
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Bulky, dendronized iridium complexes and their photoluminescence†
Guang
Zhang
a,
Felix
Hermerschmidt
b,
Anup
Pramanik
 c,
Dieter
Schollmeyer
d,
Martin
Baumgarten
*a,
Pranab
Sarkar
*c,
Emil J. W.
List-Kratochvil
be and
Klaus
Müllen
*a
c,
Dieter
Schollmeyer
d,
Martin
Baumgarten
*a,
Pranab
Sarkar
*c,
Emil J. W.
List-Kratochvil
be and
Klaus
Müllen
*a
aMax Planck Institute for Polymer Research, Ackermannweg 10, Mainz D-55128, Germany. E-mail: baumgart@mpip-mainz.mpg.de; muellen@mpip-mainz.mpg.de
bHumboldt-Universität zu Berlin, Institut für Physik, Institut für Chemie, IRIS Adlershof, Brook-Taylor-Str. 6, 12489 Berlin, Germany
cDepartment of Chemistry, Visva-Bharati University, Santiniketan 731235, India. E-mail: pranab.sarkar@visva-bharati.ac.in
dInstitute für Organische Chemie, Johannes-Gutenberg Universität, Mainz, 55128, Germany
eHelmholtz-Zentrum Berlin für Materialien und Energie GmbH, Brook-Taylor-Str. 6, 12489 Berlin, Germany
First published on 5th November 2019
Abstract
Solution-processed blue emitters are essential for low-cost organic light-emitting diodes (OLEDs) but still face challenges due to their poor color purity, low efficiency and limited operational stability. Herein, by extending the conjugation of ultraviolet-emissive, facial tris(diphenylbenzimidazolyl)iridium (Ir) (fac-(dpbic)3Ir), we introduce two new types of solution-processed emitters, i.e. triisopropylsilylethynyl(TIPSE)-substituted fac-(dpbic)3Ir (2) and fac-(dpbic)3Ir-based polyphenylene dendrimers D1 and D2. The emissions of Ir-complex 2 and the dendrimers were successfully pushed toward a pure and sky blue color, respectively, due to the dominant 3π–π* nature of their emissive excited states. As a pleasant surprise, the troublesome aggregation-induced red shift of the emission of Ir-complex 2 could be totally suppressed by the bulky TIPSE moieties. Ir-complex 2 displays pure blue emission in a solution-processed, non-doped OLED (non-optimized) with moderate efficiency and without any observed aggregation effects, which paves a way for the future design of high-performance, non-doped phosphorescent emitters. The dendrimers exhibit strong sky-blue emission at 77 K but their emission is completely quenched at ambient temperature. This is demonstrated to result from the much elongated Ir–Ccarbene bond by the strong steric hindrance of the bulky polyphenylene dendrons. The remarkably long Ir–Ccarbene bonds of the dendrimers make their TI states more easily accessible to the non-emissive 3MC state than those for fac-(dpbic)3Ir and compound 2 as supported by the quantum chemical results. This finding also promises suggestions for designing better dendrimer-based blue emitters.
Introduction
Even though OLED-based products, e.g. flat-panel displays and solid-state lighting are coming into people's lives slowly,1–3 the prices are still quite high due to the complicated fabrication of multilayer devices with costly ultrahigh vacuum (UHV) deposition and inefficient use of materials.4 Solution-processed methods, e.g. spin-coating and inkjet printing, on the other hand, could dramatically reduce the cost for the fabrication of devices, and therefore attract great attention among the communities of flexible electronics and are believed to play vital roles in the manufacture of inexpensive and next-generation OLEDs.5 Therefore, developing high-performance and solution-processable emitters has been and is still a hot topic.4,6,7Dendrimers and polymers are more popular candidates for designing solution-processed materials than small molecules which are usually prone to form crystalline rather than the preferred amorphous films for OLEDs.4 Dendrimers are monodisperse and highly-branched macromolecules with a tailor-made core, a shell and a surface structure.8 They feature several desired attributes for the design of solution-processable emitters in comparison to polymers, i.e. absolute reproducibility, high photoluminescence quantum yield (PLQY) by encapsulation of the emitter in the core, and layer-by-layer substitution to create multi-functional materials with potential for greatly reducing the complexity of the device.6,9–12
It is essential to select an appropriate core, i.e. an emitter for the development of efficient dendrimer-based light-emitting materials. Phosphorescent emitters (PEs) such as iridium and platinum complexes are much more efficient than conventional fluorescent ones (e.g. pyrene derivatives) because PEs can generate both singlet and triplet excitons in OLEDs (corresponding to 100% internal quantum yield (IQE) and 20% external quantum yield (EQE) in theory) but the other kind can only harvest singlet ones (not more than 25% IQE and 5% EQE).13–16
The neat films of small-molecular PEs are liable to experience severe self-quenching. Therefore, a matrix is usually employed to accommodate the emitters (also called dopants) to ensure high PLQYs. The matrix composed of host materials, e.g. N,N′-dicarbazolyl-3,5-benzene (mCP), serves as a medium for charge transport and energy transfer to the dopants.17 However, this method still encounters uneven dispersions of the dopants in the host with UHV depositions. Dendrimers on the other hand effectively avoid this problem.6,9–12 Within a rigid and shape-persistent dendritic architecture, a PE is well encapsulated in the core by the bulky dendrons, and the surface is functionalized with host moieties to exert charge transport and surface-to-core energy transfer. Besides, one can precisely manipulate the ratio and distances between the hosts and the PE by molecular design.10,11 On account of these merits, dendrimer-based PEs have gained significant breakthroughs within the last two decades.6,18–23 Both highly-efficient green and red dendrimer PEs with comparable OLED performances to small molecules have been reported.21–23 As to dendrimer-based blue PEs, even though a few examples are available, they all suffer from either very low device efficiencies or poor colour purities.24–28 Therefore, a high-performance, dendrimer-based and pure blue PE is still missing.
PEs based on shape-persistent polyphenylene dendrimers (PPDs) have been studied as well.29,30 For example, Qin, et al. prepared several PPDs as efficient green PEs.29 In addition, our group has demonstrated that core-surface-substituted, first-generation PPDs have a good architecture for the design of better dendrimer-based emitters by judicious selection of peripheral moieties to establish efficient surface-to-core energy transfer and intermolecular charge transport.31,32
Extending the conjugation of a PE with the dominant 3π–π* (3LC) character of the emissive excited states has been found by us to give a strongly red-shifted emission. The comparison of the fac-(dfpypy)3Ir-based dendrimer (D3) with fac-(dfpypy)3Ir (9) (red shift: 50 nm) is shown (Fig. 1).33 In addition, elongated conjugation within the ligands tends to strengthen the 3LC characteristics of its emissive excited states.34 We thought to make use of these findings to design new dendrimer-based blue PEs. Thus, utilizing a PE (with ultraviolet (UV) emission) as the core of the dendrimer could possibly push the emission to a pure blue region. Schildknecht et al. reported that fac-(dpbic)3Ir (Fig. 1) as a near UV emitter (λmax: 400 nm) furnished a PLQY of 0.19.35 We therefore envisaged fac-(dpbic)3Ir as the core to develop new dendrimer-based blue PEs. As depicted in Fig. 1, a TIPSE-substituted fac-(dpbic)3Ir (2) and two first-generation, fac-(dpbic)3Ir-based PPDs, i.e.D1 and D2 which contain peripheral carbazoles to facilitate charge transport and energy transfer are synthesized and characterized. Their photophysical properties and the OLED performances of compound 2 and D2 are discussed as well.
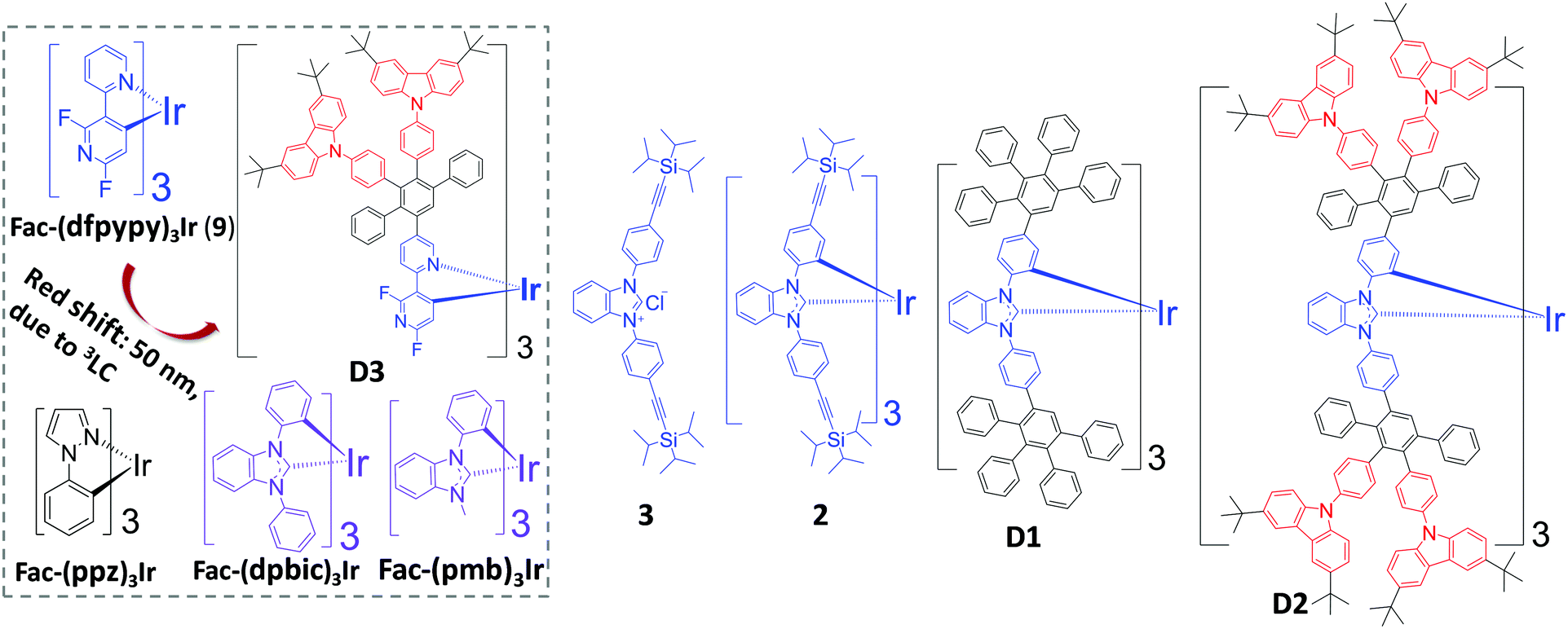 | ||
| Fig. 1 Molecular structures of fac-(dfpypy)3Ir, dendrimer D3, fac-(ppz)3Ir, fac-(dpbic)3Ir, fac-(pmb)3Ir, and the targeted compounds 3, 2, D1, and D2. | ||
Experimental section
Synthesis and characterization
Due to the final Diels–Alder reaction for building up the dendrimers, an ethynyl-substituted ligand 3 was synthesized as the starting compound (Scheme S1, ESI†). A twofold Buchwald-Hartwig coupling between 1,2-diaminobenzene and 1-bromo-4-TIPSE benzene (5) produced functionalized 1,2-diaminobenzene 4 in high yield (89%).36,37 Next, compound 4 was allowed to react with triethyl orthoformate and conc. HCl solution to afford TIPSE-functionalized 1,3-diphenyl benzimidazolium chloride 3 in high yield (86%).38,39 Thereafter, a reaction between IrCl3 and compound 3 yielded a TIPSE-functionalized carbene-chelated Ir-complex (2) (56%).40 Tetrabutylammonium fluoride (TBAF) activated the triple bonds of molecule 2 to form ethynyl-substituted Ir complex 1 in moderate yield (45%). Finally, the Diels–Alder reaction between compound 1 and tetraphenylcyclopentadienones (6 or 7) furnished the dendrimers (D1 or D2) in moderate to high yields (36% and 72% respectively). The moderate yield of D1 is probably due to the notable amount of D1 adhering to the column upon flash purification.The 1H NMR spectra (Fig. S1, ESI†) characterized Ir-complex 2 as a facial isomer with a total of eleven proton signals in the aromatic region (eleven aromatic protons in one ligand). Single crystals of compound 2 were obtained by slow addition of methanol to a dichloromethane solution. As depicted by the crystal structure in Fig. 2, the molecule adopts a quasi-octahedral geometry. Its three Ir–Ccarbene and Ir–Cphenyl bonds are slightly longer than those of fac-(pmb)3Ir34 (Table S2, ESI†) due to the bulky TIPSE moieties in Ir-complex 2.41 The non-coordinated benzene rings are highly twisted from the benzimidazole-based carbene plane due to the strong steric hindrance between the benzene ring and a carbene moiety from a nearby ligand. In contrast to fac-(dfpypy)3Ir42 and fac-(pmb)3Ir (Fig. 1),34 intermolecular π⋯π close interactions are not observed for compound 2 due to the protection by the bulky TIPSE segments. This suggests Ir-complex 2 as a promising candidate for application in non-doped solution-processed OLEDs.24–26,31,32,43,44 The reduced intermolecular interactions of Ir-complex 2 are also in accordance with its longer intermolecular Ir⋯Ir distance (∼12.90 Å) than those in fac-(dfpypy)3Ir (∼9.10 Å)42 and fac-(pmb)3Ir (∼9.38 Å).34
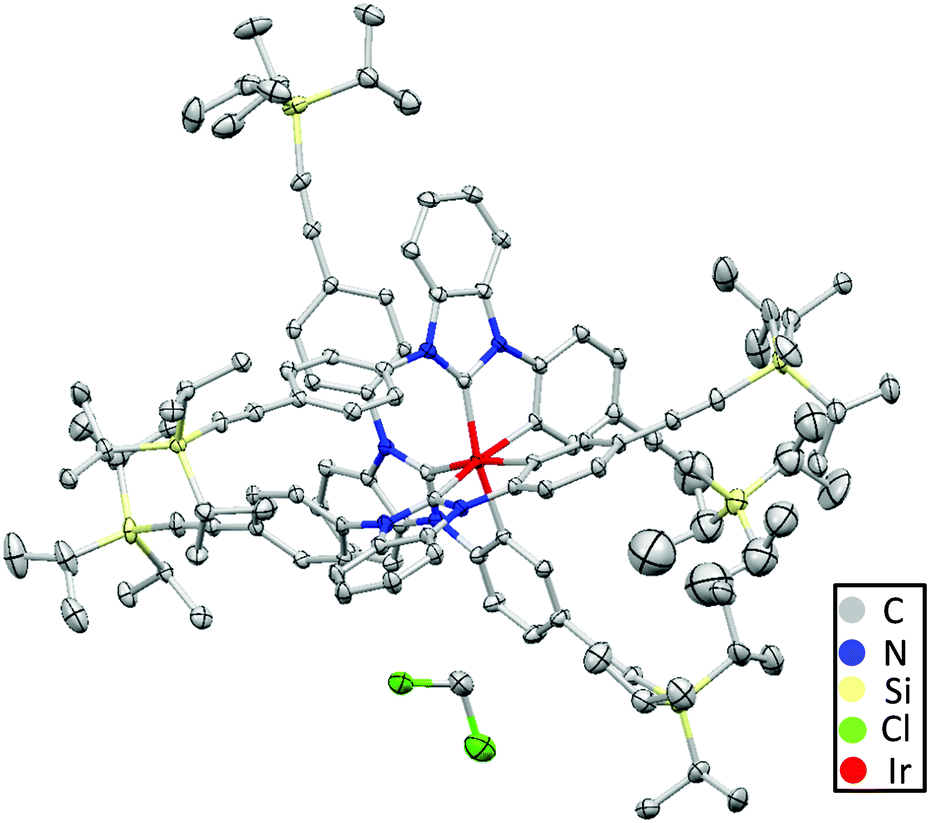 | ||
| Fig. 2 Single crystal structure of Ir-complex 2 (hydrogen atoms were eliminated for the sake of clarity). A dichloromethane solvent molecule is included in the unit cell. | ||
The dendrimers D1 and D2 were characterized by 1H and 13C NMR, MALDI-TOF mass and high-resolution mass spectroscopy. The MALDI-TOF mass spectra of each dendrimer showed a single peak of the molecular ion. In addition, the high-resolution MALDI-TOF mass spectra of the dendrimers show isotope patterns of the molecular ion in good agreement with the calculated results (Fig. S2 and S3, ESI†).
Results and discussion
Photophysical characterization
Regarding the absorption of compound 2, a major band with several shoulders in the UV region is due to the transitions of the TIPSE-functionalized, 1,3-diphenylbenzimidazole-based carbene ligand, which is consistent with the absorption of TIPSE-substituted, 1,3-diphenylbenzimidazolium chloride (3) (Fig. 1, 3 and Table 1). The small band above 350 nm is attributed to a mixture of metal-to-ligand charge transfer (MLCT) and 3LC, based on a comparison with the transitions of fac-(pmb)3Ir.34 Ir-complex 2 exhibits strong and pure-blue emission (λmax: 440 nm and 469 nm) in solution under argon protection because oxygen easily quenches the triplet states.45,46 The peak emission of compound 2 undergoes a bathochromic shift of 40 nm compared with that of (dpbic)3Ir.35 This red shift is due to the dominant 3LC nature of the emissive excited states of compound 2 together with its elongated conjugation.33 In addition, the emission of Ir-complex 2 at 77 K displays intense and nearly identical spectra to those measured at room temperature (Fig. 3b). This supports the major 3LC nature of the transition because a dominant 3MLCT-type of emission usually undergoes a hypsochromic shift in the solid matrix in contrast to that in the solution state.47,48 The PLQY was measured to be 0.19 with an integration sphere. The photoluminescence lifetime extracted from the corresponding decay curve of Ir-complex 2 (Fig. S7, ESI†) is around 0.49 μs which suggests the phosphorescent nature of Ir-complex 2.49 The phosphorescence lifetime is shorter than that of most known triplet emitters,49 and its decay pathways are currently under study. To the best of our knowledge, luminophore 2 is the first reported pure-blue Ir complex with alkynyl moieties in the ligand. Moreover, the thin film of compound 3 shows a strong and featureless deep blue emission, which is similar to those of other reported highly emissive benzimidazole-based organic salts;50,51 for instance, Boydston et al. synthesized a series of benzobis(imidazolium) salts with high PLQYs.50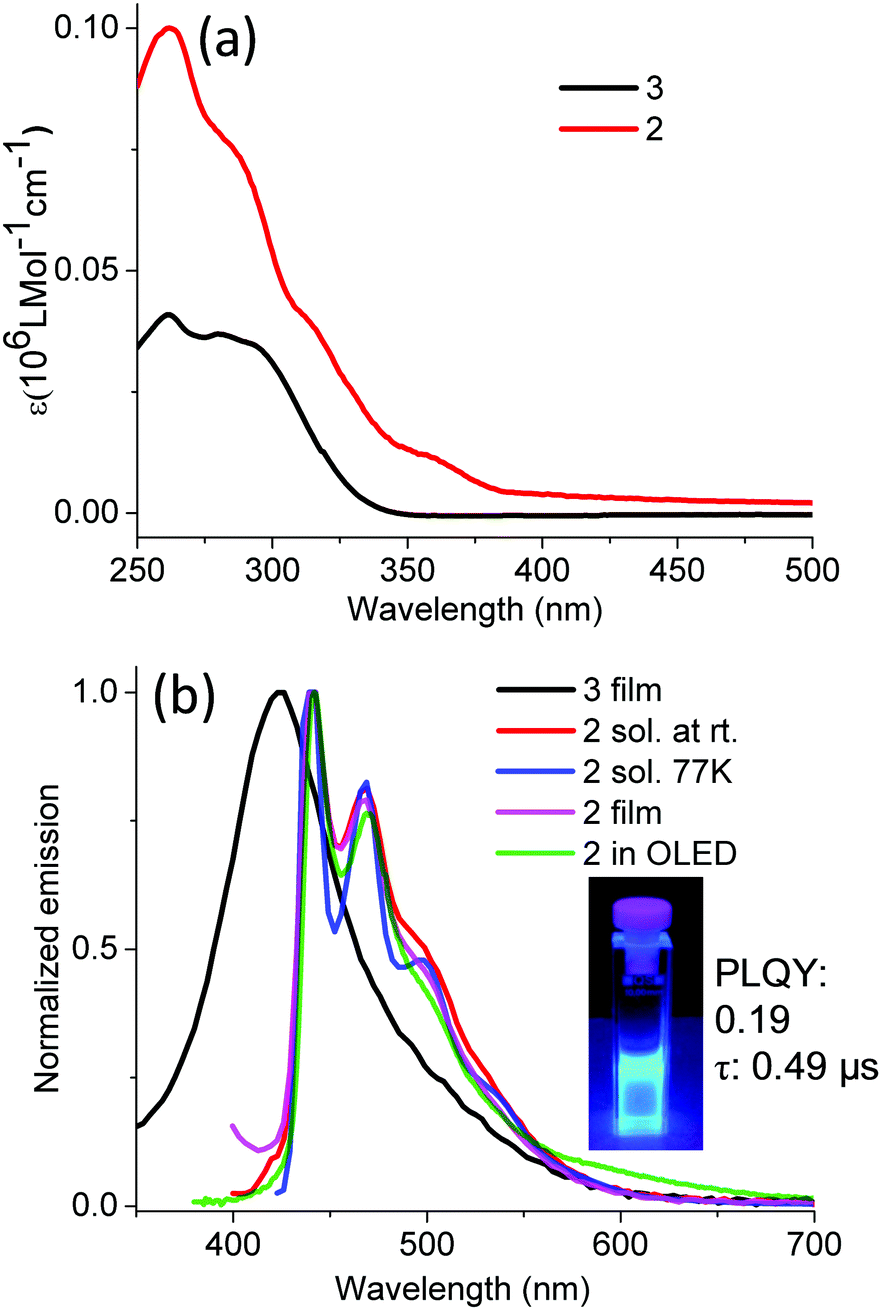 | ||
| Fig. 3 UV-vis absorption (a) and emission spectra (b) of Ir-complex 2 and bis(4-TIPSE phenyl)-substituted benzimidazolium salt 3 (10−6 M in DCM for absorption, emission: thin film of compound 3, ex: 300 nm; 2.4 × 10−4 M THF (at rt, argon protection) or 2-MeTHF (77 K) or thin films (doped in PMMA, 2 wt%) for compound 2, ex: 375 nm, inset image: emission of compound 2 at rt, ex: 365 nm, PLQY (error margin: ±5%) and photoluminescence lifetime of compound 2 were measured at 298 K under N2 in toluene at a concentration of 10−5 M; the electroluminescence spectra of compound 2 were measured in a solution-processed, non-doped device). | ||
| λ ab (nm) | λ em (nm) | E T | HOMO (eV) | LUMO (eV) | E g (eV)e | ||
|---|---|---|---|---|---|---|---|
| Sol | Sol | Film | |||||
| a Obtained from ref. 35. b The triplet energy (ET) was calculated from the highest energy peak of emission spectra (77 K), which is ∼1240/λem. c Obtained from ref. 17. d Calculated from cyclic voltammetry (CV) by comparing the first redox onset of dendrimers and the oxidation onset of ferrocene. e Calculated from the absorption edge of the longest wavelength band. f Obtained from the difference between the LUMO and Eg. | |||||||
| fac-(dpbic)3Ir | — | — | 400a | — | −5.28c | −1.35c | |
| 3 | 261, 280 | 433 | 425 | — | — | — | 3.70 |
| 2 | 261, 287, 355 | 441, 469, 504 | 441, 468, 505 | 2.81 | −5.23d | −2.11d | 3.26 |
| D1 | 281, 323 | 462, 488 (77 K) | 470, 500 | 2.68 | — | — | 3.25 |
| D2 | 297, 348 | 462, 490 (77 K) | 503 | 2.68 | −5.53d | −2.30f | 3.23 |
As to the absorption of dendrimers D1 and D2 (Fig. 4), the bands between 250 and 350 nm are due to ligand- and polyphenylene-centered transitions;52 for example, the peak at 297 nm of D2 is characteristic of a carbazole absorption.53 The weak shoulders above 350 nm are attributed to a mixture of MLCT and 3LC. The dendrimers in solutions are not emissive at room temperature even under argon protection but exhibit strong sky-blue emission at 77 K with nearly identical peak positions (λmax: ∼462 nm and 488 nm) and a slight bathochromic shift of D2 compared with D1. The quenching of the dendrimer emission at ambient temperatures could be due to the small energy barrier between the nonradiative excited state (NR) and the emissive excited state (T1), similar to the situation prevailing in other reported heavy-metal complexes, e.g. fac-Ir(ppz)3 (Fig. 1).34,54 Alternatively, the decay of the emissive excite state could be depleted by the vibrations or rotations of the polyphenylene dendrons. The latter assumption is supported by the detected weak emission of the dendrimers in thin films (Fig. 4). We claim that this is not the major origin of the phosphorescence quenching of the dendrimers at room temperature. We rather postulate that it must be ascribed to the rapid transition between T1 and NR states at ambient temperature.
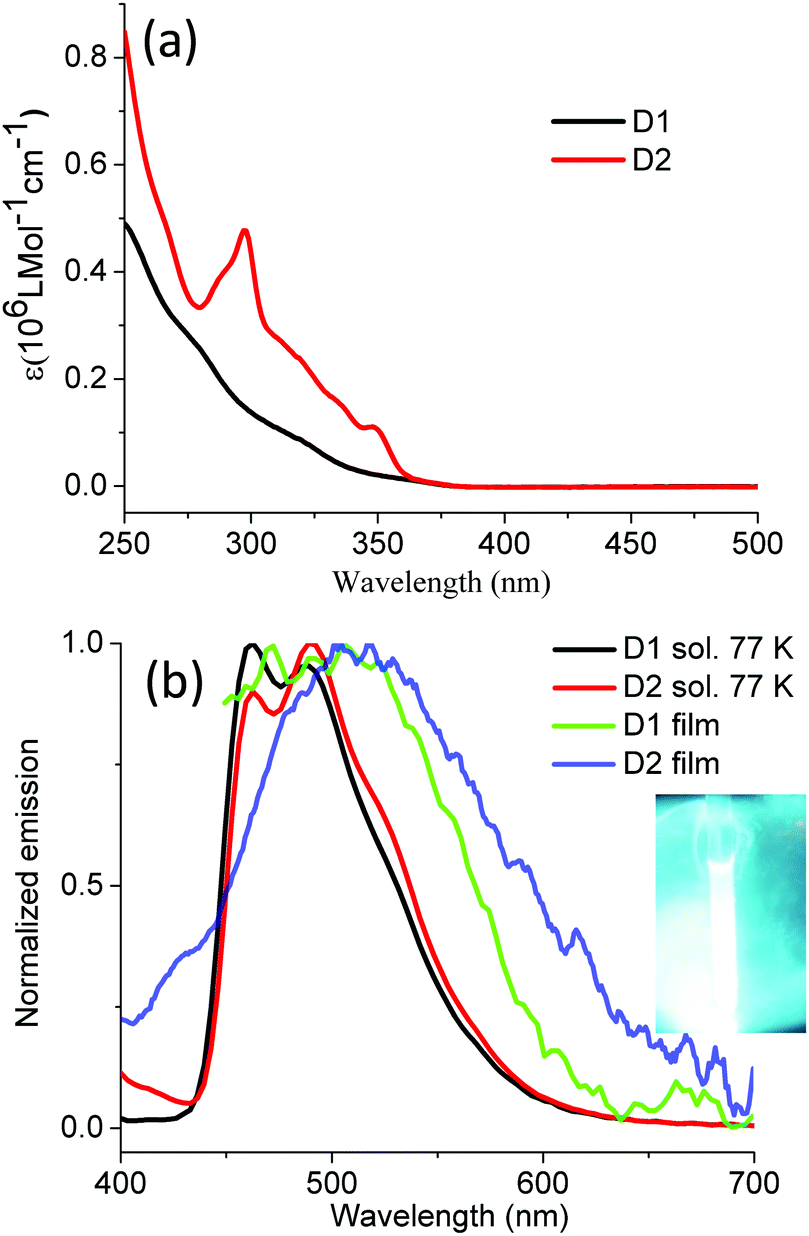 | ||
| Fig. 4 UV-vis absorption (a) and emission spectra (b) of dendrimers D1 and D2 (10−6 M in DCM for absorption, 1 mg ml−1 2-MeTHF (77 K) for emission, ex: 360 nm for both solution and thin film, inset picture: the emission of D1 at 77 K). | ||
Exploration of the emission efficiencies through theoretical calculations
Many transition-metal complexes are strongly emissive at 77 K but their emission is severely quenched at room temperature, which in many cases is ascribed to the thermal population of the nonradiative triplet metal-centered charge transfer state (3MC).54–59 For example, Sajoto et al. demonstrated that the emission efficiencies of many Ir-complexes, such as fac-(ppz)3Ir and fac-(pmb)3Ir (Fig. 1), were primarily determined by the energy gap between the T1 and NR states, i.e.3MC.54 Recently, Zhou et al. concluded that the thermal population of 3MC was the major nonradiative decay pathway for N-heterocyclic carbene-chelated Ir complexes.60 Thereby, the energy level of 3MC is an important factor for evaluating the emission efficiencies of many Ir complexes. 3MC originates from transitions between the non-degenerate d orbitals of the metal atom in heavy metal complexes as predicted by crystal field theory (five d orbitals are split into three occupied and two unoccupied ones, called t2g and eg respectively) (Fig. 5). The energy gap between t2g and eg is determined by the arrangement and type of the ligands,61 and it is known that strengthening the metal–ligand bond destabilizes the 3MC state.34 Sajoto et al. reported that fac-(pmb)3Ir (Fig. 1) showed a higher energy level of 3MC than many carbon-and-nitrogen-chelated Ir-complexes (Ir(C^N)3); this is argued from calculations and is consistent with the shorter Ir–Ccarbene bond length of fac-(pmb)3Ir than the Ir–N bond length of Ir(C^N)3, e.g. fac-(ppz)3Ir.54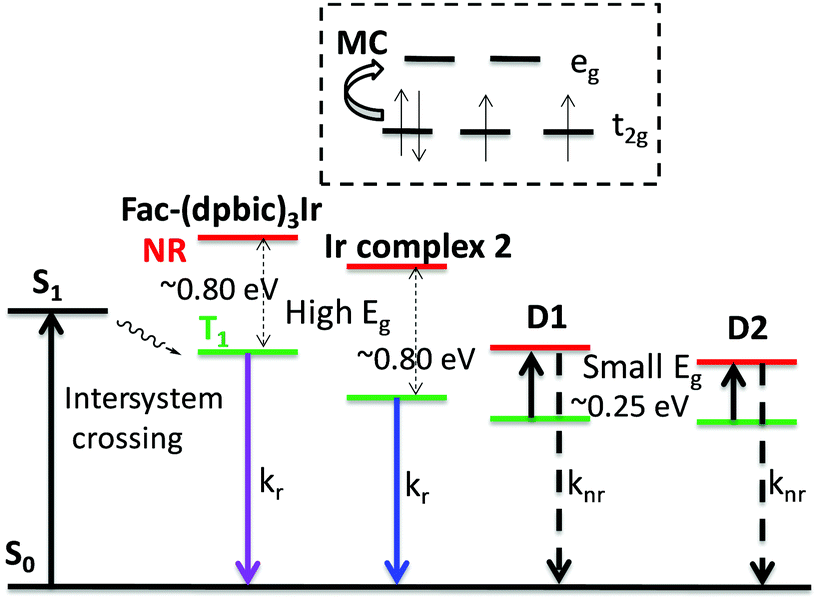 | ||
| Fig. 5 Graphic description of the metal-centered charge transfer (MC) of Ir complexes (up) and proposed energy levels of T1 (in green) and NR (in red) states for fac-(dpbic)3Ir, Ir-complex 2, D1 and D2 (bottom), together with the calculated energy barrier between T1 and NR. | ||
Therefore, comparing the bond lengths of Ir–Ccarbene among our targeted fac-(dpbic)3Ir-containing molecules is justified to assess their relative 3MC levels and thereby to explain their different emission efficiencies at room temperature. Based on the crystal structures and structural optimizations of several Ir-complexes as shown in Fig. 6(a), it is suggested that the average bond length of Ir–Ccarbene increases with more bulky ligands and the longest one is from the bulkiest dendrimer D1. Therefore, the energy level of 3MC should decrease in the order: fac-(dpbic)3Ir, Ir-complex 2 and D1. To explain the emission efficiency of these molecules, the energy barriers for the transition of T1 → 3MC were calculated through a constrained potential energy surface (PES) scan along the longest Ir–C bond length (ESI† for the detailed method of calculation).57 The energy difference between the highest point of the PES and the T1 state is the barrier height (Fig. 6(b)). The calculations reveal that, for fac-(dpbic)3Ir and Ir-complex 2, the energy barriers are about 0.80 eV, however, for dendrimer D1, the computed barrier is only 0.25 eV. Owing to the very complex structure of D2, we did not attempt a calculation of it; however, it can be inferred that D2 should possess an even lower energy of 3MC than D1 as indicated in the trend (Fig. 6a) because of the bigger size of D2 than D1; due to their very similar peak emission at 77 K (ET: 2.68 eV for both) (Fig. 4 and Table 1), we could envisage that the energy barrier (T1 → 3MC) for D2 is even smaller than that of D1 (Fig. 5). Therefore, we conclude that the lack of emission from D1 and D2 under ambient conditions is due to the easy access to the non-radiative 3MC state from the emissive state T1 (Fig. 5).
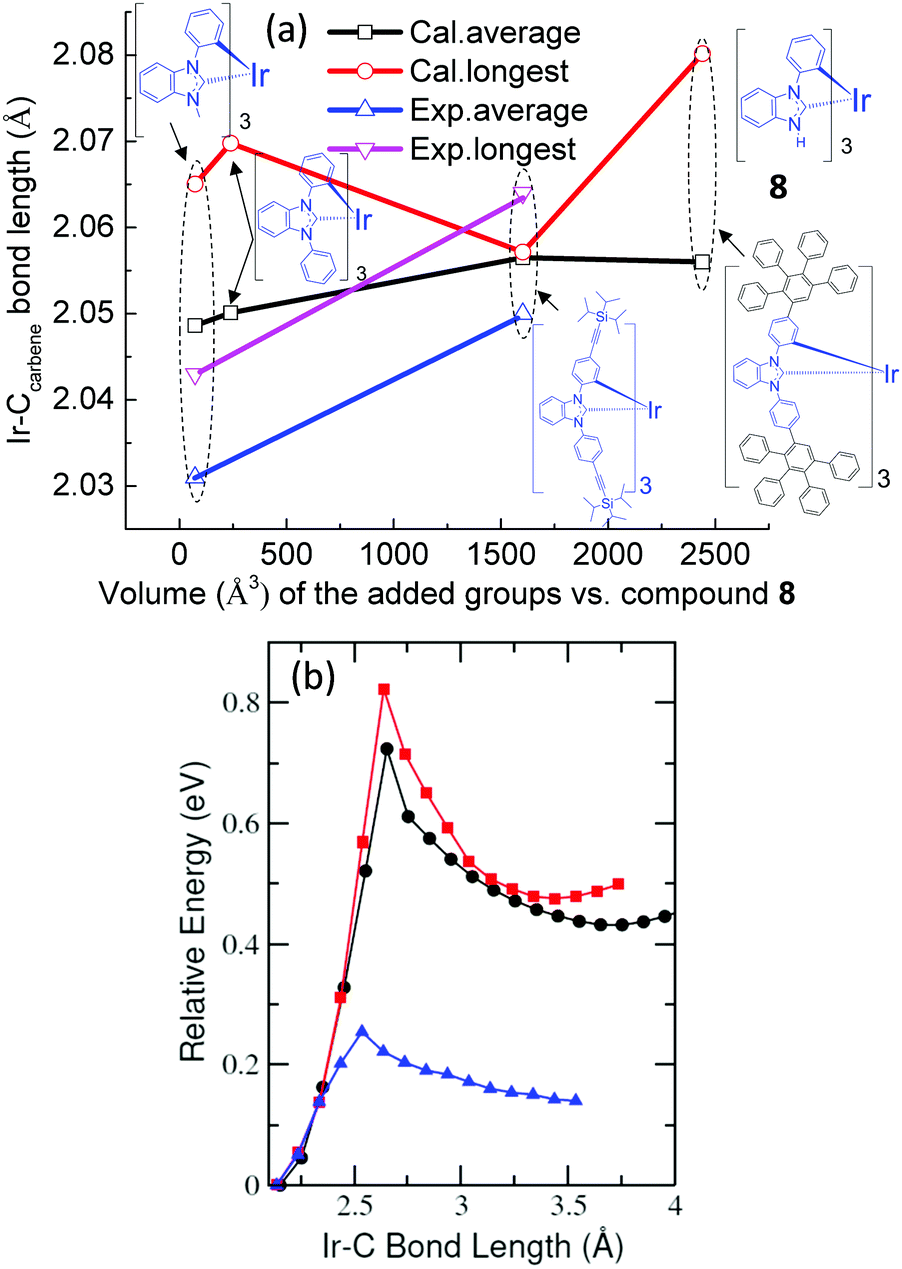 | ||
| Fig. 6 The relationship between the bulkiness of the molecules (quantified with the van der Waals volume62 of the groups in the molecule) and the Ir–Ccarbene bond length (a) (the dark and red curves represent the calculated average and longest bond length; the calculations were performed using the Gaussian software,63 DFT, the B3LYP hybrid functional and the LanL2DZ basis set; the pink and blue curves represent the experimental longest and average bond length, from the crystal structures); (b) computed PES scan plots for fac-(dpbic)3Ir (black line), Ir-complex 2 (red line) and dendrimer D1 (blue line). | ||
Electrochemistry
From the CV measurements, it is deduced that the HOMO energy (−5.23 eV) of Ir-complex 2 is increased and its LUMO energy (−2.11 eV) is decreased compared with the parent fac-(dpbic)3Ir due to the extended conjugation.17 This qualifies it for OLED applications because many available charge-transporting materials could correspond to the HOMO and LUMO energies of compound 2 for efficient charge injection and transport in OLEDs, such as PEDOT:PSS (HOMO: −5.10 eV, for hole injection and transport)31 and 4-(triphenylsilyl)phenyldiphenylphosphine oxide (TSPO1) (LUMO: −2.52 eV and ET: 3.36 eV for electron transport and exciton blocking).13 As to dendrimer D2, the HOMO energy was calculated to be −5.53 eV from the oxidation onset of the CV curve of D2. This is consistent with that of the peripheral carbazoles,53 probably owing to the large number of carbazole groups per Ir complex segment within one molecule (12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1).
:1).
OLED performance
The Ir-complex 2 and dendrimer D2 were tested in OLEDs. The device structure for Ir-complex 2 is ITO/PEDOT:PSS/Ir-complex 2/TPCz/TmPyPB/LiF/Al (TPCz: 3,6-bis(diphenylphosphoryl)-9-(4′-(diphenylphosphoryl)phenyl)carbazole; TmPyPB: 1,3,5-tri(m-pyrid-3-yl-phenyl)benzene).64 In this device, Ir-complex 2 was deposited by spin coating and the layers of TPCZ and TmPyPB were generated by vacuum evaporation (ESI†). Interestingly, Ir-complex 2 in this non-doped OLED exhibits a pure blue emission with peak emission at 442 and 469 nm, a maximum current efficiency of 1.04 cd A−1 and a CIExy (0.18, 0.18) (Table 1). Surprisingly, the emission of the Ir-complex 2 in the device is nearly the same as that measured in dilute solution and the thin film (Fig. 3). The lack of an aggregation-induced red-shifted emission is, again, due to the very bulky TIPSE moieties. As far as we know, this is the first reported non-doped small-molecule Ir-complex with pure blue emission in solution-processed OLEDs. This finding provides a new design concept for high-performance, pure-or-deep-blue emitters in solution-processed devices. For dendrimer D2, all charge transporting and emitting layers were spin coated with selected solvents, except that CuSCN was deposited by inkjet printing (ESI†). The devices have a structure of ITO/PEDOT:PSS/CuSCN/D2/Ca/Al. As shown in Table 1 and Fig. S9 (ESI†), the device displays green emission (λmax: ∼520 nm with a shoulder around 600 nm), consistent with the photoluminescence of the film. The overall poor device performances are due to the very easy access to the nonradiative T1 → 3MC transition for D2 at room temperature (Fig. 5). The OLED performance of fac-(dpbic)3Ir was reported in 2005 (Table 2).35 Due to the difficulty in finding an appropriate host material for fac-(dpbic)3Ir, utilizing essentially insulating poly(methyl methacrylate) (PMMA) as the matrix rendered a moderate efficiency and the emission was kept deep blue desirably.| V on (V) | η (cd A−1) | L max (cd m−2) | CIE (x, y) | |
|---|---|---|---|---|
| Device structure:a ITO/PEDOT:PSS/Ir-complex 2/TPCz/TmPyPB/LiF/Al.b ITO/PEDOT:PSS/CuSCN/D2/Ca/Al.c ITO/PEDOT:PSS/fac-(dpbic)3Ir:PMMA/BCP/LiF/Au cited from the literature.32d In EQE. | ||||
| Molecule 2a | 3.7 | 1.04 | 460 | (0.18, 0.18) |
| D2 | 5.5 | 0.034 | 80 | (0.32, 0.50) |
| fac-(dpbic)3Irc | 6 | 1.5%d | — | (0.16, 0.06) |
Conclusions
TIPSE-functionalized Ir-complex 2 is a blue phosphorescent emitter under ambient conditions, whereas dendrimers are barely emissive at room temperature. The failure of the dendrimers to emit at room temperature is related to the much reduced energy level of 3MC states. This, in turn, is due to the observed notable lengthening of the Ir–Ccarbene bonds induced by the bulky polyphenylene dendrons. This explanation is further supported by our PES calculations, which reveal that the barrier height for the transition from the emissive T1 state to the non-emissive 3MC state is only 0.25 eV for the dendrimer. Therefore, we conclude that, even though the introduction of bulky moieties into a PE can effectively inhibit excimer and triplet–triplet annihilation,65 the bulky groups are detrimental to the PLQY of a PE if the steric hindrance is too high. Therefore, the impact of the bulkiness of the molecules on the emission of metal–organic complex-based phosphorescent emitters is significant for the design of highly emissive dendrimer-based PEs by proposing: (i) to adjust the steric hindrance by the dendrons and (ii) to utilize more rigid phosphorescent emitters as the core of a dendrimer. Examples would be square-planar platinum complexes which minimize the impact of the steric hindrance.66,67 In addition, Ir-complex 2, as the first-reported pure-blue small-molecular PE in solution-processed and non-doped OLEDs, stimulates the design of new small-molecular phosphorescent materials.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors gratefully thank the Deutsche Forschungsgemeinschaft (SFB625) for providing financial support for this research. The authors also gratefully thank Prof. Lixiang Wang, Prof. Shumeng Wang and their group for measuring the PLQY, decay lifetime and device properties of Ir-complex 2, Prof. Hartmut Yersin for valuable discussions about photoluminescent lifetime, Ms Hien Thu Vu and Mr Paul Zybarth for their contribution to the OLED characterization of dendrimer D2, Dr Manfred Wegner for NMR characterizations, Dr Wen Zhang for MALDI-TOF mass characterizations and Inuru GmbH for the fabrication of the CuSCN layers. This work was carried out in the framework of the Joint Lab GEN_FAB and was supported by the HySPRINT innovation lab at Helmholtz-Zentrum Berlin. Open Access funding provided by the Max Planck Society.References
- O. Samsung, http://www.oled-info.com/samsung-oled.
- OLED-LG, http://www.oled-info.com/lg-oled.
- O. lighting, https://www.oled-info.com/oled-lighting.
- N. Sharma, M. Y. Wong, I. D. W. Samuel and E. Zysman-Colman, Highly Efficient OLEDs: Materials Based on Thermally Activated Delayed Fluorescence, 2018, ch. 14 Search PubMed.
- M. Eslamian, Nano-Micro Lett., 2017, 9, 3 CrossRef PubMed.
- X. B. Xu, X. L. Yang, J. Zhao, G. J. Zhou and W. Y. Wong, Asian J. Org. Chem., 2015, 4, 394–429 CrossRef CAS.
- C. Sekine, Y. Tsubata, T. Yamada, M. Kitano and S. Doi, Sci. Technol. Adv. Mater., 2014, 15, 034203 CrossRef PubMed.
- A. W. Bosman, H. M. Janssen and E. W. Meijer, Chem. Rev., 1999, 99, 1665–1688 CrossRef CAS PubMed.
- P. W. Wang, Y. J. Liu, C. Devadoss, P. Bharathi and J. S. Moore, Adv. Mater., 1996, 8, 237–241 CrossRef CAS.
- P. L. Burn, S. C. Lo and I. D. W. Samuel, Adv. Mater., 2007, 19, 1675–1688 CrossRef CAS.
- S. C. Lo and P. L. Burn, Chem. Rev., 2007, 107, 1097–1116 CrossRef CAS PubMed.
- S. H. Hwang, C. N. Moorefield and G. R. Newkome, Chem. Soc. Rev., 2008, 37, 2543–2557 RSC.
- S. O. Jeon, S. E. Jang, H. S. Son and J. Y. Lee, Adv. Mater., 2011, 23, 1436–1441 CrossRef CAS PubMed.
- X. C. Hang, T. Fleetham, E. Turner, J. Brooks and J. Li, Angew. Chem., Int. Ed., 2013, 52, 6753–6756 CrossRef CAS PubMed.
- X. Y. Li, J. Y. Zhang, Z. F. Zhao, L. D. Wang, H. N. Yang, Q. W. Chang, N. Jiang, Z. W. Liu, Z. Q. Bian, W. P. Liu, Z. H. Lu and C. H. Huang, Adv. Mater., 2018, 30, 1705005 CrossRef PubMed.
- C. Yao, J. X. Li, J. S. Wang, X. J. Xu, R. H. Liu and L. D. Li, J. Mater. Chem. C, 2015, 3, 8675–8683 RSC.
- D. Wagner, S. T. Hoffmann, U. Heinemeyer, I. Munster, A. Kohler and P. Strohriegl, Chem. Mater., 2013, 25, 3758–3765 CrossRef CAS.
- J. M. Lupton, I. D. W. Samuel, M. J. Frampton, R. Beavington and P. L. Burn, Adv. Funct. Mater., 2001, 11, 287–294 CrossRef CAS.
- J. P. J. Markham, S. C. Lo, S. W. Magennis, P. L. Burn and I. D. W. Samuel, Appl. Phys. Lett., 2002, 80, 2645–2647 CrossRef CAS.
- S. C. Lo, T. D. Anthopoulos, E. B. Namdas, P. L. Burn and I. D. W. Samuel, Adv. Mater., 2005, 17, 1945–1948 CrossRef CAS.
- D. M. Stoltzfus, W. Jiang, A. M. Brewer and P. L. Burn, J. Mater. Chem. C, 2018, 6, 10315–10326 RSC.
- L. Zhao, S. M. Wang, J. H. Lu, J. Q. Ding and L. X. Wang, J. Mater. Chem. C, 2017, 5, 9753–9760 RSC.
- S. M. Russell, A. M. Brewer, D. M. Stoltzfus, J. Saghaei and P. L. Burn, J. Mater. Chem. C, 2019, 7, 4681–4691 RSC.
- S. C. Lo, R. N. Bera, R. E. Harding, P. L. Burn and I. D. W. Samuel, Adv. Funct. Mater., 2008, 18, 3080–3090 CrossRef CAS.
- S. C. Lo, R. E. Harding, C. P. Shipley, S. G. Stevenson, P. L. Burn and I. D. W. Samuel, J. Am. Chem. Soc., 2009, 131, 16681–16688 CrossRef CAS PubMed.
- S. C. Lo, G. J. Richards, J. P. J. Markham, E. B. Namdas, S. Sharma, P. L. Burn and I. D. W. Samuel, Adv. Funct. Mater., 2005, 15, 1451–1458 CrossRef CAS.
- D. B. Xia, B. Wang, B. Chen, S. M. Wang, B. H. Zhang, J. Q. Ding, L. X. Wang, X. B. Jing and F. S. Wang, Angew. Chem., Int. Ed., 2014, 53, 1048–1052 CrossRef CAS PubMed.
- C. H. Lee, M. C. Tang, Y. C. Wong, M. Y. Chan and V. W. W. Yam, J. Am. Chem. Soc., 2017, 139, 10539–10550 CrossRef CAS PubMed.
- T. S. Qin, J. Q. Ding, L. X. Wang, M. Baumgarten, G. Zhou and K. Mullen, J. Am. Chem. Soc., 2009, 131, 14329–14336 CrossRef CAS PubMed.
- T. S. Qin, J. Q. Ding, M. Baumgarten, L. X. Wang and K. Mullen, Macromol. Rapid Commun., 2012, 33, 1036–1041 CrossRef CAS PubMed.
- G. Zhang, M. Baumgarten, M. Auer, R. Trattnig, E. J. W. List-Kratochvil and K. Mullen, Macromol. Rapid Commun., 2014, 35, 1931–1936 CrossRef CAS PubMed.
- G. Zhang, M. Auer-Berger, D. W. Gehrig, P. W. M. Blom, M. Baumgarten, D. Schollmeyer, E. J. W. List-Kratochvil and K. Muellen, Molecules, 2016, 1400 CrossRef PubMed.
- G. Zhang, M. Baumgarten, D. Schollmeyer and K. Mullen, ACS Omega, 2018, 3, 13808–13816 CrossRef CAS PubMed.
- T. Sajoto, P. I. Djurovich, A. Tamayo, M. Yousufuddin, R. Bau, M. E. Thompson, R. J. Holmes and S. R. Forrest, Inorg. Chem., 2005, 44, 7992–8003 CrossRef CAS PubMed.
- C. Schildknecht, G. Ginev, A. Kammoun, T. Riedl, W. Kowalsky, H. H. Johannes, C. Lennartz, K. Kahle, M. Egen, T. Geßner, M. Bold, S. Nord and P. Erk, Proc. SPIE, 2005, 5937, 59370E CrossRef.
- H. Wang, Y. Y. Xia, S. Lv, J. L. Xu and Z. H. Sun, Tetrahedron Lett., 2013, 54, 2124–2127 CrossRef CAS.
- A. R. Chianese, A. Mo and D. Datta, Organometallics, 2009, 28, 465–472 CrossRef CAS.
- F. M. Rivas, U. Riaz, A. Giessert, J. A. Smulik and S. T. Diver, Org. Lett., 2001, 3, 2673–2676 CrossRef CAS PubMed.
- D. M. Khrarnov and C. W. Bielawski, J. Org. Chem., 2007, 72, 9407–9417 CrossRef PubMed.
- K. Tsuchiya, S. Yagai, A. Kitamura, T. Karatsu, K. Endo, J. Mizukami, S. Akiyama and M. Yabe, Eur. J. Inorg. Chem., 2010, 926–933, DOI:10.1002/ejic.200900936.
- C. A. Tolman, Chem. Rev., 1977, 77, 313–348 CrossRef CAS.
- S. J. Lee, K. M. Park, K. Yang and Y. Kang, Inorg. Chem., 2009, 48, 1030–1037 CrossRef CAS PubMed.
- Z. H. Xiang, C. Q. Fang, S. H. Leng and D. P. Cao, J. Mater. Chem. A, 2014, 2, 7662–7665 RSC.
- S. C. Lo, R. E. Harding, E. Brightman, P. L. Burn and I. D. W. Samuel, J. Mater. Chem., 2009, 19, 3213–3227 RSC.
- C. Grewer and H. D. Brauer, J. Phys. Chem., 1994, 98, 4230–4235 CrossRef CAS.
- L. K. Patterson, G. Porter and M. R. Topp, Chem. Phys. Lett., 1970, 7, 612–614 CrossRef CAS.
- M. Wrighton and D. L. Morse, J. Am. Chem. Soc., 1974, 96, 6 Search PubMed.
- A. J. Lees, Comments Inorg. Chem., 1995, 17, 319–346 CrossRef CAS.
- H. Yersin, A. F. Rausch, R. Czerwieniec, T. Hofbeck and T. Fischer, Coord. Chem. Rev., 2011, 255, 2622–2652 CrossRef CAS.
- A. J. Boydston, C. S. Pecinovsky, S. T. Chao and C. W. Bielawski, J. Am. Chem. Soc., 2007, 129, 14550–14551 CrossRef CAS PubMed.
- A. J. Boydston, P. D. Vu, O. L. Dykhno, V. Chang, A. R. Wyatt, A. S. Stockett, E. T. Ritschdbrff, J. B. Shear and C. W. Bielawski, J. Am. Chem. Soc., 2008, 130, 3143–3156 CrossRef CAS PubMed.
- S. Bernhardt, M. Kastler, V. Enkelmann, M. Baumgarten and K. Mullen, Chem. – Eur. J., 2006, 12, 6117–6128 CrossRef CAS PubMed.
- J. Q. Ding, B. H. Zhang, J. H. Lu, Z. Y. Xie, L. X. Wang, X. B. Jing and F. S. Wang, Adv. Mater., 2009, 21, 4983–4986 CrossRef CAS PubMed.
- T. Sajoto, P. I. Djurovich, A. B. Tamayo, J. Oxgaard, W. A. Goddard and M. E. Thompson, J. Am. Chem. Soc., 2009, 131, 9813–9822 CrossRef CAS PubMed.
- A. Islam, N. Ikeda, K. Nozaki, Y. Okamoto, B. Gholamkhass, A. Yoshimura and T. Ohno, Coord. Chem. Rev., 1998, 171, 355–363 CrossRef CAS.
- F. Barigelletti, D. Sandrini, M. Maestri, V. Balzani, A. Vonzelewsky, L. Chassot, P. Jolliet and U. Maeder, Inorg. Chem., 1988, 27, 3644–3647 CrossRef CAS.
- S. Urinda, G. Das, A. Pramanik and P. Sarkar, J. Phys. Chem. A, 2018, 122, 7532–7539 CrossRef CAS PubMed.
- S. Urinda, G. Das, A. Pramanik and P. Sarkar, Phys. Chem. Chem. Phys., 2017, 19, 29629–29640 RSC.
- S. Urinda, G. Das, A. Pramanik and P. Sarkar, J. Phys. Chem. C, 2019, 123, 14216–14222 CrossRef CAS.
- X. W. Zhou and B. J. Powell, Inorg. Chem., 2018, 57, 8881–8889 CrossRef CAS PubMed.
- C. Housecroft and A. G. Sharpe, Inorganic Chemistry, 2012, 4th edn, ch. 20 Search PubMed.
- Y. H. Zhao, M. H. Abraham and A. M. Zissimos, J. Org. Chem., 2003, 68, 7368–7373 CrossRef CAS PubMed.
- M. Frisch, G. Trucks, H. B. Schlegel, G. Scuseria, M. Robb, J. Cheeseman, G. Scalmani, V. Barone, B. Mennucci and G. Petersson, Inc., Wallingford, CT, 2009, vol. 200.
- S. Wang, L. Zhao, B. Zhang, J. Ding, Z. Xie, L. Wang and W. Y. Wong, iScience, 2018, 6, 128–137 CrossRef CAS PubMed.
- X. L. Yang, G. J. Zhou and W. Y. Wong, Chem. Soc. Rev., 2015, 44, 8484–8575 RSC.
- A. F. Rausch, L. Murphy, J. A. G. Williams and H. Yersin, Inorg. Chem., 2012, 51, 312–319 CrossRef CAS PubMed.
- D. A. K. Vezzu, J. C. Deaton, J. S. Jones, L. Bartolotti, C. F. Harris, A. P. Marchetti, M. Kondakova, R. D. Pike and S. Q. Huo, Inorg. Chem., 2010, 49, 5107–5119 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Instrumentation, synthetic procedures, methods for extracting single-crystal structures, methods for the fabrication and characterization of OLEDs, methods of computation, and characterization data of the targeted molecules. CCDC 1948965. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9tc04748d |
| This journal is © The Royal Society of Chemistry 2019 |