overdosage, Br. Med. J., 1966, 2(5512), 506–507 CrossRef CAS PubMed.
- J. R. Mitchell, D. J. Jollow, W. Z. Potter, D. C. Davis, J. R. Gillette and B. B. Brodie, Acetaminophen-induced hepatic necrosis. I. Role of drug metabolism, J. Pharmacol. Exp. Ther., 1973, 187(1), 185–194 CAS.
- W. Z. Potter, D. C. Davis, J. R. Mitchell, D. J. Jollow, J. R. Gillette and B. B. Brodie, Acetaminophen-induced hepatic necrosis. 3. Cytochrome P-450-mediated covalent binding in vitro, J. Pharmacol. Exp. Ther., 1973, 187(1), 203–210 CAS.
- W. Z. Potter, S. S. Thorgeirsson, D. J. Jollow and J. R. Mitchell, Acetaminophen-induced hepatic necrosis. V. Correlation of hepatic necrosis, covalent binding and glutathione depletion in hamsters, Pharmacology, 1974, 12(3), 129–143 CrossRef CAS PubMed.
- A. R. Buckpitt, D. E. Rollins and J. R. Mitchell, Varying effects of sulfhydryl nucleophiles on acetaminophen oxidation and sulfhydryl adduct formation, Biochem. Pharmacol., 1979, 28(19), 2941–2946 CrossRef CAS PubMed.
- L. F. Prescott, P. Roscoe, N. Wright and S. S. Brown, Plasma-paracetamol half-life and hepatic necrosis in patients with paracetamol overdosage, Lancet, 1971, 1(7698), 519–522 CrossRef CAS.
- B. H. Rumack and H. Matthew, Actaminophen poiosning and toxicity, Paediatrics, 1975, 55, 871–876 CAS.
- W. Bernal and J. Wendon, Acute liver failure, N. Engl. J. Med., 2013, 369(26), 2525–2534 CrossRef CAS PubMed.
- A. L. Chiew, C. Gluud, J. Brok and N. Buckley, Interventions for paracetamol (acetaminophen) overdose, Cochrane Database Syst. Rev., 2018,(2), CD003328 Search PubMed.
- N. Pakravan, K. J. J. Simpson, W. S. S. Waring, C. M. M. Bates and D. N. N. Bateman, Renal injury at first presentation as a predictor for poor outcome in severe paracetamol poisoning referred to a liver transplant unit, Eur. J. Clin. Pharmacol., 2009, 65(2), 163–168 CrossRef CAS PubMed.
- B. H. Rumack and D. N. Bateman, Acetaminophen and acetylcysteine dose and duration: Past, present and future, Clin. Toxicol., 2012, 50(2), 91–98 CrossRef CAS PubMed.
- J. R. Mitchell, S. S. Thorgeirsson, W. Z. Potter, D. J. Jollow and H. Keiser, Acetaminophen-induced hepatic injury: protective role of glutathione in man and rationale for therapy, Clin. Pharmacol. Ther., 1974, 16(4), 676–684 CrossRef CAS PubMed.
- J. O. Miners, R. Drew and D. J. Birkett, Mechanism of action of paracetamol protective agents in mice in vivo, Biochem. Pharmacol., 1984, 33(19), 2995–3000 CrossRef CAS PubMed.
- L. F. Prescott, R. W. Newton, C. P. Swainson, N. Wright, A. R. Forrest and H. Matthew, Successful treatment of severe paracetamol overdosage with cysteamine, Lancet, 1974, 1(7858), 588–592 CrossRef CAS.
- L. F. Prescott, G. R. Sutherland, J. Park, I. J. Smith and A. T. Proudfoot, Cysteamine, methionine, and penicillamine in the treatment of paracetamol poisoning, Lancet, 1976, 2(7977), 109–113 CrossRef CAS.
- L. F. Prescott, J. Park, A. Ballantyne, P. Adriaenssens, A. T. Proudfoot and R. N. Illingworth, et al., Treatment of paracetamol (acetaminophen) poisoning with N-acetylcysteine, Lancet, 1977, 2(8035), 432–434 CrossRef CAS.
- L. F. Prescott, R. N. Illingworth, J. A. Critchley, M. J. Stewart, R. D. Adam and A. T. Proudfoot, Intravenous N-acetylcystine: the treatment of choice for paracetamol poisoning, Br. Med. J., 1979, 2(6198), 1097–1100 CrossRef CAS PubMed.
- J. A. Vale, T. J. Meredith and R. Goulding, Treatment of acetaminophen poisoning. The use of oral methionine, Arch. Intern. Med., 1981, 141, 394–396 CrossRef CAS PubMed.
- M. J. Smilkstein, G. L. Knapp, K. W. Kulig and B. H. Rumack, Efficacy of oral N-acetylcysteine in the treatment of acetaminophen overdose. Analysis of the national multicenter study (1976 to 1985), N. Engl. J. Med., 1988, 319(24), 1557–1562 CrossRef CAS PubMed.
- Benefit risk profile of acetylcysteine in the management of paracetamol overdose [Internet]. Medicines and Healthcare products Regulatory Agency, 2012. Available from: http://www.mhra.gov.uk/home/groups/pl-p/documents/drugsafetymessage/con184609.pdf.
- L. F. Prescott, The chief scientist reports...prevention of hepatic necrosis following paracetamol overdosage, Health Bull., 1978, 36(4), 204–212 CAS.
- P. Routledge, J. A. Vale, D. N. Bateman, G. D. Johnston, A. Jones and A. Judd, et al., Paracetamol (acetaminophen) poisoning, Br. Med. J., 1998, 317(7173), 1609–1610 CrossRef CAS PubMed.
- A. D. Vliegenthart, R. A. Kimmitt, J. H. Seymour, N. Z. Homer, J. I. Clarke and M. Eddleston, et al., Circulating acetaminophen metabolites are toxicokinetic biomarkers of acute liver injury, Clin. Pharmacol. Ther., 2017, 101, 531–540 CrossRef CAS PubMed.
- K. J. Heard, Acetylcysteine for Acetaminophen Poisoning, N. Engl. J. Med., 2008, 359, 285–292 CrossRef CAS PubMed.
- J. T. Slattery, J. M. Wilson, T. F. Kalhorn and S. D. Nelson, Dose-dependent pharmacokinetics of acetaminophen: evidence of glutathione depletion in humans, Clin. Pharmacol. Ther., 1987, 41(4), 413–418 CrossRef CAS.
- J. T. Slattery, S. D. Nelson and K. E. Thummel, The complex interaction between ethanol and acetaminophen, Clin. Pharmacol. Ther., 1996, 60(3), 241–246 CrossRef CAS . Available from: http://www.ncbi.nlm.nih.gov/pubmed/8841146.
- H. A. Sampson, A. Munoz-Furlong, R. L. Campbell, N. F. Adkinson Jr., S. A. Bock and A. Branum, et al., Second symposium on the definition and management of anaphylaxis: summary report–Second National Institute of Allergy and Infectious Disease/Food Allergy and Anaphylaxis Network symposium, J. Allergy Clin. Immunol., 2006, 117(2), 391–397 CrossRef PubMed.
- A. D. B. Vliegenthart, R. A. Kimmitt, J. H. Seymour, N. Z. Homer, J. I. Clarke and M. Eddleston, et al., Circulating acetaminophen metabolites are toxicokinetic biomarkers of acute liver injury, Clin. Pharmacol. Ther., 2017, 101(4), 531–540 CrossRef CAS PubMed.
- A. Ramachandran and H. Jaeschke, Acetaminophen Hepatotoxicity, Semin. Liver Dis., 2019 DOI:10.1055/s-0039-1679919 , [Epub ahead of print].
- L. F. Prescott, J. W. Donovan, D. R. Jarvie and A. T. Proudfoot, The disposition and kinetics of intravenous N-acetylcysteine in patients with paracetamol overdosage, Eur. J. Clin. Pharmacol., 1989, 37(5), 501–506 CrossRef CAS PubMed.
- L. Borgstrom, B. Kagedal and O. Paulsen, Pharmacokinetics of N-acetylcysteine in man, Eur. J. Clin. Pharmacol., 1986, 31(2), 217–222 CrossRef CAS PubMed.
- J. Zhou, L. D. Coles, R. V. Kartha, N. Nash, U. Mishra and T. C. Lund, et al., Intravenous Administration of Stable-Labeled N-Acetylcysteine Demonstrates an Indirect Mechanism for Boosting Glutathione and Improving Redox Status, J. Pharm. Sci., 2015, 104(8), 2619–2626 CrossRef CAS PubMed.
- H. K. R. Thanacoody, A. Gray, J. W. W. Dear, J. Coyle, E. A. A. Sandilands and D. J. J. Webb, et al., Scottish and Newcastle antiemetic pre-treatment for paracetamol poisoning study (SNAP), BMC Pharmacol. Toxicol., 2013, 14(20), 1–12 Search PubMed.
- D. G. Cairney, H. K. S. Beckwith, K. Al-Hourani, M. Eddleston, D. N. Bateman and J. W. Dear, Plasma paracetamol concentration at hospital presentation has a dose-dependent relationship with liver injury despite prompt treatment with intravenous acetylcysteine, Clin. Toxicol., 2016, 54, 405–410 CrossRef CAS PubMed.
- R. G. Hendrickson, What is the most appropriate dose of N-acetylcysteine after massive acetaminophen overdose?, Clin. Toxicol., 2019, 1–6 Search PubMed.
- M. L. Sivilotti, A. M. Good, M. C. Yarema, D. N. Juurlink and D. W. Johnson, A new predictor of toxicity following acetaminophen overdose based on pretreatment exposure, Clin. Toxicol., 2005, 43(4), 229–234 CrossRef CAS.
- M. L. A. Sivilotti, T. J. Green, C. Langmann, M. Yarema, D. Juurlink and D. Johnson, Multiplying the serum aminotransferase by the acetaminophen concentration to predict toxicity following overdose, Clin. Toxicol., 2010, 48, 793–799 CrossRef CAS PubMed.
- A. Wong, M. L. A. Sivilotti, P. I. Dargan, D. M. Wood and S. L. Greene, External validation of the paracetamolaminotransferase multiplication product to predict hepatotoxicity from paracetamol overdose, Clin. Toxicol., 2015, 53, 353 Search PubMed.
- A. Wong, A. Graudins, F. Kerr and S. L. Greene, Paracetamol toxicity: what would be the implications of a change in Australian treatment guidelines?, Emerg. Med. Australas., 2014, 26(2), 183–187 CrossRef PubMed.
- L. E. Schmidt, T. T. Knudsen, K. Dalhoff and F. Bendtsen, Effect of acetylcysteine on prothrombin index in paracetamol poisoning without hepatocellular injury, Lancet, 2002, 360(9340), 1151–1152 CrossRef CAS.
- I. M. Whyte, N. A. Buckley, D. M. Reith, I. Goodhew, M. Seldon and A. H. Dawson, Acetaminophen causes an increased international normalized ratio by reducing functional factor VII, Ther. Drug Monit., 2000, 22, 742–748 CrossRef CAS PubMed.
- N. Pakravan, W. S. Waring, S. Sharma, C. Ludlam, I. Megson and D. N. Bateman, Risk factors and mechanisms of anaphylactoid reactions to acetylcysteine in acetaminophen overdose, Clin. Toxicol., 2008, 46(8), 697–702 CrossRef CAS PubMed.
- D. N. Bateman, J. W. Dear, H. K. R. Thanacoody, S. H. L. Thomas, M. Eddleston and E. A. Sandilands, et al., Reduction of adverse effects from intravenous acetylcysteine treatment for paracetamol poisoning: A randomised controlled trial, Lancet, 2014, 383(9918), 697–704 CrossRef CAS.
- D. J. J. Antoine, J. W. W. Dear, P. S. S. Lewis, V. Platt, J. Coyle and M. Masson, et al., Mechanistic biomarkers provide early and sensitive detection of acetaminophen-induced acute liver injury at first presentation to hospital, Hepatology, 2013, 58(2), 777–787 CrossRef CAS PubMed.
- J. W. Dear, J. I. Clarke, B. Francis, L. Allen, J. Wraight and J. Shen, et al., Risk stratification after paracetamol overdose using mechanistic biomarkers: results from two prospective cohort studies, Lancet Gastroenterol. Hepatol., 2018, 3, 104–113 CrossRef.
- EMA. 2016. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Other/2016/09/WC500213479.pdf.
- FDA. 2016. Available from: http://www.fda.gov/Drugs/DevelopmentApprovalProcess/ucm434382.htm.
- N. G. Walton, T. A. Mann and K. M. Shaw, Anaphylactoid reaction to N-acetylcysteine, Lancet, 1979, 2(8155), 1298 CrossRef CAS.
- T. G. Mant, J. H. Tempowski, G. N. Volans and J. C. Talbot, Adverse reactions to acetylcysteine and effects of overdose, Br. Med. J., 1984, 289(6439), 217–219 CrossRef CAS PubMed.
- A. V. Appelboam, P. I. Dargan and J. Knighton, Fatal anaphylactoid reaction to N-acetylcysteine: caution in patients with asthma, Emerg. Med. J., 2002, 19(6), 594–595 CrossRef CAS.
- D. N. N. Bateman, K. W. W. Woodhouse and M. D. D. Rawlins, Adverse reactions to N-acetylcysteine, Hum. Toxicol., 1984, 3(5), 393–398 CrossRef CAS.
- W. S. Waring, A. F. Stephen, O. D. Robinson, M. A. Dow and J. M. Pettie, Lower incidence of anaphylactoid reactions to N-acetylcysteine in patients with higher acetaminophen concentrations after overdose, Clin. Toxicol., 2008, 46, 496–500 CrossRef CAS.
- L. E. Schmidt and K. Dalhoff, Risk factors in the development of adverse reactions to N-acetylcysteine in patients with paracetamol poisoning, Br. J. Clin. Pharmacol., 2001, 51(1), 87–91 CrossRef CAS.
- N. Buckley and D. Gunnell, Does restricting pack size of paracetamol (acetaminophen) reduce suicides?, PLoS Med., 2007, 4, e152 CrossRef PubMed.
- D. N. Bateman, R. Carroll, J. Pettie, T. Yamamoto, M. E. Elamin and L. Peart, et al., Effect of the UK's revised paracetamol poisoning management guidelines on admissions, adverse reactions and costs of treatment, Br. J. Clin. Pharmacol., 2014, 78(3), 610–618 CrossRef PubMed.
- M. J. Smilkstein, A. C. Bronstein, C. Linden, W. L. Augenstein, K. W. Kulig and B. H. Rumack, Acetaminophen overdose: a 48-hour intravenous N-acetylcysteine treatment protocol, Ann. Emerg. Med., 1991, 20(10), 1058–1063 CrossRef CAS PubMed.
- N. A. Buckley, I. M. Whyte, D. L. O'Connell and A. H. Dawson, Oral or intravenous N-acetylcysteine: which is the treatment of choice for acetaminophen (paracetamol) poisoning?, J. Toxicol., Clin. Toxicol., 1999, 37(6), 759–767 CrossRef CAS PubMed.
- M. C. Yarema, D. W. Johnson, R. J. Berlin, M. L. Sivilotti, A. Nettel-Aguirre and R. F. Brant, et al., Comparison of the 20-hour intravenous and 72-hour oral acetylcysteine protocols for the treatment of acute acetaminophen poisoning, Ann. Emerg. Med., 2009, 54(4), 606–614 CrossRef PubMed.
- J. Pettie, E. A. Sandilands, M. A. Dow, E. Macrae, E. Brogan and A. Veiraiah, et al., Treatment of acetaminophen overdose with a 12 h acetylcysteine regimen: The first report of safety and efficacy in routine clinical practice, Clin. Toxicol., 2016, 54, 666 Search PubMed.
- A. Wong, R. McNulty, D. Taylor, M. Sivilotti, S. Greene and N. Gunja, et al., The NACSTOP Trial: A Multicenter, Cluster-Controlled Trial of Early Cessation of Acetylcysteine in Acetaminophen Overdose, Hepatology, 2019, 69(2), 774–784 CrossRef CAS PubMed.
- D. M. Rissin, B. López-Longarela, S. Pernagallo, H. Ilyine, A. D. B. Vliegenthart, J. W. Dear and J. J. Díaz-Mochón, et al., Polymerase-free measurement of microRNA-122 with single base specificity using single molecule arrays: Detection of drug-induced liver injury, PLoS One, 2017, 12(7), e0179669 CrossRef PubMed.
- R. Keays, P. M. Harrison, J. A. Wendon, A. Forbes, C. Gove and G. J. Alexander, et al., Intravenous acetylcysteine in paracetamol induced fulminant hepatic failure: a prospective controlled trial, Br. Med. J., 1991, 303(6809), 1026–1029 CrossRef CAS PubMed.
- D. N. Bateman and J. W. Dear, Should we treat very large paracetamol overdose differently?, Br. J. Clin. Pharmacol., 2017, 83, 1163–1165 CrossRef PubMed.
- J. W. W. Dear, K. J. J. Simpson, M. P. Nicolai, J. H. H. Catterson, J. Street and T. Huizinga, et al., Cyclophilin A is a damage-associated molecular pattern molecule that mediates acetaminophen-induced liver injury, J. Immunol., 2011, 187(6), 3347–3352 CrossRef CAS PubMed.
- J. W. Dear on behalf of the P trial investigators, Randomised Open Label Exploratory, Safety and Tolerability Study with Calmangafodipir in Patients Treated with the 12-hour Regimen of N-Acetylcysteine for Paracetamol Overdose: The PP100-01 for Overdose of Paracetamol Trial (POP Trial): study protocol for, BMC Trials, 2018, 20, 27 Search PubMed.
- A. Wong and A. Graudins, Simplification of the standard three-bag intravenous acetylcysteine regimen for paracetamol poisoning results in a lower incidence of adverse drug reactions, Clin. Toxicol., 2016, 54(2), 115–119 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2019 |