
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Molecular dynamics simulation of α-unsubstituted oligo-thiophenes: dependence of their high-temperature liquid-crystalline phase behaviour on molecular length†
Flora D.
Tsourtou
 a,
Stavros D.
Peroukidis
ab and
Vlasis G.
Mavrantzas
*ac
a,
Stavros D.
Peroukidis
ab and
Vlasis G.
Mavrantzas
*ac
aDepartment of Chemical Engineering, University of Patras & FORTH/ICE-HT, Patras GR 26504, Greece. E-mail: vlasis@chemeng.upatras.gr
bSchool of Science/Technology, Natural Sciences, Hellenic Open University, GR-26335, Patras, Greece
cParticle Technology Laboratory, Department of Mechanical and Process Engineering, ETH Zürich, CH-8092 Zürich, Switzerland. E-mail: vlasiosm@mat.ethz.ch
First published on 25th July 2019
Abstract
Despite that α-unsubstituted oligo-thiophenes (α-nTs) have been synthesized a long time ago and studied for many years, their phase behavior (which governs electronic performance, and thus their application in thin-film transistors) is still not fully understood. Here, we employ molecular dynamics simulations to study the high-temperature phase behaviour of α-nTs with n = 5–8 as a function of their molecular length using a recently proposed fully-flexible, united-atom model. We follow a methodology already developed for simulating the liquid-crystalline behaviour of α-sexithiophene, which we extend here to three other members of the family of α-nTs characterized by n = 5 (α-quinquethiophene), n = 7 (α-septithiophene) and n = 8 (α-octamer). Upon cooling fully pre-equilibrated bulk structures of these molecules from their amorphous, isotropic (Iso) phase at a high enough temperature down to lower temperatures, successive spontaneous phase transitions are observed leading to liquid crystalline phases. For n > 5, we observe first the formation of a nematic (Nem) phase and then the formation of two different types of smectic (Sm) phases. For n = 5, on the other hand, only one type of smectic phase is observed (no Nem was detected). We find that the type of the Sm phase formed is determined by the parity of the molecule: odd-numbered α-nTs display an orthogonal smectic A (SmA) phase whereas even-numbered α-nTs display both SmA and smectic C (SmC) phases, demonstrating a unique odd–even effect for these systems. In the SmC phase, the direction of the tilt angle is uniform, characteristic of a synclinic structure. The odd–even effect is also evident in the simulated crystalline phases of the studied α-nTs but also in other properties such as the density and the tilt angle which are found to alternate between high and low values for even- and odd-numbered α-nTs, respectively. It is interpreted here in terms of intra-molecular configurational changes taking place at the phase transition points.
1. Introduction
Poly-thiophenes (PTs) and their finite oligomers, α-conjugated oligo-thiophenes (OTs), have received considerable attention over the years due to their remarkable optical and electrical properties which render them promising candidate materials for application in devices such as organic thin film transistors (OTFTs),1–7 light-emitting diodes (OLEDs)8,9 and photovoltaic cells.10,11 A key factor controlling performance in such devices is the molecular ordering of thiophene molecules (denoted here as nTs, with n being the number of thiophene rings per molecule) affecting directly charge carrier mobility. In nT-based thin film transistors, in particular, carrier mobility increases with the chemical purity of the layer, number of thiophene rings and structural ordering.1,12,13 Liquid crystalline (LC) behaviour of α-unsubstituted nTs (e.g., see chemical structure of α-6T in Fig. 1) enhances self-organization, which could be an efficient avenue for obtaining defect-free lower-temperature crystalline states, accompanied by improved charge properties. The higher the degree of ordering in the smectic layer, the higher the charge mobility exhibited.14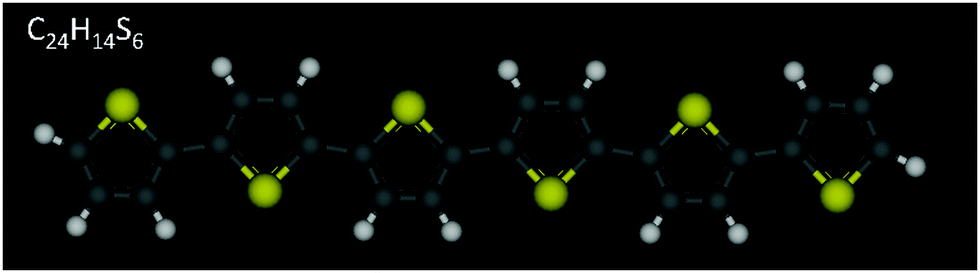 | ||
| Fig. 1 Chemical structure of an α-6T oligomer. Sulfur atoms are shown in yellow, carbon atoms in grey, and hydrogen atoms in white. | ||
Interestingly, there are several experimental studies in the earlier and recent literature where evidence for mesophase behaviour in α-nTs was reported. Taliani et al.15 carried out differential scanning calorimetry (DSC) and polarized optical microscopy (POM) measurements and observed that the α-6T (α-sexithiophene) crystal forms a nematic (Nem) phase when heated above 585 K, but determination of the clearing point (or isotropization temperature) was not successful. Using X-ray diffraction (XRD) and differential scanning calorimetry (DSC), Destri et al.16 detected a mesophase above 578 K upon heating a high-temperature (HT) 6T crystal, but they could assign this neither as a Nem nor as a smectic (Sm) one. For 6T films deposited on substrates at room temperature, two different liquid crystalline (LC) phases are typically observed: a Sm phase near the substrate and a Nem phase in the bulk.17 Optical micrographs of the Nem phase of α-6T have also been reported.18 According to Kuiper et al.,19 α-5T exhibits only a crystal-to-isotropic (Iso) transition at 525 K while its decomposition at high temperatures renders its crystallization unfeasible upon cooling down from the Iso phase. Furthermore, DSC measurements and optical microscopy observations by Mellucci et al.20 did not provide evidence for LC behaviour for α-5T. On the other hand, experimental studies based on Scanning Force Microscopy (SFM) measurements21 and Fourier-Transform Infrared (FTIR) spectroscopy17 have reported that 5T at room temperature substrates self-organizes, most probably, into a Sm phase. Polarizing microscopy and DSC measurements for α-3T have verified that it does not exhibit a LC behaviour.22 The DSC analysis of Destri et al.16 have further indicated that differences in the enthalpies associated with the two processes, Crystal-to-mesophase transition (5–15 kcal mol−1) and mesophase-to-Iso transition (0.1–0.5 kcal mol−1), are relatively small. With respect to this, Facchetti et al.23 have reported that their DSC measurements for α-nTs with n = 2–6 did not show any second peak that could indicate the presence of a mesophase. For α-7T and α-8T, we further mention that, despite that they have not been widely studied, their melting points have been measured and found oddly high.24,25 Still, though, no clear evidence of LC behaviour has ever been reported for them. Only in the case of α-8T, Fichou et al.24 have reported a second endothermic DSC peak below its melting point that most likely corresponded to a structural transition (either solid-to-solid or solid-to-LC).
To exploit the advantages offered by LC phases and overcome several of the issues described above, functionalized thiophene-based compounds have been synthesized favouring the formation of mesophases.18,23,26–33 For example, end-functionalization of nTs (n = 3–7) with alkylamide, alkyl or branched alkyl groups favours liquid crystallinity.26–33 Nevertheless, functionalization may subvert the preferred molecular orbitals for achieving the desired electrical and optical properties; moreover, the accurate characterization of the phase behaviour of functionalized compounds is not easy. The POM and DSC analysis of Ponomarenko and Kirchmeyer,33 for example, has demonstrated that 4T, 5T and 6T with terminal alkyl-substituents display high temperature-ordered Sm phases, whose type, however, is not easy to identify.
Clearly, the accurate experimental detection and characterization of LC phases exhibited by nTs is a very challenging task. In this respect, computer simulations can play an important role, since they can address issues related with self-organization and molecular ordering rather efficiently because of the relatively high temperatures involved, rendering possible the robust exploration of phase space. Pizzirusso et al.,34 for example, used all-atom Molecular Dynamics (MD) simulations to address the phase behaviour of α-6T by starting from its low-temperature (LT) crystal phase and gradually heating it to observe the following phase sequence: (a) LT crystal-to-smectic A (SmA), (b) SmA-to-Nem, (c) Nem-to-Iso. Their findings were verified recently by large-scale MD simulations by our group35 using an optimized, fully flexible, united-atom model. Our simulations demonstrated the spontaneous formation of several ordered phases by cooling a bulk model of α-6T from its Iso phase down to lower temperatures. They also revealed the possibility of Sm phase polymorphism. In particular, the following phase sequence was obtained: (a) Iso-to-Nem, (b) Nem-to-SmA, (c) SmA-to-SmC, and (d) SmC-to-crystal. The SmC phase, in particular, was observed not only in the MD simulations but also in simulations with a state-of-the-art Monte Carlo (MC) algorithm using a stiff version (fixed bond lengths and angles) of the developed united-atom model.
In the present study, we extend the simulations reported in ref. 35 for α-6T to three other members of the family of α-nTs characterized by n = 5, 7 and 8, our aim being to understand the dependence of phase behaviour and the type of LC phases formed on the molecular length of the α-nT molecule. As we will see below, our study will reveal that it is the parity of the molecule (i.e., the odd or even number of thiophene rings per molecule) that mainly determines phase behaviour in these systems, pointing to an important odd–even effect.
In the literature, odd–even effects have been reported for alkanes,36 liquid crystals (here the parity refers to the number of carbon atoms in the alkyl chains),37,38 crystal phases39 and films of nTs.17 In alkanes, the number of carbon atoms in the chain affects their packing pattern, thus also their physical properties such as melting point.36 In LC polymers, flexible molecules (e.g., flexible methylene spacers) link mesogenic units (e.g., phenyl rings) along the chain which can significantly impact phase behaviour.37a Both melting (Tm) and isotropization (Ti) temperatures decrease in a non-monotonic, zig-zag manner as the molecular length of the spacer increases. In particular, the zig-zag pattern of the Ti temperature is related to the parity in the number of carbons in the alkyl spacer. In LC dimers,37b the odd–even effect has been observed for molecules comprising two rod- or disk-like mesogenic units linked via an alkyl chain. Quite recently, a new Nemx phase (consisting of chiral domains) was experimentally identified in symmetric LC dimers38 with an odd number of alkyl spacers. The same LC dimers with an even number of alkyl spacers exhibit only the typical Nem phase. Hence, the odd–even effect is reflected not only in the phase transition temperatures but also in the type of phases manifested. An odd–even effect associated with the parity in the number of terminal units has also been observed for Sm phases.37c
In all of the above-mentioned cases, the odd–even effect is related to the length of the alkyl chains (either spacers or terminal groups) rather than to the number of rings of the mesogenic groups. In our work, we will report another type of odd–even effect which, to the best of our knowledge, has never been mentioned before referring to the dependence of the type of the Sm phases formed by α-nT molecules (that consist solely of thiophene rings) on the parity in the total number of thiophene rings in the molecule. This new odd–even effect is different, for example, to the effect reported in the literature for the density of the crystalline states of OTs and its alternation with the parity in the number of thiophene rings.39 The same phenomenon is supported by XRD patterns of OT films deposited at low temperature substrates. It is found that, overall, even-numbered OT films self-organize in well-ordered structures showing large carrier mobility, while in odd-numbered OT films two different crystalline polymorphs co-exist, which lowers carrier mobility.17 Also dependent on the parity in the number of thiophene rings in crystalline OTs are their emission properties: for odd-numbered OTs, the purely electronic emission is absent, which should be contrasted with the corresponding behaviour for even-numbered OT crystals.40 This feature has been interpreted in terms of the different point groups between odd- and even-numbered nT chains. Another odd–even effect reported recently is related to the excited state properties of a series of thiophene-based compounds with truxene as end-capping units in solution,41 and has been attributed to the different symmetry properties characterizing odd- and even-numbered molecules.
Clearly, investigating and understanding the relationship between molecular ordering of α-nTs and parity in their number of thiophene rings is important for controlling, improving or optimizing their optoelectronic properties for several technological applications nowadays.12,40
The rest of our paper is organized as follows: Section 2 describes in detail the molecular systems studied in this work and the simulation methodology followed. Results from the MD simulations concerning phase transitions and self-organization in the simulated α-nTs as a function of temperature (with emphasis on the detailed characterization of the LC phases formed) are reported in Section 3. The paper concludes with Section 4 summarizing the most significant results of our work and briefly outlining future plans.
2. Simulation methodology and details
Our MD simulations were carried out with bulk systems of α-nT molecules characterized by n = 5, 7 and 8. Large cubic simulation cells were utilized (subject to periodic boundary conditions along all three space directions) containing a big number of α-nT molecules (7040 molecules for α-5T, 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 200 molecules for α-7T, and 2000 molecules for α-8T) in order to minimize errors due to finite system size effects. In all cases, the initial configuration of the system was constructed using the Amorphous Builder module42 integrated in the Scienomics MAPS software.43 This was subjected to a potential energy minimization for removing undesired atom–atom overlaps, followed by a long MD simulation in the isothermal–isobaric (NPT) ensemble at a high enough temperature (e.g., T = 900 K in the case of α-8T) and P = 1 atm to fully relax structural and conformational properties. The final structure from the NPT MD simulation was used as initial configuration for the subsequent cooling MD simulations at progressively lower temperatures. For the α-6T system, all simulation data were borrowed from our previous study35b where a very large simulation cell (it contained 8960 α-6T molecules) had also been used.
200 molecules for α-7T, and 2000 molecules for α-8T) in order to minimize errors due to finite system size effects. In all cases, the initial configuration of the system was constructed using the Amorphous Builder module42 integrated in the Scienomics MAPS software.43 This was subjected to a potential energy minimization for removing undesired atom–atom overlaps, followed by a long MD simulation in the isothermal–isobaric (NPT) ensemble at a high enough temperature (e.g., T = 900 K in the case of α-8T) and P = 1 atm to fully relax structural and conformational properties. The final structure from the NPT MD simulation was used as initial configuration for the subsequent cooling MD simulations at progressively lower temperatures. For the α-6T system, all simulation data were borrowed from our previous study35b where a very large simulation cell (it contained 8960 α-6T molecules) had also been used.
The MD simulations were carried out with GROMACS44 using the Nosé–Hoover thermostat45 coupled with the Parrinello–Rahman barostat46 (with corresponding time constants equal to 10 fs and 350 fs) to maintain the temperature and pressure constant at their desired values. The equations of motion were integrated with the leap-frog algorithm, with an integration time step equal to 1.0 fs. In our simulations, we utilized the fully flexible united-atom (UA) model reported recently,35a based on the standard Dreiding force field.47 To calculate van der Waals interactions, a uniform cutoff of 12 Å was used, together with standard tail corrections for the energy and pressure. Typical conformations of α-8T and α-7T molecules in the united-atom representation adopted in our MD simulations are provided in Fig. 2.
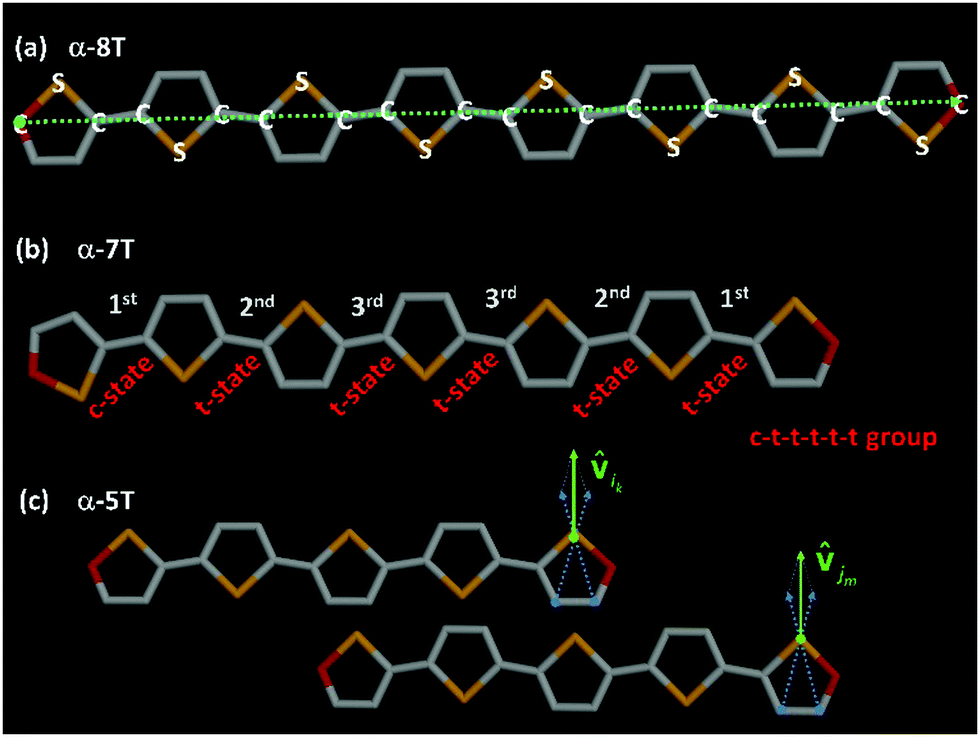 | ||
Fig. 2 (a) Schematic illustration of an α-8T chain in the united-atom representation adopted in this work and the corresponding direction of the end-to-end vector (green dashed line). (b) Typical atomistic configuration of a c–t–t–t–t–t conformational group corresponding to an α-7T chain, with inter-ring bonds denoted according to their local index (i.e., 1st, 2nd and 3rd). (c) Schematic illustration of two α-5T chains in the united-atom representation adopted in our MD simulations showing the definition of unit vectors ![[v with combining circumflex]](https://www.rsc.org/images/entities/b_char_0076_0302.gif) ik and jm that appear in eqn (6). In all panels, sulfur (S) atoms are shown in yellow, carbon (C) atoms in grey, and end-carbon (C) atoms in red. ik and jm that appear in eqn (6). In all panels, sulfur (S) atoms are shown in yellow, carbon (C) atoms in grey, and end-carbon (C) atoms in red. | ||
To quantify orientational order in the mesophases observed in our simulations we made use of the orientational order parameter (S+) corresponding to the largest eigenvalue of the ordering matrix defined as35b
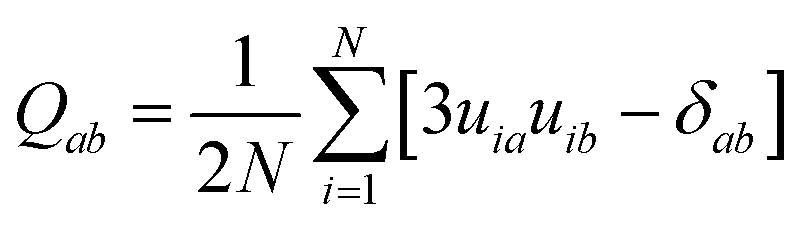 | (1) |
![[n with combining circumflex]](https://www.rsc.org/images/entities/b_char_006e_0302.gif) of the phase. In our simulations, the mean value of S+ is calculated as an ensemble average over all equilibrated molecular configurations for the system under study.
of the phase. In our simulations, the mean value of S+ is calculated as an ensemble average over all equilibrated molecular configurations for the system under study.
Molecular structure within each phase with respect to positional order was examined by using a set of three special pair correlation functions of the chains’ centers-of-mass (CoM). The first is the usual radial distribution function
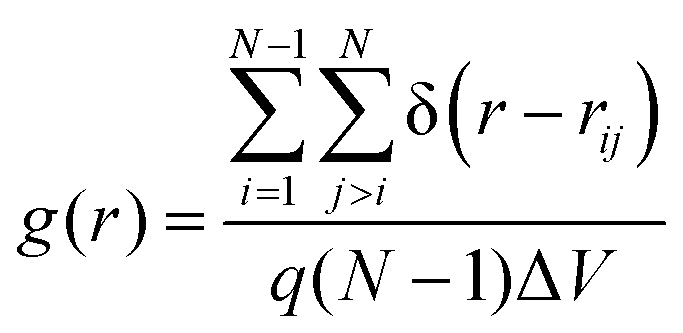 | (2) |
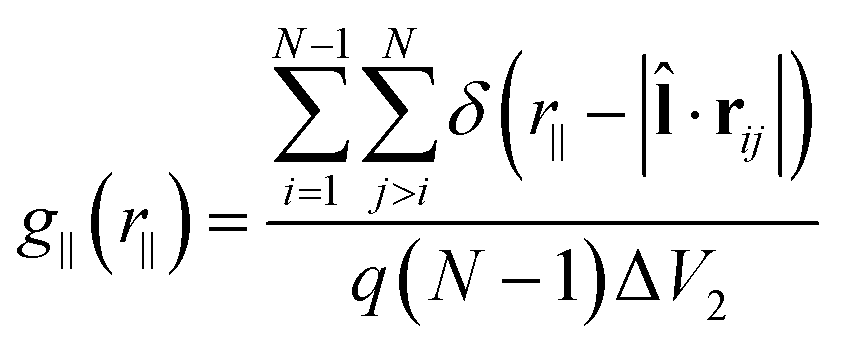 | (3) |
![[l with combining circumflex]](https://www.rsc.org/images/entities/b_char_006c_0302.gif) , with |·rij| being the projection of the intermolecular vector rij onto . For the calculation of the layer normal vector , see ref. 35b. To examine the phase transitions from either an Iso or Nem phase to Sm phase, the layer normal vector can be replaced by the director . The third special pair correlation function is the perpendicular correlation function g⊥(r⊥) used to obtain information about positional ordering within the smectic layers. It is defined as
, with |·rij| being the projection of the intermolecular vector rij onto . For the calculation of the layer normal vector , see ref. 35b. To examine the phase transitions from either an Iso or Nem phase to Sm phase, the layer normal vector can be replaced by the director . The third special pair correlation function is the perpendicular correlation function g⊥(r⊥) used to obtain information about positional ordering within the smectic layers. It is defined as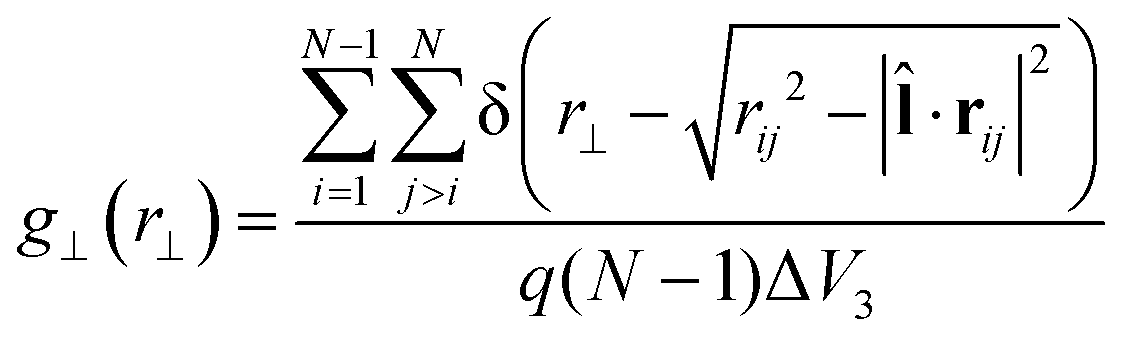 | (4) |
To further analyze structural order in the liquid crystalline (LC) and crystal (Cry) phases of the simulated α-nTs, we calculated the inter-molecular pair correlation function of the thiophene rings’ centers-of-mass (CoM-ring):
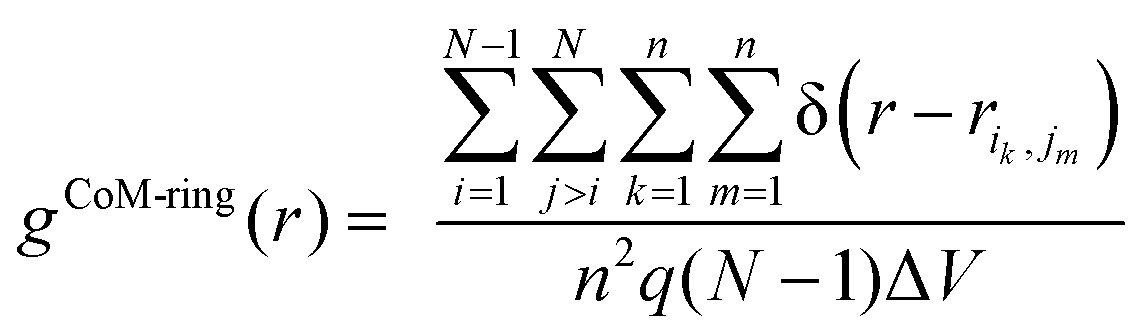 | (5) |
We have also examined directional preferences in the layered conformations of the simulated α-nT systems by calculating the following quantity:
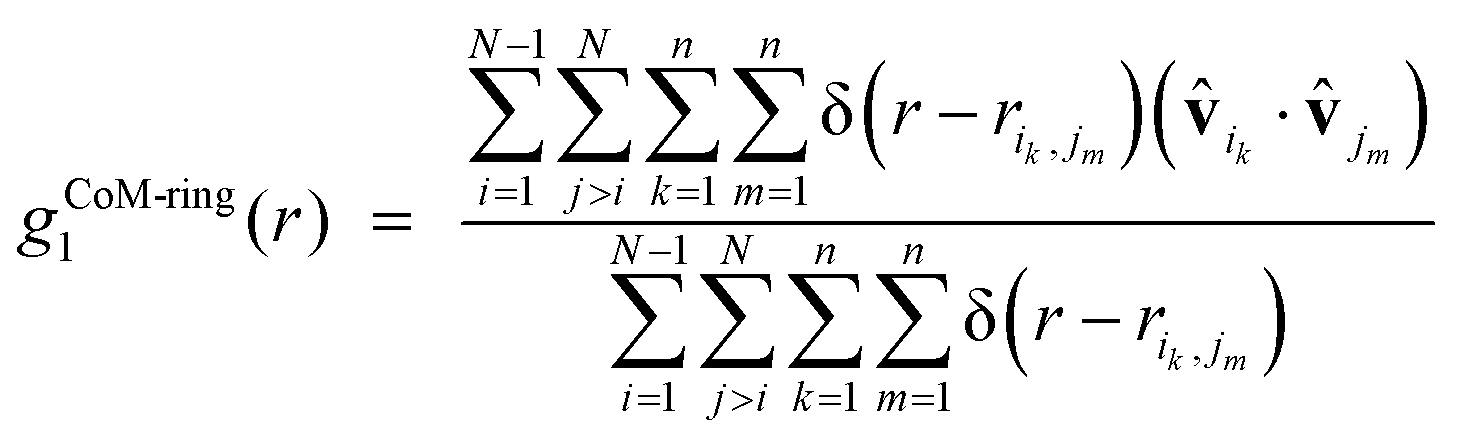 | (6) |
ik and jm appear referring to the i-th and j-th α-nT molecules, respectively; these are defined as shown in Fig. 2c.
3. Results and discussion
3.1. Mesophase behaviour of α-nTs
For each of the α-nT systems with n = 5–8 studied in this work, phase behaviour was examined by performing cooling MD runs under isobaric conditions at progressively lower temperatures starting from their Iso phase at a high enough temperature. The phase transitions were identified from the discontinuities (abrupt jumps) in the density (ρ) and order parameter (S+) curves as a function of temperature. The ρ-vs.-T and S+-vs.-T diagrams obtained from our simulations are shown in Fig. 3a and b, respectively. Error bars on the simulation data smaller than the size of the symbols are not shown (in general, these error bars are very small owing to the large simulation cells utilized in our work).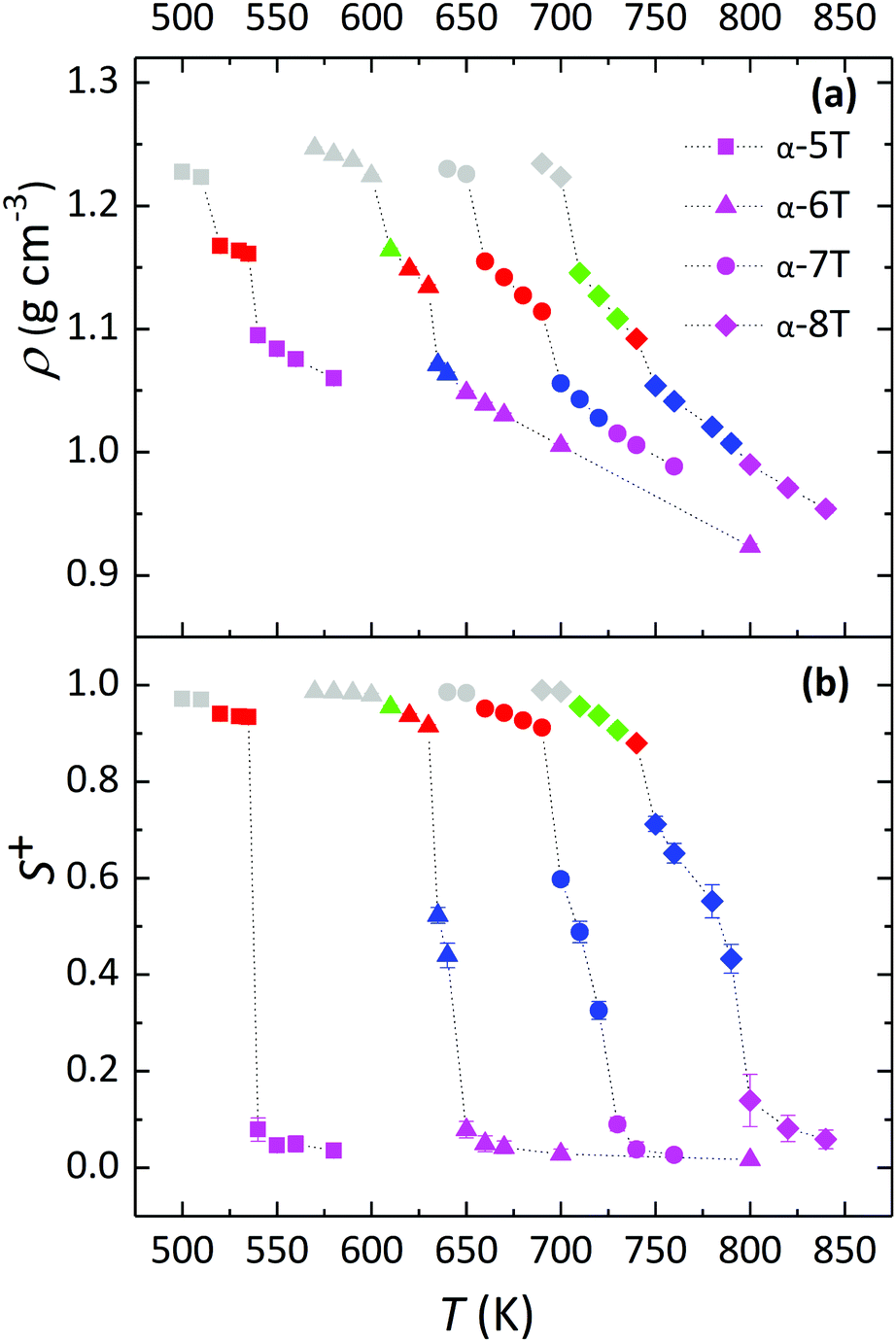 | ||
| Fig. 3 MD predictions for: (a) the density (ρ), and (b) the order parameter (S+) versus temperature (T) for the simulated α-nT systems with n = 5–8, at P = 1 atm. The data for α-6T have been borrowed from the simulations of ref. 35b with the 8960-chain system discussed there. Squares, triangles, circles and diamonds refer to α-5T, α-6T, α-7T and α-8T, respectively. Colour codes for ρ and S+ branches (with the type of the phase shown in parenthesis) are as follows: magenta (Iso), blue (Nem), red (SmA), green (SmC) and gray (Cry). | ||
For all α-nTs, there exists a range of (high enough) temperatures where the order parameter S+ is less than 0.2, indicative of complete lack of any long-range orientational order. In addition, in this temperature regime, the radial pair correlation function g(r) (see Fig. 4a–d) is structureless at long intermolecular distances confirming the uniform distribution of the centers-of-mass of the simulated α-nT systems. Clearly, all systems at this high temperature range live in their amorphous, Iso phase.
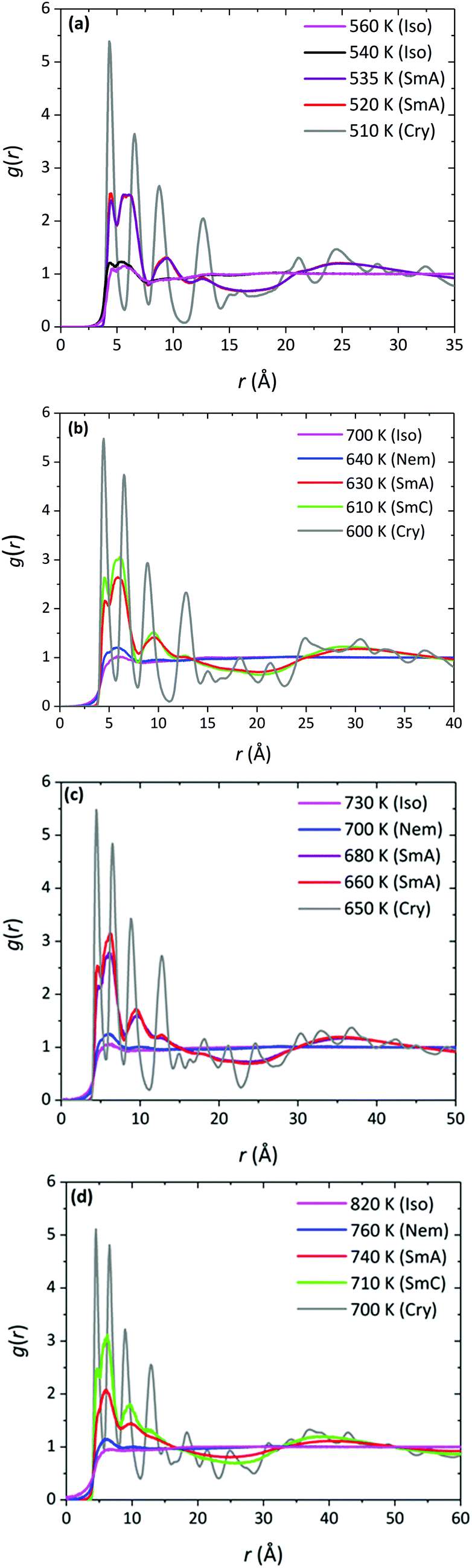 | ||
| Fig. 4 Radial pair correlation function g(r) of the chains’ centers-of-mass for: (a) α-5T, (b) α-6T, (c) α-7T, and (d) α-8T chain system, as obtained from our NPT MD simulations at various temperatures (in K) and P = 1 atm. | ||
As the temperature is lowered, spontaneous phase transitions occur resulting in the formation of several mesophases. The range of temperatures where each of these mesophases appears is illustrated with different colours in the ρ and S+ plots of Fig. 3a and b. Color alteration in these plots is typically accompanied by a discontinuity in ρ and S+. Representative simulation snapshots showing how α-nT molecules are organized at temperatures close to the discontinuity points are shown in Fig. 5, 6 and Fig. S5 (ESI†). Overall, α-nT molecules appear to have an overall rod-like shape (even in the Iso phase), to be oriented along a common direction, and/or to organize in layers within the ordered phases.
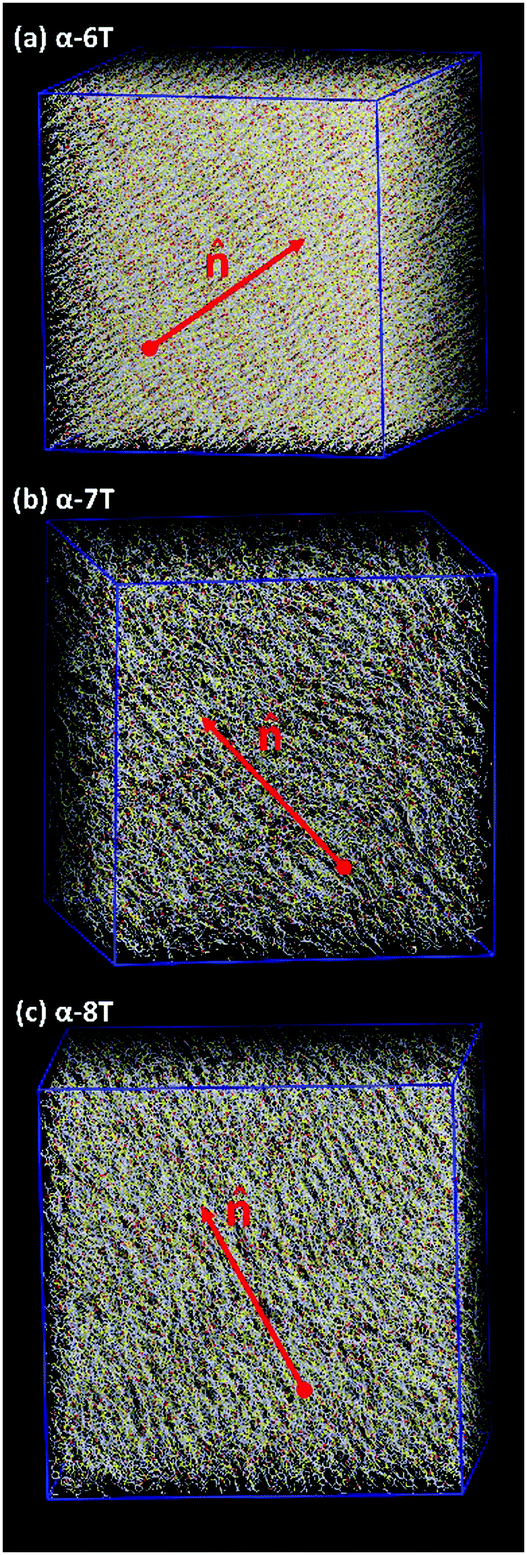 | ||
| Fig. 5 Characteristic atomistic snapshots of the Nem phases observed in our MD simulations with: (a) α-6T at T = 630 K, (b) α-7T at 710 K, and (c) α-8T at 780 K. In all cases, P = 1 atm. Sulfur, carbon and end-carbon atoms of thiophene rings are represented with yellow, white and red colour, respectively. The vector indicates the unit vector along the phase director. | ||
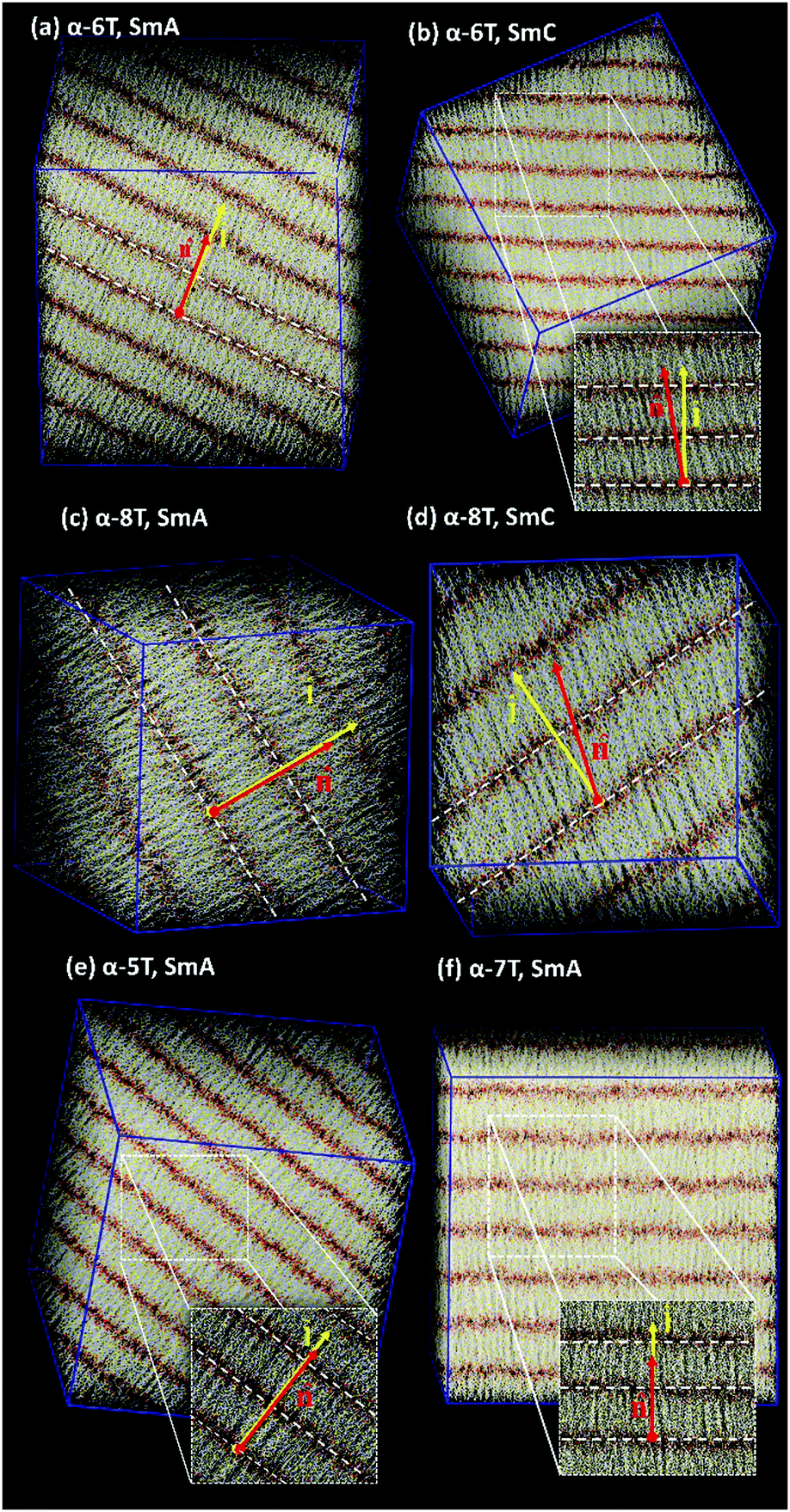 | ||
| Fig. 6 Characteristic atomistic snapshots of: (a) α-6T at T = 620 K (SmA), (b) α-6T at T = 610 K (SmC), (c) α-8T chain system at T = 740 K (SmA), (d) α-8T at T = 730 K (SmC), (e) α-5T at T = 535 K (SmA), and (f) α-7T at T = 690 K (SmA). In all cases, P = 1 atm. Sulfur, carbon and end-carbon atoms of thiophene rings are represented with yellow, white and red colour, respectively. The vectors and indicate the unit vectors along the layer normal and the phase director, respectively. | ||
Upon cooling from the Iso phase, α-nTs with n = 6–8 undergo an Iso-to-Nem phase transition as is evident from the jump in the value of S+ shown in Fig. 3b; this is also accompanied by a smooth increase in the mass density ρ (Fig. 3a) of the system, characteristic of a weak first order phase transition. In the Nem phase, the maximum values of S+ are between 0.6 and 0.7, whereas the calculated g(r) as well as the longitudinal g‖(r‖) graphs confirm the lack of any long range positional order (see Fig. 4 and Fig. S1, ESI†). Characteristic configurations of the various α-nT systems in their Nem phase from the simulations are provided in Fig. 5. Molecules adopt a rod-like shape and exhibit a tendency to align along a common direction as defined by the vector representing the average direction of the long molecular axis of α-nT oligomers. From Fig. 3b and 4, but also by visually examining several simulation snapshots, it is found that α-5T does not form a Nem phase. Therefore, α-6T can be considered as the shortest α-nT molecule exhibiting a Nem phase.
By further lowering the temperature, an abrupt change in the density ρ is observed for the systems with n = 6–8. At the same time, S+ jumps to a value between 0.8 and 0.9 indicating a rather high degree of orientational order. By further examining the characteristic configuration snapshots shown in Fig. 6, we understand that these α-nTs self-organize into periodic layers expanding all over the simulation cell. α-5T, on the other hand, exhibits a direct Iso-to-Sm transition. For this system, the values of the pair (ρ, S+) shift abruptly from (1.09 g cm−3, 0.08) at T = 540 K to (1.14 g cm−3, 0.88) at T = 535 K, indicating clearly (Fig. 6e) the emergence of a layered structure. This particular behaviour of the α-5T system is difficult to verify experimentally, because it might be preceded by sample decomposition at high temperatures.19,24,48,49 Indeed, experimentally, Sm phases of α-5T at room temperature have been observed only on substrates.17,21 Kuiper et al.,19 on the other hand, have reported a direct Cry-to-Iso phase transition but they could not detect signs of crystallization of α-5T upon further cooling, most likely because of earlier sample decomposition at the higher temperature molten state. Baker et al.48 have also contended that some p-phenylene oligomers can decompose before the Nem-to-Iso transition.
To thoroughly characterize the structure of α-5T in the LC phases, we make use of several pair correlation functions. One of these is g‖(r‖) monitoring the formation of layers and their spacing by computing the distance separating successive peaks in the corresponding diagram (see, e.g., Fig. S1, ESI†). For all the α-nTs studied here, computed values of the layer spacing come out to be close to the corresponding molecular lengths as estimated from the square root of the average square end-to-end distance 〈Ree2〉 plus the van der Waals radii of the two end carbon atoms. This means that no inter-digitation of molecules takes place, which agrees with previous simulation studies.34,35b The 〈Ree2〉-vs.-T results for the simulated α-nTs are presented in Table 1. The calculation of the end-to-end distance has been based on the magnitude of the vector connecting the end carbon atoms along an α-nT molecule (see Fig. 2a). Additionally, the g⊥(r⊥) correlation function is observed to be structureless at long intermolecular distances (see Fig. S2, ESI†), which confirms the absence of any long-range positional correlations within the Sm phase layers.
| Phase | α-5T | α-6T | α-7T | α-8T | ||||
|---|---|---|---|---|---|---|---|---|
| T (K) | 〈Ree2〉 (Å2) | T (K) | 〈Ree2〉 (Å2) | T (K) | 〈Ree2〉 (Å2) | T (K) | 〈Ree2〉 (Å2) | |
| Iso | 540 | 300.0 ± 0.2 | 700 | 423.0 ± 0.5 | 730 | 572.0 ± 0.7 | 800 | 733.0 ± 2.6 |
| Nem | — | — | 640 | 434.0 ± 0.3 | 720 | 578.0 ± 0.7 | 790 | 747.0 ± 2.1 |
| 635 | 435.0 ± 0.3 | 700 | 590.0 ± 0.4 | 750 | 773.0 ± 1.4 | |||
| SmA | 530 | 309.0 ± 0.3 | 630 | 452.0 ± 0.2 | 690 | 619.0 ± 0.2 | 740 | 801.0 ± 1.4 |
| 520 | 309.0 ± 0.1 | 620 | 456.0 ± 0.2 | 660 | 633.0 ± 0.3 | |||
| SmC | — | — | 610 | 459.0 ± 0.2 | — | — | 730 | 811.0 ± 1.0 |
| 710 | 829.0 ± 0.9 | |||||||
| Cry | 510 | 310.0 ± 0.2 | 600 | 462.0 ± 0.1 | 650 | 641 ± 0.2 | 700 | 848.0 ± 0.6 |
| 500 | 310.0 ± 0.1 | 590 | 463.0 ± 0.1 | 640 | 642 ± 0.2 | 690 | 851.0 ± 0.5 | |
Visual inspection of snapshots (compare, e.g., Fig. 6c and d for α-8T) suggests that, at the low temperature Sm phases, the long molecular axes of even-numbered α-nTs organize in such a way that leads to a distinctive tilt with respect to the layer normal vector . The magnitude of the tilt is quantified by computing the average value of the corresponding tilt angle through 〈θ〉 = 〈cos−1(·)〉. Numerical results for the variation of 〈θ〉 with temperature for the α-5T, α-7T and α-8T oligo-thiophenes are reported in Table 2.
| Phase | T (K) | 〈θ〉 (°) |
|---|---|---|
| (a) | ||
| SmA | 535 | 0.63 ± 0.23 |
| SmA | 530 | 0.54 ± 0.24 |
| SmA | 520 | 0.77 ± 0.47 |
| Cry | 510 | 7.96 ± 0.17 |
| Cry | 500 | 8.78 ± 0.16 |
| (b) | ||
| SmA | 690 | 0.96 ± 0.09 |
| SmA | 680 | 0.15 ± 0.08 |
| SmA | 670 | 0.34 ± 0.16 |
| SmA | 660 | 0.14 ± 0.08 |
| Cry | 650 | 11.76 ± 0.11 |
| Cry | 640 | 12.43 ± 0.12 |
| (c) | ||
| SmA | 740 | 2.01 ± 0.91 |
| SmC | 730 | 3.87 ± 1.52 |
| SmC | 720 | 7.20 ± 0.61 |
| SmC | 710 | 7.49 ± 0.50 |
| Cry | 700 | 13.9 ± 0.18 |
| Cry | 690 | 18.0 ± 0.30 |
Values of 〈θ〉 equal to 1° with a standard deviation of the same order indicate the formation of orthogonal Sm phases denoted as SmA, while values of 〈θ〉 considerably larger than 1° indicate the formation of a SmC phase comprised of oligo-thiophenes with a distinct tilt with respect to the layer normal vector. Actually, our calculations (see Fig. 6) indicate this to be synclinic tilt, because α-nTs in all Sm layers are inclined towards the same direction with respect to the unit normal-to-the-layer vector. From the data reported in Table 2a–c and Table 3 of ref. 35b it appears that α-5T and α-7T form only a SmA phase at low temperatures, in contrast to α-6T and α-8T that form both a SmA and a SmC phase.
It is a remarkable result that the type of the Sm phase(s) formed by α-nTs depends on their parity, suggesting a structural odd–even effect. To the best of our knowledge, this is the first report of such a unique phenomenon for the LC phases formed by such rather stiff (rod-like) molecules consisting solely of ring moieties. As we already discussed in the Introduction, in typical systems of liquid crystals functionalized with alkyl chains (either as spacers or as terminal groups), the odd–even effect is related to the parity in the number of carbon atoms of these chains that appears to control the clearing temperatures and/or the structure of the Nem and Sm phases.37,38 We discuss again the particular odd–even effect identified in the present study in Section 3.2.
At even lower temperatures, α-nTs undergo a phase transition from the Sm (either SmA or SmC depending on the parity of the system) to the Cry phase. Representative snapshots of the crystalline states formed by the simulated α-nT systems are depicted in Fig. S5 (ESI†). The SmA/SmC-to-Cry phase transition is accompanied by a sharp jump in the value of ρ and S+ (see gray branches in Fig. 3a and b). The crystalline character of this phase was further confirmed through calculations of relevant pair correlation functions which revealed long-range positional correlations within the layers (see Fig. 4, Fig. S1 and S2, ESI†).
At this point, we recall the structural odd–even effect observed experimentally for the Cry phases of α-nTs,39 manifested as an alternation in the density of the Cry phase between large and small values depending on the parity of the α-nT molecule (see Fig. 7a). Interestingly, our simulations (see Fig. 7a) are in full support of this observation, especially for the second crystalline state point observed in the ρ-vs.-T plot. A similar alternation is observed in the magnitude of the average tilt angle with respect to the number of thiophene rings shown in Fig. 7b. Even though our model cannot precisely reproduce the structure of the ideal α-nT crystals formed at low temperatures (such as the herringbone arrangement of the molecules and the density),1,24,39 it is capable of qualitatively describing their odd–even effect.
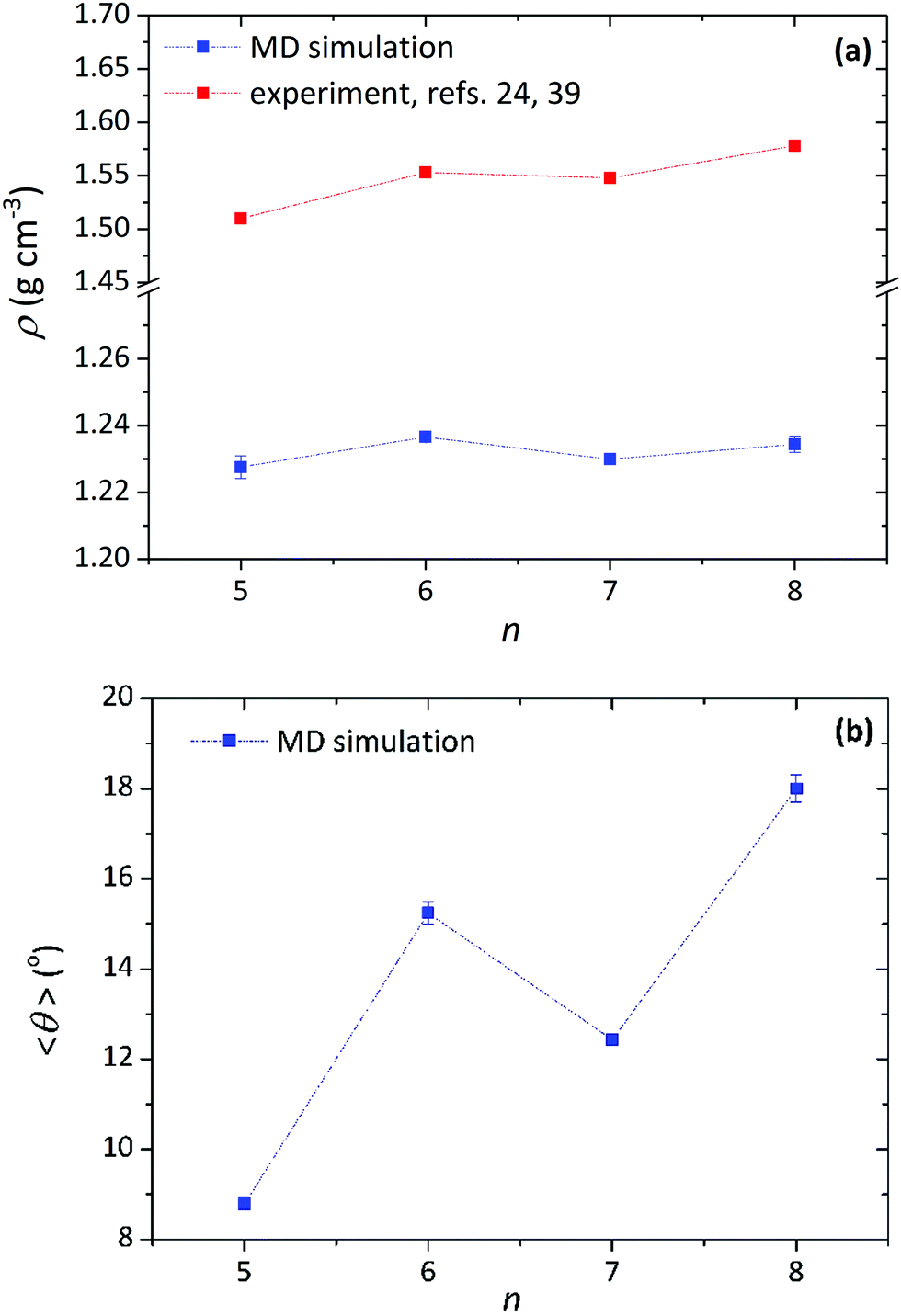 | ||
| Fig. 7 MD results (blue symbols) concerning the dependence of: (a) mass density, and (b) average tilt angle on number of thiophene rings (n) per molecule in the Cry phase of α-nT molecules at different temperatures (at P = 1 atm). Some experimental data at around room temperature (red symbols) are also shown in (a).24,39 | ||
Our MD predictions for the phase behaviour of α-nTs with n = 5–8 are summarized in Fig. 8. For n = 5, 7 and 8, it is the first time (to our knowledge) that their mesophase behaviour is reported and quantitatively analyzed. In the past, clear evidence of structural transitions and manifestation of mesophases have been provided only for α-6T,15,16,34,35b together with a report for an undefined structural transition for α-8T from DSC analysis.24
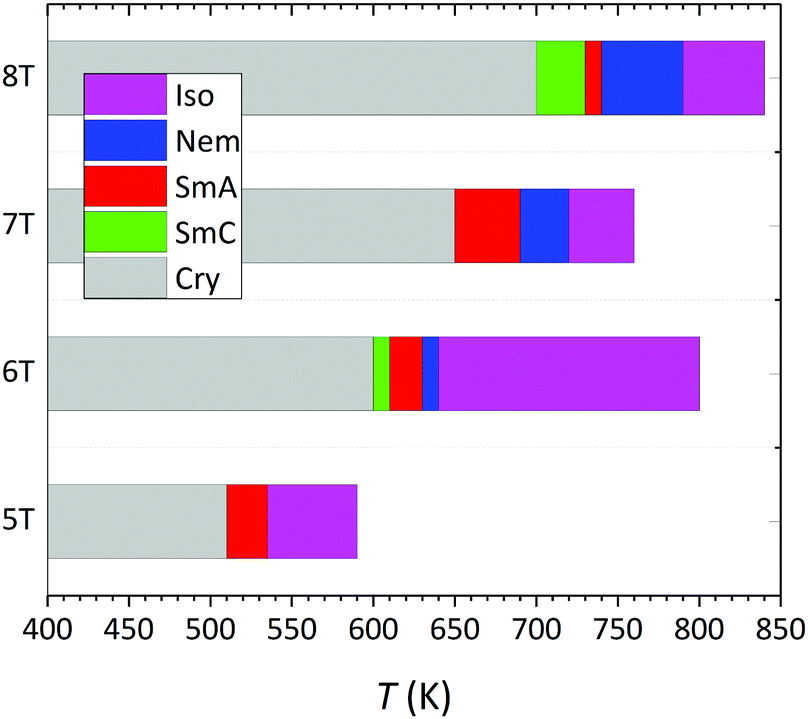 | ||
| Fig. 8 Phase transition diagram describing the overall behaviour of α-nTs with n = 5–8 from the current NPT MD simulations, at P = 1 atm. The data for α-6T have been taken from ref. 35b. | ||
In our simulations, the Nem phase is observed only for α-nTs with n = 6–8 (not for n = 5), and its temperature range widens as the number of thiophene rings increases. At the same time, the range of temperatures for which the SmC phase is observed (only for even-numbered α-nTs) is broader for α-8T. In the literature, dependence of the temperature range of mesophases on the number of thiophene rings has been reported also for other all-aromatic compounds.19,48,49 It is also important to note that all phase transitions in our simulations occurred spontaneously upon gradually cooling the system from its Iso phase at a high enough temperature down to lower temperatures. In the literature, mesophase formation through simulations is typically studied by employing heating runs starting from a well-ordered or crystal structure and gradually increasing the temperature. From a methodological point of view, employing cooling and not heating simulations to study phase behaviour is very challenging, because there is always the possibility that the system can freeze to intermediate amorphous/glassy states as the temperature decreases.
Fig. 9 shows the LC phase transition temperatures that are found from our MD simulations for all α-nT systems addressed here showing a clear dependence on the number of thiophene rings. The melting points and the clearing temperatures increase as the number of thiophene rings increases. Interestingly, the clearing temperature can be described as a linear function with respect to the number of thiophene rings. A similar trend has been reported by Ponomarenko and Kirchmeyer for α,α′-didecyloligothiophene chains consisting of n = 4–6 thiophene rings.33 The united-atom approximation employed in this work seems to be capable of capturing fairly well the transition points from the Sm phase to the Cry phase (i.e., the melting points) compared to the experimentally measured melting points, particularly for the shorter α-5T and α-6T molecules. For example, Fig. 9 shows that the predicted melting point of α-5T is 510 K while the corresponding experimentally estimated value is 525 K.23–25 It is also interesting that the experimental melting point is close to the Iso-to-SmA transition point (535 K) predicted from the simulations. Similar reasonable deviations (on the order of 50 to 60 K) between simulation and experimental data are noticed for the melting points of α-7T and α-8T. An explanation for these deviations is that the calculated melting points from the simulations are obtained by gradually cooling the sample from higher to lower temperatures. Moreover, the crystal structure from the simulations is not identical to the ideal crystal structure of α-nT (typically characterized by a herringbone arrangement of chains within the layers). On the other hand, the experimental melting points are obtained by melting such ideal crystal structures. Therefore, not only the process (heating vs. cooling) but also the starting structures for the calculation of the melting points between our simulations and the experiments are different, which offers an additional explanation for the deviations between the two approaches. Overall, however, and taking into consideration that even independent experimental measurements24,25 from different research groups show similar differences (see Fig. 9), the comparison is quite favourable. We further note that the experimental observations indicate a saturation of the melting point with increasing n. Our simulations also suggest that the melting points approach a constant value with n but with a smaller rate.
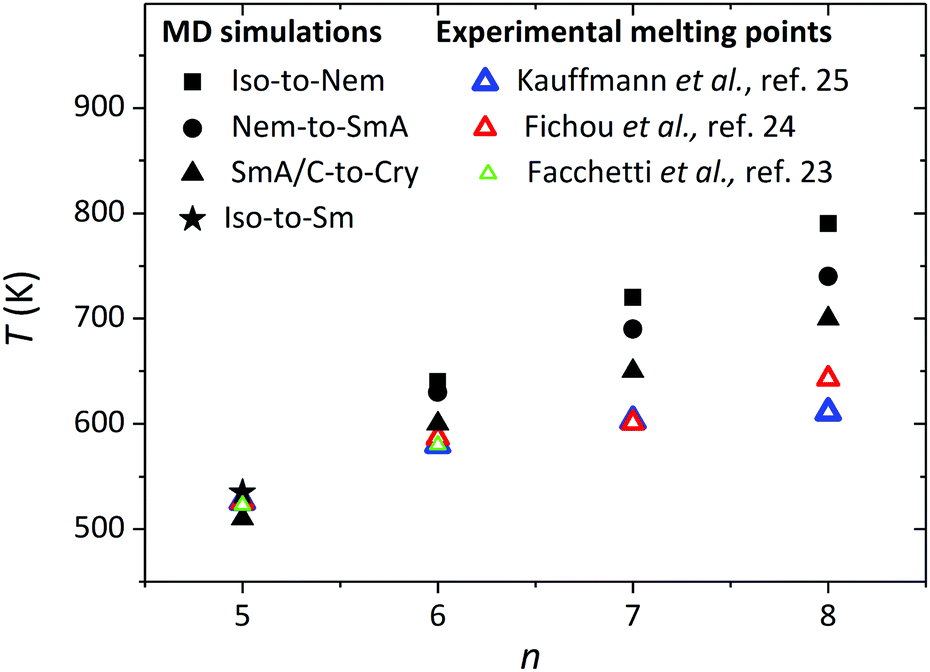 | ||
| Fig. 9 Liquid crystalline phase transition temperatures as a function of the number of thiophene rings (n) from the present NPT MD simulations (P = 1 atm). The Iso-to-Nem, Nem-to-SmA, and SmA/SmC-to-Cry transitions are marked with filled squares, circles and triangles, respectively. The Iso-to-SmA phase transition in α-5T is shown with a star. The open triangles (green, red and blue) illustrate experimentally measured melting points.23–25 The simulation data for α-6T have been taken from ref. 35b. | ||
3.2. Interpretation of the phase behaviour based on detailed structural and conformational analysis
To further analyze molecular organization in the observed LC phases of the α-nTs addressed in our work, we carried out a detailed investigation of their conformational properties based on the sequence of characteristic values for the inter-ring torsional angle along their backbone.The type of conformational state of successive thiophene rings along an α-nT molecule can be categorized based on the calculation of the inter-ring dihedral angle ϕS–C–C–S, as follows:35 if ϕS–C–C–S ∈ [−90°, 90°], then two successive rings along the molecule are considered to be in the syn-SS-parallel configuration denoted as a c-state; if, on the other hand, ϕS–C–C–S ∈ [−180°, −90°) ∪ (90°, 180°], then, the two successive rings are considered to be in the anti-SS-parallel configuration denoted as a t-state (see Fig. 2). Consequently, different conformations of α-nT chains can be distinguished by counting the number of t- and c-states of successive thiophene rings along inter-ring bonds of the chain. Each group consists of a number of conformations that occur by mutual permutation of the constituent c- and t-states. For a molecule with n rings, n conformational groups can appear. For example, an α-7T molecule has seven (7) different conformational groups; one of them, the c–t–t–t–t–t group, is illustrated in Fig. 2b. Calculated percentages of the possible conformational groups appearing in the studied α-nT systems as a function of temperature are reported in Fig. 10. Several conclusion can be drawn from the data shown in Fig. 10: (a) the relative population of conformational groups containing at least one c-state is very high in all mesophases formed (Iso, LC and Cry); (b) the percentage of the all-trans (all t-states) conformational group increases with decreasing temperature, especially in the well-ordered phases; (c) the percentage of the conformational groups that contain two c-states increases as the number of thiophene rings increases; nevertheless, it becomes very small below the first positionally ordered phase formed; and (d) conformational groups that contain more than two c-states are the least probable.
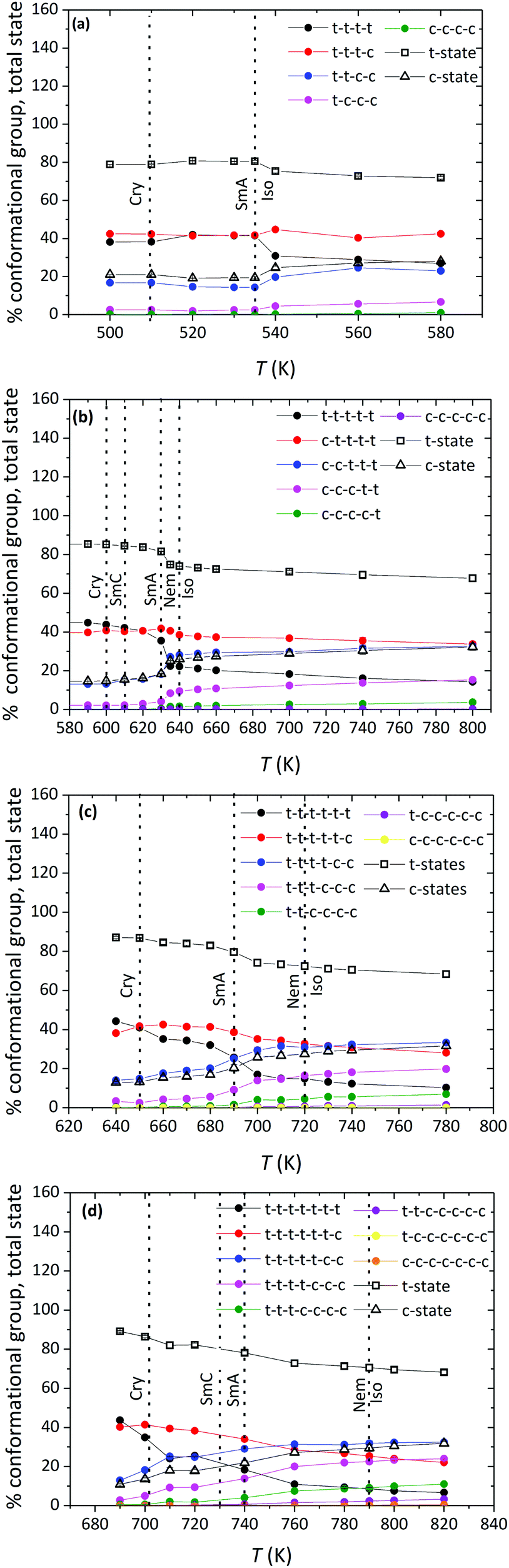 | ||
| Fig. 10 Simulation predictions for the percentage of n conformational groups (filled symbols) and the total percentage of c- and t-states (open symbols) of α-nTs as a function of temperature, at P = 1 atm. Plots (a), (b), (c) and (d) correspond to α-5T, α-6T, α-7T and α-8T, respectively. The data for α-6T have been taken from ref. 35b. Dotted lines indicate roughly the borders between the various phases. | ||
An important characteristic of α-nTs is that t-states are dominant over the entire range of temperatures studied. This is confirmed by calculating the total percentage of t- (or c-states) irrespective of the conformational group they belong to, by using the formula35a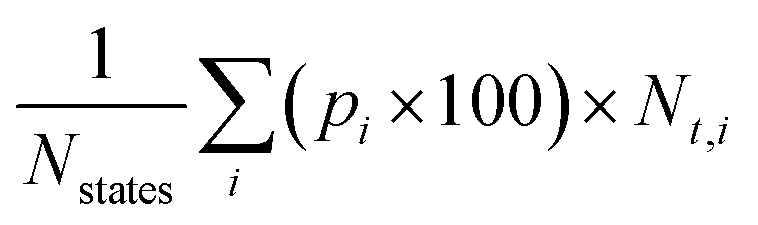 , where Nstates = n − 1 denotes the total number of conformational groups (equal to the number of rings per α-nT molecule), the index i runs over all conformational groups, pi × 100 is the percentage of the i-th conformational group and Nt,i denotes the number of t-states in the i-th conformational group. According to Fig. 10, for all α-nT molecules studied, the total percentage of t-states increases with decreasing temperature, with the corresponding total percentage of c-states decreasing accordingly.
, where Nstates = n − 1 denotes the total number of conformational groups (equal to the number of rings per α-nT molecule), the index i runs over all conformational groups, pi × 100 is the percentage of the i-th conformational group and Nt,i denotes the number of t-states in the i-th conformational group. According to Fig. 10, for all α-nT molecules studied, the total percentage of t-states increases with decreasing temperature, with the corresponding total percentage of c-states decreasing accordingly.
The above conformational analysis is clearly related to the phase behaviour of the molecules, because the transition from the Iso to ordered phases is accompanied by significant changes in their conformational properties. In particular, for α-nTs with n = 6–8, the percentage of the all-trans group exhibits a smooth increase at the Iso-to-Nem phase transition whereas that of groups containing more than two c-states decreases. Interestingly, more striking conformational changes for α-nTs with n = 6–8 are observed at the Nem-to-SmA phase transitions (see Fig. 10) where a distinct jump shows up in the percentage of the all-trans group. In the case of α-5T, a small jump is discernible also at the Iso-to-SmA phase transition for the all-trans group. Increased all-trans populations together with reduced conformations containing two or more c-states facilitate chain packing of α-nT molecules, thus also their self-assembly into layers. This happens because conformational groups with at least two c-states contain a mixture of different molecular configurations (in terms of the exact location of c- and t-states along the chain) which favours structural defects thereby facilitating disorder. It appears therefore that conformational changes occurring at the intra-molecular level play a key role in the formation of ordered phases. For example, the increase in the population of all-trans conformations is related to the extension of the molecule, with the values of 〈Ree2〉 increasing as the temperature decreases (see Table 1) revealing a strong tendency for more elongated structures; obviously, this promotes macroscopic orientational order.
To investigate even further structural ordering in the LC and Cry phases of α-nTs, we calculated the gCoM-ring(r) and gCoM-ring1(r) pair correlation functions of the thiophene rings’ centers-of-mass (see eqn (5) and (6)) in the simulated systems. Characteristic plots of gCoM-ring(r) and gCoM-ring1(r) at the first stable SmA and Cry phases formed are shown in Fig. S3 and S4 (ESI†), respectively. In the SmA phase, gCoM-ring1(r) is nearly flat implying that neighboring thiophene rings between different chains exhibit either an inter-SS-antiparallel configuration where successive sulfur (S) atoms are pointing to opposite directions or an inter-SS-parallel configuration where all sulfur S atoms are pointing to the same direction. SS-antiparallel and -parallel conformations appear randomly, which prevents one or the other to dominate (see Fig. S3, ESI†). On the other hand, in the Cry phase, the non-vanishing positive values of gCoM-ring1(r) for values r corresponding to the peaks in the gCoM-ring(r) plot indicate that there is a slight tendency of inter-molecular neighboring thiophene rings to point to the same direction (inter-SS-parallel configuration) (see Fig. S4, ESI†).
The above detailed analysis demonstrates that α-nTs with n = 5–8 share many common structural and conformational characteristics irrespective of their parity (odd or even number of thiophene rings). In particular, conformational groups containing a mixture of c- and t-states contribute practically equally to self-organization (in the corresponding phases) for all members in the family. Nevertheless, there is a striking difference: the internal symmetry of the all-trans conformation in even-numbered α-nTs is not the same as the corresponding all-trans conformation in odd-numbered α-nTs, because the former possess a C2h point group and the latter a C2v point group.39,40 The main difference lies in the fact that even-numbered α-nT molecules exhibit a center of inversion located at the CoM of the molecules (i.e., at the middle point of the central inter-ring bond) while odd-numbered ones possess a mirror plane that intersects the central thiophene ring.39 This molecular symmetry renders even-numbered α-nTs in the all-trans group sterically non-polar as successive pairs of rings follow an anti-SS-parallel arrangement along the chain; odd α-nTs, on the other hand, are sterically polar, since they contain always one thiophene ring that remains un-paired along the chain. This difference favours chain inclination within layers (which promotes the formation of SmC phases) and results in larger tilt angles in the Cry phase for even-numbered α-nTs than for odd-numbered ones.
4. Conclusions
Our work offers the first systematic computational study of the mesomorphic behaviour of α-unsubstituted thiophene-based oligomers (α-nTs) with the help of a validated, fully flexible united-atom model. Our MD simulations indicate that α-nTs with n = 5–8 exhibit a rich phase behaviour characterized by the appearance of a nematic phase and of two types of smectic phases (SmA and SmC). In the SmC phase, the tilt-direction is uniform between adjacent layers, which points to the development of a synclinic structure.Moreover, our simulations demonstrate that it is mainly the parity of the molecule (the even or odd number of thiophene rings) that determines the type of smectic phases formed. Even-numbered α-nTs form both SmA and SmC phases whereas odd-numbered α-nTs form only a SmA phase. This points to a unique odd–even structural phenomenon for chain molecules consisting solely of rings, which occurs in positionally ordered liquid crystals (herein, the smectic phases). Such a phenomenon appears also in the Cry phases: depending on the parity of the molecule, α-nTs are characterized by density values that alternate between large (for even-numbered) and small (for odd-numbered) values; even-numbered chains are thus self-organizing into more dense states. A similar trend has also been observed in some experiments.39 Additionally, in the same phase, the value of the calculated tilt angle (the angle between the layer normal vector and the director of the phase) alternates in a zig-zag manner: even-numbered α-nTs exhibit larger tilt angles while odd-numbered ones exhibit smaller tilt angles.
A detailed analysis of chain conformations revealed that t-states in α-nT oligomers (corresponding to an anti-SS-parallel configuration of successive thiophene rings) are the dominant ones. The total number of t-states is always higher than the total number of c-states for all temperatures studied (extending from the Iso to the Cry phases), with their difference increasing as the temperature is decreased.
Configurations of α-nT oligomers were further categorized into groups according to their number of c- and t-states along the molecule. It was observed, then, that significant changes in the percentage of all conformational groups occur at the phase transition points. The group, in particular, that contains one c-state is the most abundant one at all temperatures studied. The percentage of the all-trans conformational group is also significant, exhibiting distinct jumps at the transition points. In contrast, conformational groups that contain two or more c-states are the least probable. Variations in the intra-chain conformation as a function of temperature are accompanied by non-negligible variations in the average end-to-end distance of the molecule, which tends to increase as the temperature is decreased.
The observed intra-chain conformational changes with decreasing temperature are common to all α-nTs studied here, and appear to be the main driving force for their self-assembly into ordered phases. We also mention that the all-trans configuration of even-numbered α-nTs has a different internal molecular symmetry than that of odd-numbered ones, giving rise to a unique, structural odd–even effect.
The melting points predicted by our MD simulations are in good agreement with experimentally measured ones, especially for the shorter α-nTs (α-5T and α-6T) studied. It appears that α-nTs are characterized by relatively short LC temperature ranges and high transition temperatures, similar to other all-aromatic molecules;19,48,49 moreover, their clearing temperature increases linearly with number of thiophene rings in the molecule.33
We hope that our work will stimulate new experimental studies of these molecules, particularly as far as the detailed characterization of their mesophase behaviour is concerned. We also hope that the structural changes and transitions demonstrated by our study for short α-nT molecules will motivate additional studies for other type of rod-like molecules (such as liquid crystals consisting of rings). Extending the present study to significantly longer (i.e., higher molecular weight) α-nT molecules would also be very important.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support for this study has been provided by General Secretariat for Research and Technology (GSRT) and Hellenic Foundation for Research and Innovation (HFRI) under the scholarship announcement ‘First HFRI call for the financial support of doctoral candidates’, code: 1460. Financial support has also been provided through the National Project titled: General method for the simulation of self-organization in nanostructured polymeric systems (GENESIS, code 1010 – D655) which is part of the Action ‘‘ARISTEIA – Education and Lifelong Learning’’ Programme and is supported both by the European Union (European Social Fund-ESF) and national funds. We acknowledge the Greek Research & Technology Network (GRNET) for generous allocation of computational time on the National HPC facility-ARIS, based in Athens, Greece, under project THIOSIM (pr005043). We feel grateful to Dr Dimitris Dellis from GRNET for his support on several technical aspects of the work.Notes and references
- D. Fichou, Handbook of Oligo- and Polythiophenes, Wiley-VCH, Weinheim, Germany, 1999 Search PubMed.
- G. Horowitz and M. E. Hajlaoui, Adv. Mater., 2000, 12, 1046–1050 CrossRef CAS.
- C. D. Dimitrakopoulos and P. R. L. Malefant, Adv. Mater., 2002, 14, 99–117 CrossRef CAS.
- H. E. Katz and Z. Bao, J. Phys. Chem. B, 2000, 104, 671–678 CrossRef CAS.
- G. Horowitz, J. Mater. Chem., 1999, 9, 2021–2026 RSC.
- G. Horowitz, Adv. Mater., 1998, 10, 365–377 CrossRef CAS.
- A. J. Lovinger and L. J. J. Rothberg, Mater. Res., 1996, 11, 1581–1592 CrossRef CAS.
- H. S. Nalwa, Handbook of Advanced Electronic and Photonic Materials and Devices, Academic Press, 2000 Search PubMed.
- J. H. Burroughes, D. D. C. Bradley, A. R. Brown, R. N. Marks, K. Mackay, R. H. Friend, P. L. Burns and A. B. Holmes, Nature, 1990, 347, 539–541 CrossRef CAS.
- H. Spanggaard and F. C. Krebs, Sol. Energy Mater. Sol. Cells, 2004, 83, 125–146 CrossRef CAS.
- A. P. Smith, R. R. Smith, B. E. Taylor and M. F. Durstock, Chem. Mater., 2004, 16, 4687–4692 CrossRef CAS.
- S. Nagamatsu, K. Kaneto, R. Azumi, M. Matsumoto, Y. Yoshida and K. Yase, J. Phys. Chem. B, 2005, 109, 9374–9378 CrossRef CAS PubMed.
- X. Yang, L. Wang, C. Wang, W. Long and Z. Shuai, Chem. Mater., 2008, 20, 3205–3211 CrossRef CAS.
- K. Oikawa, H. Monobe, J. Takahashi, K. Tsuchiya, B. Heinrich, D. Guillon and Y. Shimizu, Chem. Commun., 2005, 5337–5339 RSC.
- C. Taliani, R. Zamboni, G. Ruani, S. Rossini and R. Lazzaroni, J. Mol. Electron., 1990, 6, 225–226 CAS.
- S. Destri, M. Mascherpa and W. Porzio, Adv. Mater., 1993, 5, 43–46 CrossRef CAS.
- M. Kramer and V. Hoffmann, Opt. Mater., 1998, 9, 65–69 CrossRef CAS.
- M. Melucci, G. Barbarella, M. Zambianchi, P. Di Pietro and A. Bongini, J. Org. Chem., 2004, 69, 4821–4828 CrossRef CAS PubMed.
- (a) S. Kuiper, W. F. Jager, T. J. Dingemans and S. J. Picken, Liq. Cryst., 2009, 36, 389–396 CrossRef CAS; (b) S. Kuiper, Synthesis, Mesophase Behaviour and Charge Mobility of Phenyl-Thiophene Pentamers, PhD thesis, Delft University of Technology, 2011.
- M. Melucci, M. Gazzano, G. Barbarella, M. Cavallini, F. Biscarini, P. Maccagnani and P. Ostoja, J. Am. Chem. Soc., 2003, 125, 10266–10274 CrossRef CAS PubMed.
- E. Müller and C. Ziegler, J. Mater. Chem., 2000, 10, 47–53 RSC.
- T. Yamada, R. Azumi, H. Tachibana, M. Abe, P. Bäuerle and M. Matsumoto, Chem. Lett., 2001, 1022–1023 CrossRef CAS.
- A. Facchetti, M.-H. Yoon, C. L. Stern, G. R. Hutchison, M. A. Ratner and T. J. Marks, J. Am. Chem. Soc., 2004, 126, 13480–13501 CrossRef CAS PubMed.
- D. Fichou, M.-P. Teulade-Fichou, G. Horowitz and F. Demanze, Adv. Mater., 1997, 9, 75–80 CrossRef CAS.
- (a) T. Kaufmann, Angew. Chem., 1979, 18, 1–19 CrossRef; (b) T. Kaufmann and H. Lexy, Chem. Ber., 1981, 114, 3674–3683 CrossRef.
- F. Garnier, A. Yassar, R. Hajlaoui, G. Horowitz, F. Deloffre, B. Servet, S. Ries and P. Alnot, J. Am. Chem. Soc., 1993, 115, 8716–8721 CrossRef CAS.
- R. Azumi, G. Götz and P. Bäuerle, Synth. Met., 1999, 101, 544–545 CrossRef CAS.
- M. Funahashi and J.-I. Hanna, Appl. Phys. Lett., 2000, 76, 2574–2576 CrossRef CAS.
- P. Liu, H. Nakano and Y. Shirota, Liq. Cryst., 2001, 28, 581–589 CrossRef CAS.
- A. Dell’Aquila, P. Mastrorilli, C. F. Nobile, G. Romanazzi, G. P. Suranna, L. Torsi, M. C. Tanese, D. Acierno, E. Amendola and P. Morales, J. Mater. Chem., 2006, 16, 1183–1191 RSC.
- J. Wang, W. H. de Jeu, U. Ziener, M. S. Polinskaya, S. A. Ponomarenko, U. Ruecker, M. A. Ruderer, E. M. Herzig, P. Muller-Buschbaum, M. Moeller and A. Mourran, Langmuir, 2014, 30, 2752–2760 CrossRef CAS PubMed.
- A. Kreyes, S. Ellinger, K. Landfester, M. Defaux, D. A. Ivanov, A. Elschner, T. Meyer-Friedrichsen and U. Ziener, Chem. Mater., 2010, 22, 2079–2092 CrossRef CAS.
- S. Ponomarenko and S. Kirchmeyer, J. Mater. Chem., 2003, 13, 197–202 RSC.
- A. Pizzirusso, M. Savini, L. Muccioli and C. Zannoni, J. Mater. Chem., 2011, 21, 125–133 RSC.
- (a) F. T. Tsourtou, S. D. Peroukidis, L. D. Peristeras and V. G. Mavrantzas, Macromolecules, 2018, 51, 8406–8423 CrossRef CAS; (b) F. D. Tsourtou, E. Skountzos, S. D. Peroukidis and V. G. Mavrantzas, Soft Matter, 2018, 14, 8253–8266 RSC.
- (a) R. Boese, H.-C. Weiss and D. Bläser, Angew. Chem., Int. Ed., 1999, 38, 988–992 CrossRef CAS; (b) R. Thalladi, R. Boese and H.-C. Weiss, Angew. Chem., Int. Ed., 2000, 39, 918–922 CrossRef.
- (a) D. Demus, J. Goodby, G. W. Gray, H.-W. Spiess and V. Vill, Handbook of Liquid Crystals, Wiley-VCH, 1998, ch. I, vol. 1 Search PubMed; (b) D. Demus, J. Goodby, G. W. Gray, H.-W. Spiess and V. Vill, Handbook of Liquid Crystals, Wiley-VCH, 1998, ch. X, vol. 2B Search PubMed; (c) D. Demus, J. Goodby, G. W. Gray, H.-W. Spiess and V. Vill, Handbook of Liquid Crystals, Wiley-VCH, 1998, ch. VI, vol. 2B Search PubMed.
- (a) V. Borshch, Y.-K. Kim, J. Xiang, M. Gao, A. Jakli, V. P. Panov, J. K. Vij, C. T. Imrie, M. G. Tamba, G. H. Mehl and O. D. Lavrentovich, Nat. Commun., 2013, 4, 2635–2638 CrossRef CAS PubMed; (b) D. Chen, J. H. Porada, J. B. Hooper, A. Klittnick, Y. Shen, M. R. Tuchband, E. Korblova, D. Bedrov, D. M. Walba, M. A. Glaser, J. E. Maclennan and N. A. Clark, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 15931–15936 CrossRef CAS PubMed.
- R. Azumi, M. Goto, K. Honda and M. Matsumoto, Bull. Chem. Soc. Jpn., 2003, 76, 1561–1567 CrossRef CAS.
- F. Meinardi, M. Cerminara, A. Sassella, A. Borghesi, P. Spearman, G. Bongiovanni, A. Mura and R. Tubino, Phys. Rev. Lett., 2002, 89, 157403 CrossRef CAS PubMed.
- X. Wang, G. He, Y. Li, Z. Kuang, Q. Guo, J.-L. Wang, J. Pei and A. Xia, J. Phys. Chem. C, 2017, 121, 7659–7666 CrossRef.
- J. Ramos, L. D. Peristeras and D. N. Theodorou, Macromolecules, 2007, 40, 9640–9650 CrossRef CAS.
- Scienomics, MAPS Platform, Version 3.4.2, France (2015). See also: http://scienomics.com/.
- http://www.gromacs.org/ .
- S. Nosé, Prog. Theor. Phys. Suppl., 1991, 130, 1–46 CrossRef.
- M. Parrinello and A. Rahman, J. Appl. Phys., 1981, 52, 7182–7190 CrossRef CAS.
- S. L. Mayo, B. O. Olafson and W. A. Goddard, J. Phys. Chem., 1990, 94, 8897–8909 CrossRef CAS.
- K. N. Baker, A. V. Fratini, T. Resch, H. C. Knachel, W. W. Adams, E. P. Socci and B. L. Farmer, Polymer, 1993, 34, 1571–1587 CrossRef CAS.
- N. A. Zafiropoulos, E.-J. Choi, T. Dingemans, W. Lin and E. T. Samulski, Chem. Mater., 2008, 20, 3821–3831 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Calculated inter-molecular longitudinal, perpendicular and thiophene rings’ centers-of-mass pair correlation functions (Fig. S1–S4); atomistic snapshots of crystal phases (Fig. S5). See DOI: 10.1039/c9tc02374g |
| This journal is © The Royal Society of Chemistry 2019 |