
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStable antiferromagnetic nanocrystals for room temperature applications: the case of iron nitride†
Iwona Agnieszka
Kowalik
 a,
Nevill
Gonzalez Szwacki
b,
Miguel Ángel
Niño
c,
Francisco Jesús
Luque
d and
Dimitri
Arvanitis
*e
a,
Nevill
Gonzalez Szwacki
b,
Miguel Ángel
Niño
c,
Francisco Jesús
Luque
d and
Dimitri
Arvanitis
*e
aInstitute of Physics, Polish Academy of Sciences, al. Lotników 32/46, 02-668 Warsaw, Poland
bInstitute of Theoretical Physics, Faculty of Physics, University of Warsaw, 00-681 Warsaw, Poland
cIMDEA Nanociencia, Campus de Cantoblanco, E-28049 Madrid, Spain
dDepto. de Física de la Materia Condensada, Universidad Autónoma de Madrid, E-28049, Madrid, Spain
eDepartment of Physics and Astronomy, Uppsala University, P.O. Box 516, 751 20 Uppsala, Sweden. E-mail: dimitri.arvanitis@physics.uu.se
First published on 10th July 2019
Abstract
We characterise the magnetism of self-assembled FenN nanocrystals, combining core level spectroscopy with first-principles theory. Not only ferromagnetic but also antiferromagnetic iron nitride nanocrystals are identified, exhibiting stable magnetic properties at room temperature. New stable magnetic phases are found, previously believed to order magnetically only well below room temperature. As determined by the growth conditions, several phases of magnetic FenN nanocrystals are identified in the near surface region of GaN based thin films, with typical dimensions from 50 to 100 nm, embedded in the (Ga,Fe)N lattice or residing on the GaN surface. We determine, at room temperature, Fe4N and Fe3N ferromagnetic nanocrystals, as well as Fe2N and FeN nanocrystals in an antiferromagnetic state, which is not stable at room temperature in their bulk phases.
1 Introduction
The miniaturization of devices requires new materials with a size in the nanometer range, exhibiting stable magnetic properties. One of the challenges material designers face is to stabilize the magnetic moments against temperature fluctuations, whose impact is increasing as the material size is reduced. Here we show that iron nitride nanocrystals on a widely used semiconductor, gallium nitride, offer a rich potential for novel magnetic materials in the nanometer range.The iron–nitrogen system, FenN, has attracted a lot of attention because of its intricate phase diagram and its potential for applications.1–3 For doped nitrides with transition metal (TM) impurities, nanocomposites formed during the epitaxy process show often ferromagnetic (FM) signatures persisting to above the room temperature.4,5 Such nanocomposites containing metallic magnetic nanostructures, have a potential for applications in electronics, spintronics, photovoltaics, plasmonics, and thermoelectrics.5 Methods have been elaborated to position the nanocrystals in predefined planes,6,7 to impose lateral order,8 and to fabricate nanocrystals in a form of dense nanocolumn arrays extending across the film thickness.5,9 We study here the case of the near surface region of GaN doped with Fe, obtained by metal–organic vapour phase epitaxy (MOVPE). For this system FenN nanocrystals are formed.10,11
We combine element specific X-ray Magnetic Circular Dichroism (XMCD), X-ray Linear Magnetic Dichroism (XLMD), and X-ray Photo Emission Electron Microscopy (XPEEM) at the Fe L-edges to assess the magnetic phases of Fe-rich nanocrystals in various families of (Ga,Fe)N samples. By combining XMCD with XLMD measurements we quantify the value of the antiferromagnetic (AFM) part of the magnetic moments. We find that the AFM moment turns out to be of the same order of magnitude as the FM one. By using XPEEM data, we find that the size distribution of the AFM and FM nanocrystals is similar. Using first principles calculations based on density functional theory and ab initio modelling of our data using multiple scattering calculations, we show that the existence of an AFM magnetisation component in these samples is due to Fe2N and FeN nanocrystals with an AFM ground state.
While this observation is consistent with magnetization data revealing a linear component in field,6 the presence of a high temperature AFM ordering is surprising as the known AFM phases, particularly ζ-Fe2N12 are stable only at low temperatures (below 10 K). However, it has been known since some time that particular hosts can impose chemical composition and a crystal structure of the phase separated nanocrystals, which is unstable and thus not observed in the free standing case. One of the examples is the phase separation in Ga1−xMnxAs, which if grown by molecular beam epitaxy above 280 °C at x = 5% decomposes into Mn-poor host and Mn-rich cubic nanocrystals,8 whereas free standing MnAs assumes only a hexagonal NiAs structure.
Our first principles calculations of FenN magnetism considering various structural and chemical settings show that FeN in the NaCl structure is a stable AFM system. Both FeN and Fe2N nanocrystals are found experimentally to be in a stable AFM state at room temperature on GaN. We suggest therefore that under appropriate growth conditions the GaN host can stabilize AFM nanocrystals. In view of the emerging field of AFM spintronics,13,14 the (Ga,Fe)N system becomes interesting, particularly in the context of fast magnetooptical switching, taking into account the progress in positioning of nanocrystals in the host.15
2 Methods
The samples as introduced into the ultra high vacuum end stations for the XAS measurements show X-ray absorption specific to C and O atom impurities, which disappears after in situ soft sputtering with Ar+ ions. For the data of Fig. 1 employing XPEEM allows for laterally resolving information. These measurements have been performed using a grazing X-ray incidence angle of 16 degrees versus the sample surface plane, at room temperature. In Fig. 1 XPEEM micrographs are shown in the direct XAS mode using linear X-rays, with the photon energy set at the maximum of the Fe L3 white line. These micrographs allow to characterise the chemical state of the near surface region, by tracing the relative amount of Fe. In the XMCD mode circular X-rays are used allowing to trace the ferromagnetic state of the nanocrystals which are resolved (Appendix, ESI†![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 16). No magnetic field is applied in this experiment.
16). No magnetic field is applied in this experiment.
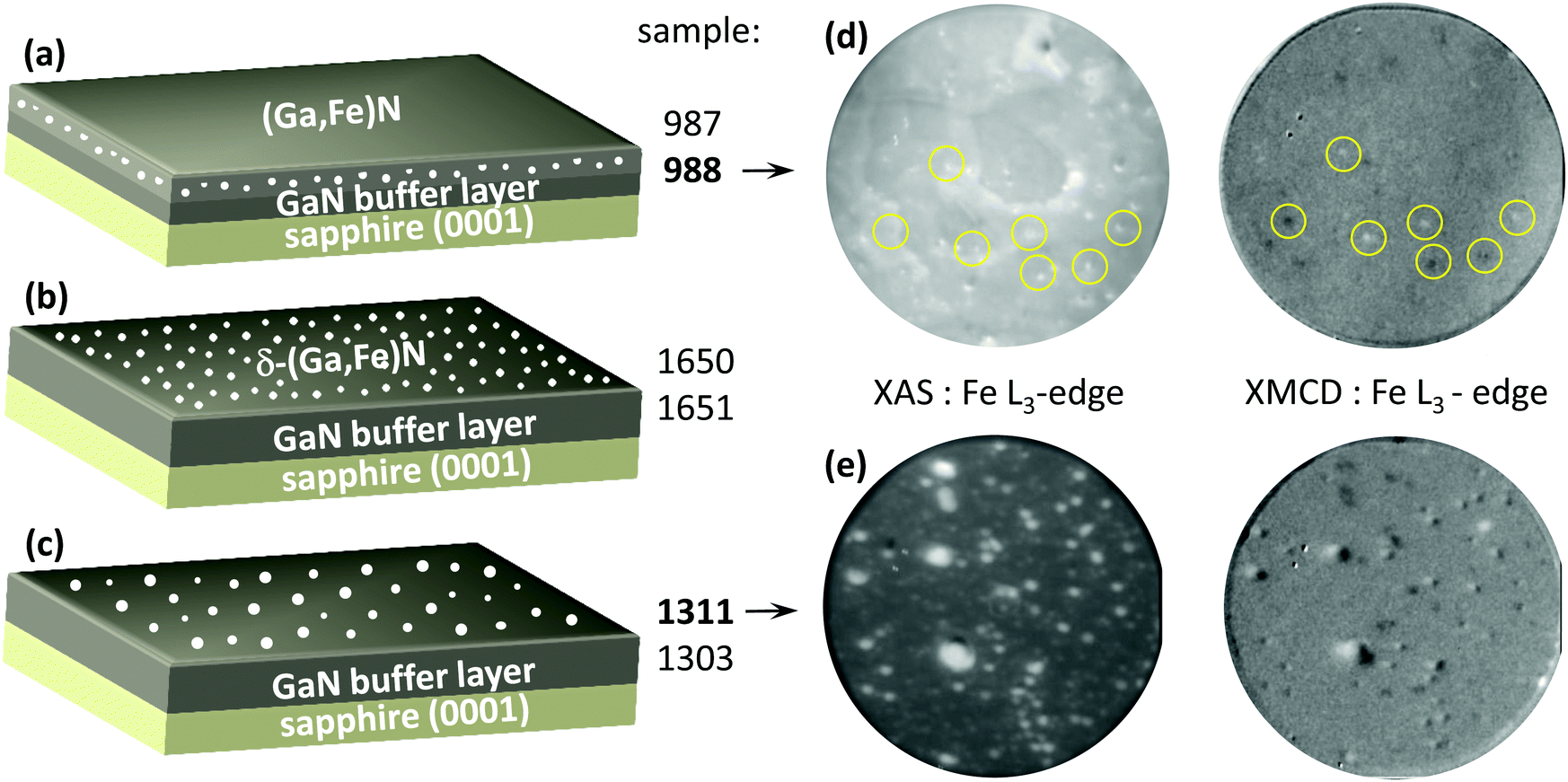 | ||
| Fig. 1 Overview of the investigated samples and their XPEEM characterisation. (a) The Fe rich nanocrystals are embedded randomly in the GaN lattice (samples 987, 988). (b) δ-Growth is leading to planar arrays of nanocrystals at a distinct depth below the surface (samples 1650, 1651). (c) The nanocrystals are located on the GaN surface (samples 1311, 1303). (d) XPEEM micrographs of sample 988 with a field of view of 2 μm, showing the same region. In the XAS mode, the Fe rich nanocrystals are visible as bright contrast regions. Only about 50% of these nanocrystals, do exhibit FM XMCD contrast (circles). (e) XPEEM micrographs of sample 1311 with a field of view of 2 μm, of the same sample region. The larger nanocrystals deviate from the single magnetic domain state in the XMCD mode. | ||
For the data of Fig. 2 no lateral resolution is applied, all the nanocrystals probed by the X-ray beam are measured. For the data of Fig. 2(a) the XMCD is probed using circular X-rays and an applied magnetic field of 6T. The angle of X-ray incidence is along the sample surface normal. Both the X-ray helicity and the magnetic field can be reversed. The exact photon energy of the Fe L-edges, its broadening and intensity are good markers of the electronic configuration of the Fe atoms. Here, the absolute photon energy and intensity is calibrated by using the bcc Fe reference sample measured in the same run within 0.1 eV accuracy. The Fe L-edges are dominated by dipole transitions to the Fe 3d final states, seen in the form of intense “white line” peaks, each followed by a continuum of final states. In the inter-peak region, for energies close to and above the L3 white line and similarly for energies above the L2 line, the Fe 4s final states are also probed but with a very small cross section. Once the spectra are properly normalised, differences in the white line intensities are indicative of a different number of Fe(3d) empty final states. The values of the magnetic moments on a per atom basis, can be obtained from the XAS and XMCD spectra by means of the XMCD magneto-optical sum rules.17
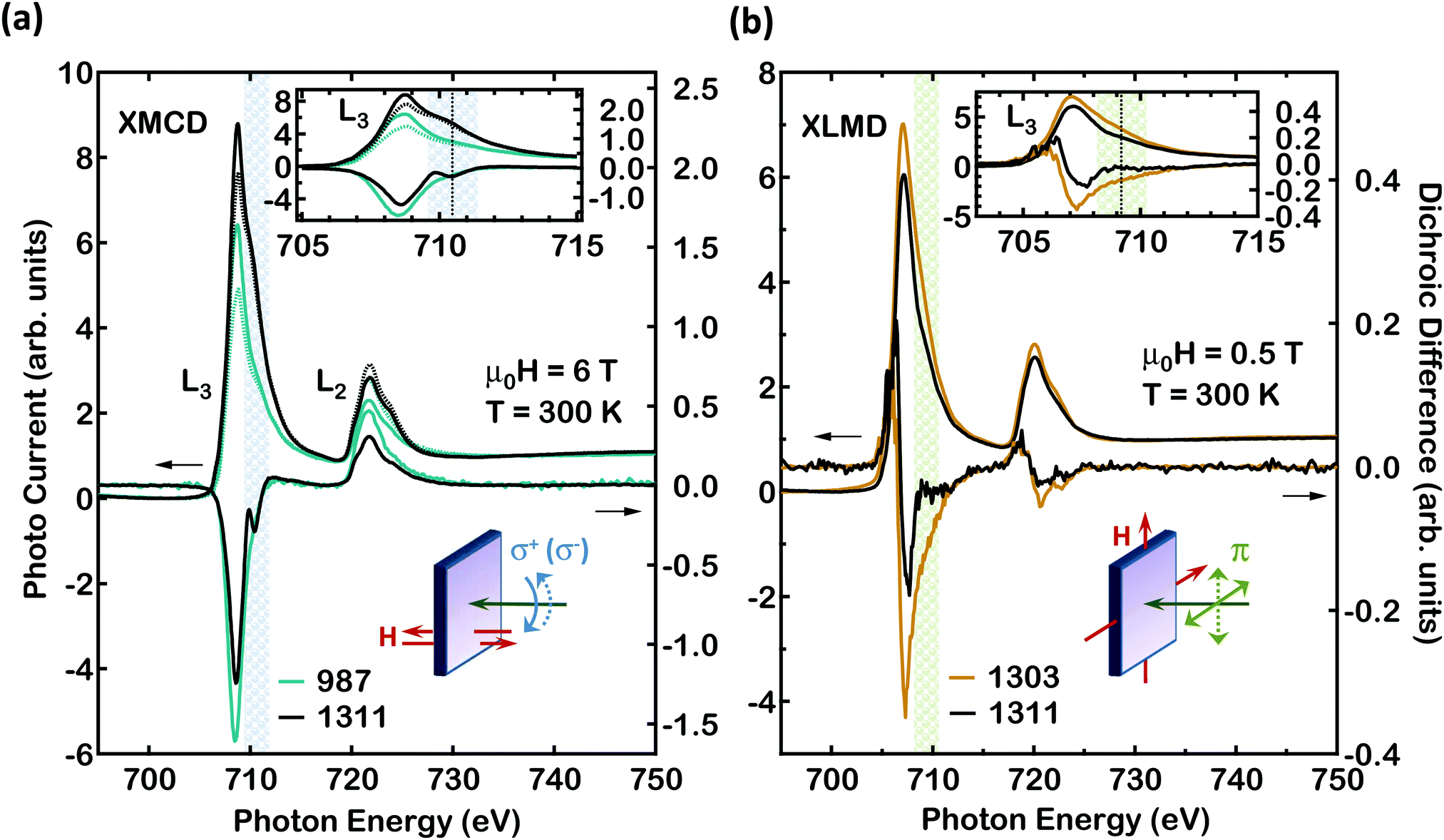 | ||
| Fig. 2 (a) Fe L-edge XAS and XMCD spectra versus photon energy for samples 987 and 1311 at 90° X-ray incidence. The XAS spectra (left scale) are obtained with circularly polarised X-rays. A magnetic field of μ0H = ±6 T serves to reverse the magnetisation direction for the evaluation of the XMCD magnitude at 300 K. In the inset the spectral region around the L3 white line is shown. For the L3 line hatched regions highlight satellite features. Here these show FM contrast. (b) XAS and XLMD spectra versus photon energy for samples 1303 and 1311 at 90° X-ray incidence. Only the XAS average spectrum is shown, as the XLMD intensity is very small. The X-ray polarisation direction is set either parallel or perpendicular to the magnetisation direction for the evaluation of the XLMD magnitude at 300 K. The XAS spectra (left scale) are obtained with linearly polarised X-rays. Here the satellite features are located at different energies. | ||
Using Fe L-edge X-ray absorption spectroscopy in the XLMD mode, it is possible to also record the Fe atom antiferromagnetic response.18,19 In an XLMD experiment the absorption coefficient is recorded with the magnetisation parallel and perpendicular to the X-ray beam polarization plane.18 The difference between the two spectra is non zero in the presence of a non zero magnetic moment on the Fe atoms even if the atomic spins between neighbouring atoms have an anti-parallel arrangement. For Fig. 2(b) linear X-rays are used, keeping the angle of X-ray incidence along the sample surface normal. For the linear X-rays the plane of X-ray polarization can be chosen to be either vertical or horizontal. The magnetic field can be applied either along the horizontal or vertical directions. For this communication we focus on the magnetic properties at room temperature, as these are most relevant for applications. Temperature dependent studies are more challenging, as the magnetic properties may be strongly influenced by structural effects, originating from the heteroepitaxial nature of the FenN nanocrystals studied here.20,21
For the theory data shown in Fig. 3 our first-principles calculations are based on DFT and are performed using a plane-wave basis and Troullier–Martins norm-conserving pseudo-potentials as implemented in the Quantum-ESPRESSO package (Appendix, ESI†16).22 We calculate the theoretical spectra of Fig. 4 using the FEFF 9 code, in its real space option.23 Here we use the calculated real space structures obtained by the structural optimisation by means of DFT, leading to the calculated magnetic moments as shown in Fig. 3(b and c).
 | ||
| Fig. 3 (a) The FM and non FM contribution to the magnetic moment per Fe atom are shown, using both the XMCD and the XLMD data. The number of 3d holes is obtained from the XAS spectra. The FM and non FM data points follow distinct trends as highlighted in the hatched areas. (b) The magnetic moment values as obtained by theory for the Fe atoms. Here the theoretical values are shown limited to those as would be observed in an XMCD and XLMD experiment. Similar trends are observed, as for the experimental data. (c) The Fe atom magnetic moment values as obtained by theory and the FenN atom structures are shown. | ||
 | ||
| Fig. 4 (a) The FEFF cluster is shown (using the VESTA software) for Fe3N along a high symmetry axis (z) and slightly off (7° and 3° rotating around x and y). The photoexcited atom is seen at the cluster center (yellow ball). (b) XAS spectra are shown in the vicinity of the Fe L3 white line, corresponding to two different families of samples. Fine structure is observed both in the low and the high energy side of the Fe L3 white line, better seen in the first derivative of the XAS signal. (c) Theoretical XAS spectra, calculated using the FEFF 9 code. The weighted average of FEFF9 FenN spectra is made, with weighting coefficients reflecting the magnetic moment determination. | ||
3 Results and discussion
3.1 Magnetism at the nanoscale
The (Ga,Fe)N films investigated here have been selected from a series of samples, whose composition, structural properties and magnetism were examined (Appendix, ESI†16).6,24 The magnetic properties were characterised for some of these samples (samples in (a) from Fig. 1).6 The size and composition of the FenN nanocrystals we study is steered by means of the sample growth parameters and procedure.4 The type of sample morphology we present is indicated in Fig. 1(a–c). MOVPE samples Nr 987 and 988 (Fig. 1(a)) contain smaller Fe-rich magnetic nanocrystals embedded in the GaN matrix6 in the near surface region, between 50 to 100 nm in average size.25 Planar arrays of Fe rich nanocrystals can be formed by MOVPE (δ-(Ga,Fe)N samples 1650 and 1651, Fig. 1(b)) deposited directly on the GaN surface.15 It is finally possible, to obtain FenN nanocrystals on the GaN surface (samples 1303 and 1311, Fig. 1(c)) by FenN film fragmentation.
For the XPEEM measurements we present results from two different types of samples as indicated in Fig. 1, panels (d) and (e). Using X-rays of photon energy leading to the maximum absorption at the Fe L3 edge, the bright regions of high intensity correspond to the Fe rich nanocrystals in the XAS micrographs. Given the lateral resolution of the instrument, we do not distinguish nanocrystals smaller than 40 nm.26–28 For sample 988 (Fig. 1(d)) lateral nanocrystal sizes identified by XPEEM are typically of the order of 50 nm. The sample FenN/GaN is laterally more inhomogenous presenting regions of smaller as well as larger FenN nanocrystals. Here we show a region containing on average larger nanocrystals (Fig. 1(e)). For the XMCD-PEEM data, the difference micrographs do characterize the magnetic state of the nanocrystals (Fig. 1(d and e) and Appendix, ESI†16). Not all nanocrystals which are resolved in the XMCD micrographs do exhibit FM contrast. For sample 988 only about 50% of the nanocrystals do contribute to the FM contrast. This is seen also for sample 1311 for the smaller nanocrystals.
We find for these samples that in a spatially integrating XMCD measurement, under magnetic saturation conditions (data of Fig. 2(a) and 3(a)) the magnetic moment obtained is only of the order of 50% of the expected saturation value. The magnetically live non FM nanocrystals are possibly in a paramagnetic or an AFM state.
3.2 Magnetic moment determination
Spatially integrated XAS and XMCD spectra taken with circular X-rays are shown in Fig. 2(a) (Appendix, ESI†16). Clear differences in the white line intensities are observed for samples 987 and 1311, indicative of a different number of Fe(3d) empty final states in each case. XAS and XLMD spectra are shown in Fig. 2(b). In Fig. 2(a) a high energy satellite to the main line is visible for both the L3 and L2 white lines. For Fe L-edge XAS data in the literature, a satellite feature has indeed been identified for Fe atoms with N atoms as first neighbours.29 The satellites to the main peak appear at different photon energies for samples 987 and 1311 (hatched regions for the L3 line in Fig. 2). Interestingly, only one of the satellites observed does exhibit XMCD contrast, which is clearly not merged with the XMCD of the main line, this is the one for sample 1311. This observation indicates that the merged satellite is characteristic of a different FM FenN phase for sample 987, versus the FM FenN phase found in sample 1311. It constitutes direct evidence for the presence of several local environments for the photo-excited Fe atoms indicative of several distinct FenN nanocrystal phases, which occur in a different manner for the two families of samples.
In order to estimate the values of the FM moments on a per atom basis, we apply the XMCD magneto optical sum rules.17 The result of this analysis is shown in Fig. 3, together with the data from a magnetic standard where Fe is in the metallic bcc state.19,25,30 It is observed that for all samples containing FM nanocrystals even in the case where a 6 T magnetic field is applied, only a FM spin magnetic moment of less than 1 μB per Fe atom is observed. For all FenN phases, based on theory, we expect values of the magnetic moment of more than 1.5 μB per Fe atom (Fig. 3(c)). This low FM spin moment value cannot be explained alone by the presence of substitutional paramagnetic Fe atoms, and is indicative of a large amount of nanocrystals in a non FM state.
Turning to the data of Fig. 2(b) we note that a substantial magnetic response is recorded for this sample also in the XLMD mode. Using Fe L-edge X-ray absorption spectroscopy in the XLMD mode, it is possible to also record the Fe atom AFM response.19 It has been previously shown that indeed the L-edge XLMD signal strength, for the 3d transition elements, follows a quadratic dependence versus the strength of the atom magnetic moments.18 Here we follow a similar approach as the one applied in this earlier work to obtain a measure of the value of the Fe atom magnetic moments. In the samples we investigate here we expect a mixture of various phases of embedded FenN nanocrystals, including Fe2N and Fe3N.6 We expect that the XLMD signal will contain contributions from both parallel and antiparallel Fe atom magnetic moments. Using the FM component determined independently in situ by means of XMCD, it is possible to extract the AFM contribution to the Fe magnetic moment using both the XLMD and XMCD results. Our findings indicate that some of the FenN nanocrystals are in an AFM state. An AFM arrangement can indeed be expected for Fe2N at low temperatures.31 The Néel temperature is probably much higher in our case as we investigate Fe2N in nanocrystal form stabilised by the GaN matrix. The data of Fig. 3(a) give the magnetic moments as determined by the experiment. The magnetic moment values from experiment together with the theoretical values shown in Fig. 3(b and c), allow to extract the contribution of the various phases of FenN nanocrystals in the samples which have been characterised. We have found both theoretically and experimentally that the individual Fe magnetic moment of FenN, first decreases with increasing nitrogen to iron ratio, followed interestingly by an increase.
3.3 First principles and ab initio theory
Our first-principles calculations are based on density functional theory (DFT) (Appendix, ESI†16).22 They have been done for ZnS- and NaCl-type FeN, and also ζ-Fe2N, ε-Fe3N, and γ′-Fe4N compounds. In all cases FM and AFM magnetic orderings have been considered. By comparison of their total energies, we have found that FM ordering is the ground state for ε-Fe3N and γ′-Fe4N, since the FM phase is lower in energy than the AFM by 271 and 148 meV per (Fe ion), respectively, whereas AFM is the preferred magnetic ordering for NaCl-type FeN (it is more favourable than the FM ordering by 250 meV (Fe ion)). The case of ζ-Fe2N is more complex, since according to our calculations the AFM ordering is preferred over the FM ordering by only 37 meV per (Fe ion), and this result is in accord with the experiment, since both magnetic phases seem to be present in our experiment and also previous experimental reports.32 Finally, the ZnS-type FeN is nonmagnetic in agreement with previous theoretical reports.33 For completeness, we have also included the bcc-Fe, that is FM in our studies with a magnetic moment of 2.31 μB, a value that is close to the experimental one. The results of our first-principles calculations are shown in Fig. 3(b and c) where we plotted the absolute magnetic moments of the Fe ions as a function of the charge transfer from Fe to N. In both considered magnetic states, FM and AFM, the amount of charge that is transferring from Fe to N is increasing with the increasing nitrogen to iron ratio. This result is in accord with the experimental picture plotted in Fig. 3(a), assuming that the increasing number of 3d holes is directly proportional to the total charge that is transferring from Fe ions to the nitrogen ions. This charge transfer has, however, a completely different influence on the behaviour of the Fe magnetic moments for the FM and AFM phases of FenN. While the FM magnetic moment decreases with the increasing N to Fe content ratio, the absolute value of the Fe magnetic moment in the AFM phase first decreases reaching its minimum for Fe2N and next increases reaching a high value of 2.17 μB for FeN. This result is independent on the type of AFM arrangement. We considered the AFM1 as well as the AFM2 phases consisting of single FM sheets alternating along the [110] and [111] directions, respectively. The AFM2 phase is energetically more favourable than the AFM1 by 44 meV per (Fe ion), but the absolute magnetic moment of AFM1 is 2.13 μB, a similar value than that obtained for AFM2. The non monotonic variation of the magnetic moment is clearly visible also in the experimental data points of Fig. 3(a). As the number of 3d holes increases, we observe a decrease of the FM moment, as indicated by the green hatched area. Beyond the value of 4.3 in 3d holes the AFM moment is observed to increase, as indicated by the hatched area in lilla.
Given the fact that various FenN nanocrystal phases are also characterised by specific white line satellite features in the XAS spectra, we can expect to be able to model the experimental XAS data, once theoretical XAS spectra can be calculated for the various FenN nanocrystal phases. We model our experimental XAS spectra using the FEFF code (Appendix, ESI†16).23 We have calculated the theoretical FEFF XAS spectra for the various FenN phases, n = 1–4. Using as a constraint the magnetic moment values of Fig. 3(a), and the theoretical magnetic values of Fig. 3(c), we have used a weighted average of the theoretical spectra as indicated in Fig. 4. In the case of FeN a 2% contraction to the lattice constant was applied, related with the presence of the GaN lattice. The weighting factors allow to match the experimental data with the theoretical ones, and to determine the content of the various nanocrystal phases present in the samples. We have not used any adjustable parameters between the theoretical FEFF spectra and the experimental ones. There is an energy shift between the absolute energy values obtained by means of the FEFF code and the experiment. Given that no adjustable parameters are used to reproduce the experimental spectra using the ab initio FEFF code the agreement between theory and experiment can be considered as excellent. In Fig. 4 the first derivatives of the spectra magnify the satellite fine structure. The theoretical modelling reproduces well also small derivative trends, a stricter criterion for the quality of the fit.
4 Conclusions
In summary direct evidence is presented for the existence of Fe4N and Fe3N nanocrystals in a ferromagnetic state in GaN at room temperature, in the near surface region for samples prepared by MOVPE. Furthermore, the present set of X-ray absorption data, based on ab initio theoretical modelling and together with first-principles calculations, allow to establish the existence of Fe2N and FeN nanocrystals in an antiferromagnetic state at room temperature in the near surface region of GaN. The magnetism of the FenN nanocrystals is stabilised by the GaN matrix. Sizeable antiferromagnetic moments are observed, due in particular to Fe2N and FeN nanocrystals in a magnetic state not stable in the bulk at room temperature. Doping GaN with Fe appears therefore as an excellent candidate system to use for spintronic applications, exploiting the antiferromagnetic component of the magnetic moment of low dimensional magnetic objects.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to acknowledge the Polish National Science Centre (NSC) under Grant No. 2011/03/D/ST3/02654 for the main support. We acknowledge the access to the computing facilities of the Interdisciplinary Centre of Modelling at the University of Warsaw. Additional financial support is acknowledged from the Carl Tryggers foundation for Science Research under No. CTS 16:32. MAN acknowledeges the support of the MICCINN/MINECO (Spain) through the programmes MAT2013-49893-EXP and MAT2014-59315-R. We acknowledge Andrea Navarro-Quezada, Bogdan Faina, Mauro Rovezzi and Alberta Bonanni for providing the samples and for interesting discussions. We acknowledge Tomasz Dietl for reviewing the manuscript and for interesting discussions. The research leading to these results has received funding through the MAX IV-lab and Elettra Synchrotrons from the European Community's Seventh Framework Programme (FP7/2007-2013) under grant agreement no. 226716 (ELISA) and 312284 (CALIPSO). I. A. Kowalik acknowledges the support from the Baltic Science Link project coordinated by the Swedish Research Council, VR. Some of the XAS and XMCD experiments were performed at the beamline 29 at ALBA Synchrotron with the collaboration of ALBA staff. We acknowledge the Nanospectroscopy beamline staff for carrying out the XMCD-PEEM measurements.Notes and references
- A. Zerr, R. Riedel, T. Sekine, J. Lowther, W. Y. Ching and I. Tanaka, Adv. Mater., 2006, 18, 2933 CrossRef CAS.
- P. Kroll, B. Eck and R. Dronskowski, Adv. Mater., 2000, 12, 307 CrossRef CAS.
- J. M. D. Coey and P. A. I. Smith, J. Magn. Magn. Mater., 1999, 200, 405 CrossRef CAS.
- A. Bonanni and T. Dietl, Chem. Soc. Rev., 2010, 39, 528 RSC.
- T. Dietl, K. Sato, T. Fukushima, A. Bonanni, M. Jamet, A. Barski, S. Kuroda, M. Tanaka, P. N. Hai and H. Katayama-Yoshida, Rev. Mod. Phys., 2015, 87, 1311 CrossRef CAS.
- A. Navarro-Quezada, W. Stefanowicz, T. Li, B. Faina, M. Rovezzi, R. T. Lechner, T. Devillers, G. Bauer, M. Sawicki, T. Dietl and A. Bonanni, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 81, 205206 CrossRef.
- J. Sadowski, J. Z. Domagala, R. Mathieu, A. Kovacs and P. Dluzewski, J. Phys., 2013, 25, 196005 Search PubMed.
- P. N. Hai, S. Yada and M. Tanaka, J. Appl. Phys., 2011, 109, 073919 CrossRef.
- M. Jamet, A. Barski, T. Devillers, V. Poydenot, R. Dujardin, P. Bayle-Guillemaud, J. Rothman, E. Bellet-Amalric, A. Marty, J. Cibert, R. Mattana and S. Tatarenko, Nat. Mater., 2006, 5, 653 CrossRef CAS PubMed.
- A. Bonanni, A. Navarro-Quezada, T. Li, M. Wegscheider, Z. Matěj, V. Holý, R. T. Lechner, G. Bauer, M. Rovezzi, F. D'Acapito, M. Kiecana, M. Sawicki and T. Dietl, Phys. Rev. Lett., 2008, 101, 135502 CrossRef CAS PubMed.
- N. Gonzalez-Szwacki, J. A. Majewski and T. Dietl, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 83, 184417 CrossRef.
- T. Hinomura and S. Nasu, Nuovo Cimento, 1996, 18, 253 CrossRef.
- P. Wadley, B. Howells, J. Zelezny, C. Andrews, V. Hills, R. P. Campion, V. Novak, K. Olejnik, F. Maccherozzi, S. S. Dhesi, S. Y. Martin, T. Wagner, J. Wunderlich, F. Freimuth, Y. Mokrousov, J. Kunes, J. S. Chauhan, M. J. Grzybowski, A. W. Rushforth, K. W. Edmonds, B. L. Gallagher and T. Jungwirth, Science, 2016, 351, 587 CrossRef CAS PubMed.
- T. Jungwirth, X. Marti, P. Wadley and J. Wunderlich, Nat. Nanotechnol., 2016, 11, 231 CrossRef CAS PubMed.
- A. Navarro-Quezada, T. Devillers, T. Li and A. Bonanni, Appl. Phys. Lett., 2012, 101, 081911 CrossRef.
- I. A. Kowalik, N. Gonzalez Szwacki, M. Á. Niño, F. J. Luque and D. Arvanitis, ESI†.
- P. Carra, B. T. Thole, M. Altarelli and X. Wang, Phys. Rev. Lett., 1993, 70, 694 CrossRef CAS PubMed.
- M. M. Schwickert, G. Y. Guo, M. A. Tomaz, W. L. O'Brien and G. R. Harp, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 58, R4289 CrossRef CAS.
- I. A. Kowalik, Acta Phys. Pol., A, 2015, 127, 831 CrossRef CAS.
- N. Ji, V. Lauter, X. Zhang, H. Ambaye and J.-P. Wang, Appl. Phys. Lett., 2013, 102, 072411 CrossRef.
- Y. Jiang, V. Dabade, M. P. Brady, O. Rios, R. D. James and J.-P. Wang, J. Appl. Phys., 2014, 115, 17A758 CrossRef.
- P. Giannozzi, S. Baroni, N. Bonini, M. Calandra, R. Car, C. Cavazzoni, D. Ceresoli, G. L. Chiarotti, M. Cococcioni, I. Dabo, A. Dal Corso, S. de Gironcoli, S. Fabris, G. Fratesi, R. Gebauer, U. Gerstmann, C. Gougoussis, A. Kokalj, M. Lazzeri, L. Martin-Samos, N. Marzari, F. Mauri, R. Mazzarello, S. Paolini, A. Pasquarello, L. Paulatto, C. Sbraccia, S. Scandolo, G. Sclauzero, A. P. Seitsonen, A. Smogunov, P. Umari and R. M. Wentzcovitch, J. Phys.: Condens. Matter, 2009, 21, 395502 CrossRef PubMed.
- J. J. Rehr and R. C. Albers, Rev. Mod. Phys., 2000, 72, 621 CrossRef CAS.
- A. Bonanni, M. Kiecana, C. Simbrunner, T. Li, M. Sawicki, M. Wegscheider, M. Quast, H. Przybylinska, A. Navarro-Quezada, R. Jakiela, A. Wolos, W. Jantsch and T. Dietl, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 75, 125210 CrossRef.
- I. A. Kowalik, A. Persson, M. Á. Niño, A. Navarro-Quezada, B. Faina, A. Bonanni, T. Dietl and D. Arvanitis, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 85, 184411 CrossRef.
- A. Locatelli and E. Bauer, J. Phys.: Condens. Matter, 2008, 20, 093002 CrossRef.
- T. O. Mentes, G. Zamborlini, A. Sala and A. Locatelli, Beilstein J. Nanotechnol., 2014, 5, 1873 CrossRef CAS PubMed.
- S. D. Col, S. Jamet, N. Rougemaille, A. Locatelli, T. O. Mentes, B. S. Burgos, R. Afid, M. Darques, L. Cagnon, J. C. Toussaint and O. Fruchart, Phys. Rev. B: Condens. Matter Mater. Phys., 2014, 89, 180405(R) CrossRef.
- Y. Takagi, K. Isami, I. Yamamoto, T. Nakagawa and T. Yokoyama, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 81, 035422 CrossRef.
- I. A. Kowalik, G. Öhrwall, B. N. Jensen, R. Sankari, E. Wallén, U. Johansson, O. Karis and D. Arvanitis, J. Phys.: Conf. Ser., 2010, 211, 012030 CrossRef.
- M. Kano, T. Nakagawa, T. A. Yamamoto and M. Katsura, J. Alloys Compd., 2001, 327, 43 CrossRef CAS.
- H. Naganuma, Y. Endo, R. Nakatani, Y. Kawamura and M. Yamamoto, Sci. Technol. Adv. Mater., 2004, 5, 83 CrossRef CAS.
- Z. Zhao, K. Bao, D. Duan, F. Tian, B. Liu and T. Cui, RSC Adv., 2015, 5, 31270 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9tc02334h |
| This journal is © The Royal Society of Chemistry 2019 |