
Aggregation-induced photodimerization of an alkynylpyrene derivative as a photoresponsive fluorescent ink†
 *a
and
Shengyu
Feng
a
*a
and
Shengyu
Feng
a
Abstract
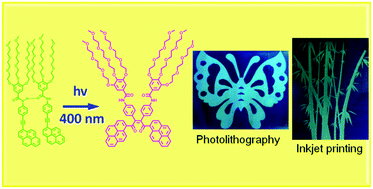
Smart luminescent materials can substantially alter their physicochemical properties with various stimuli and thus have long been a point of interest for the development and manufacture of various commodities. In particular, photoresponsive materials have seen much commercial success as sensors and for applications in the paint and coating industry. Herein, we report the design, synthesis, and material processing of a novel alkynylpyrene derivative. The aggregation behavior of this compound can be fine tuned in response to different conditions, such as solvent polarity, concentration, and ratio of good solvents to poor solvents. In addition, the concentration-dependent 1H NMR spectroscopic study and single crystal structure of the model compound revealed that multiple weak intermolecular interactions might be responsible for the formation of highly ordered aggregates. With the formation of highly ordered aggregates through molecular self-assembly, photodimerization can be realized effectively with 400 nm LED irradiation. Owing to the invisible and controlled printable characteristics of the aggregates and the conversion process of the luminescent material, we have demonstrated that our platform can act as a smart luminescent system towards confidential information encryption and decryption with various high-resolution patterns by photolithography and inkjet-printing techniques. In addition, the inherent molecular packing of the materials allows us to quench the luminescence of the as-printed characters easily upon blue visible light irradiation and realize the irreversible erasure of the luminescence signal for multiple information rewritable processes. As a result, the present luminescent material will find applications in the fields of optical information storage, information security protection, and erase/rewrite systems and provide a proof-of-principle application.
 Please wait while we load your content...
Please wait while we load your content...

