
A spiro-silafluorene–phenazasiline donor-based efficient blue thermally activated delayed fluorescence emitter and its host-dependent device characteristics†
Seung-Je
Woo‡
a,
Youheon
Kim‡
b,
Yun-Hi
Kim
 *b,
Soon-Ki
Kwon
*c and
Jang-Joo
Kim
*a
*b,
Soon-Ki
Kwon
*c and
Jang-Joo
Kim
*a
aDepartment of Materials Science and Engineering, Seoul National University, Seoul 151-742, South Korea. E-mail: jjkim@snu.ac.kr
bDepartment of Chemistry and RIGET, Gyeongsang National University, Jinju 660-701, South Korea
cDepartment of Materials Engineering and Convergence Technology and ERI, Gyeongsang National University, Jinju 660-701, South Korea
First published on 6th March 2019
Abstract
We report a blue thermally activated delayed fluorescence compound based on a spiro-silicon-connected silafluorene–phenazasiline donor. By incorporating triphenyltriazine as an acceptor, the compound exhibited well-separated highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) levels and charge transfer-type emission, confirmed by density functional theory (DFT) calculations and its solvatochromic characteristics. The compound exhibited blue emission with a photoluminescence (PL) peak at 469 nm, a high photoluminescence quantum yield (PLQY) of 68%, and a single-triplet energy gap of 0.20 eV in a DPEPO host matrix. Due to its co-planar silafluorene and triphenyltriazine moieties, the compound exhibited a high horizontal emission dipole ratio of 79%. Various electroluminescent devices of single host, mixed host, and interface exciplex structures were fabricated using the compound as the emitter. The external quantum efficiency (EQE), maximum brightness, and Commission Internationale de L’Eclairage (CIE) y-values ranged from 0.63 to 20.6%, 210 to 6930 nit, and 0.120 to 0.206, respectively, depending on the host materials and device structures. Among the fabricated EL devices, the DPEPO host device exhibited the highest EQE of 20.6% with CIE color coordinates of (0.150, 0.184).
1. Introduction
Blue thermally activated delayed fluorescence (TADF) emitters that could harvest triplet excitons via reverse intersystem crossing (RISC) have attracted a great deal of attention in the field of organic light-emitting diodes (OLEDs) due to their potential for replacing the current, relatively inefficient blue fluorescence emitters in display applications.1–4 TADF emitters typically incorporate donor–acceptor structures to minimize the singlet–triplet gap by separating the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO).5 The emission color of TADF emitters can be modulated by selecting donors and acceptors with appropriate HOMO and LUMO levels, respectively. Therefore, blue TADF emitters generally employ a relatively weak donor or acceptor moiety. Adachi et al. reported the first pure blue TADF emitter based on a diphenylsulfone acceptor and a dimethylcarbazole donor exhibiting a high external quantum efficiency of 9.9%.6 Since then, various acceptor moieties, such as triazine,7,8 pyrimidine,9,10 oxadiazole,11,12 cyanobenzene,2,13,14 benzophenone,15,16 phosphine oxide,17 mesitylboron,18,19 indolocarbazole,20,21 and their derivatives,22–25 have been used to develop efficient blue TADF emitters. In contrast, only a few donor moieties, including carbazole,2,7,13,14 acridine,8,9 phenoxazine15,17,26 and their alkyl-substituted derivatives,6,10,13,27–30 have been used to develop blue TADF emitters. Therefore, significant improvements in blue TADF emission systems require the further development and characterization of new donor moieties.Silicon-based compounds are widely used as both the host and light-emitting material in blue OLEDs. High bandgap and triplet energy hosts based on tetraarylsilanes,31–33 and blue light emitting materials including oligomers and polymers based on silafluorene or spiro-silafluorene scaffolds34–39 are examples. In addition, the introduction of silicon atoms into azaheterocycles has resulted in blue OLEDs with many desirable characteristics. For example, phenazasiline, which is a silicon-bridged diphenylamine, has been successfully applied as the host and emitter material in blue OLEDs due to its high triplet level, high bandgap, deep HOMO, and rigid molecular structure.40–42 However, only a few TADF emitters containing silicon atoms have been reported to date.16,43 Moreover, to the best of our knowledge, a spiro-silicon scaffold has not been used for a TADF emitter. This is partly due to synthetic difficulties, including the silicon-heterocyclization of a donor (or an acceptor) followed by the cyclization of silafluorene.41,42
We report herein the first TADF emitter based on a spiro-silicon scaffold (Fig. 1). We connected silafluorene and phenazasiline moieties through a spiro-silicon as a donor moiety. The resulting donor, spiro[phenazasiline-10(5H),9′-[9H-9]silafluorene], was combined with a triphenyltriazine acceptor into an efficient blue TADF molecule. The newly designed TADF emitter, SAzTrz, exhibited a blue emission with a peak wavelength of 469 nm, a high photoluminescence quantum yield (PLQY) of 68%, and a high horizontal emission dipole ratio of 79% in a DPEPO host. OLEDs based on SAzTrz as the emitter and DPEPO as the host showed a high external quantum efficiency of 20.6% with blue Commission Internationale de L’Eclairage (CIE) color coordinates of (0.150, 0.184). Furthermore, we investigated the photophysical and electroluminescent (EL) properties of SAzTrz in various host materials to provide guidance regarding further applications of spiro-silafluorene–phenazasiline as the donor moiety in blue TADF emitters.
 | ||
| Fig. 1 Molecular structure, ground state geometry, highest occupied molecular orbital–lowest unoccupied molecular orbital (HOMO–LUMO) distribution, and excited state energies of SAzTrz. | ||
2. Results and discussion
2.1 Synthesis and characterization
The synthetic route of SAzTrz is illustrated in Scheme 1. Intermediate (1), 5′-(4-methoxybenzyl)-5′H-spiro[dibenzo[b,d]silole-5,10′-dibenzo[b,e][1,4]azasiline], was synthesized by in situ nucleophilic substitution of 2-bromo-N-(2-bromophenyl)-N-(4-methoxybenzyl)aniline using 2,2′-dibromo-1,1′-biphenyl. Intermediate (2), 5′H-spiro[dibenzo[b,d]silole-5,10′-dibenzo[b,e][1,4]azasiline], was obtained through a de-protection reaction using DDQ. The final product (3), 5′-(4-(4,6-diphenyl-1,3,5-triazin-2-yl)-2-methylphenyl)-5′H-spiro[dibenzo[b,d]silole-5,10′-dibenzo[b,e][1,4]azasiline] (SAzTrz), was synthesized by Buchwald–Hartwig amination. The obtained intermediates and SAzTrz were characterized by proton nuclear magnetic resonance (1H-NMR), carbon-13 NMR (13C-NMR), and electron ionization (EI) mass spectrometry. Detailed synthesis and characterization procedures are described in the ESI.† The thermal stability of SAzTrz was analyzed by thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC) (Fig. S1, ESI†). SAzTrz was thermally stable up to 360 °C with a glass transition temperature of 245 °C. The HOMO level of SAzTrz was determined as −5.64 eV from the cyclovoltametric measurement and the LUMO level was determined as −2.69 eV by adding the optical bandgap (2.95 eV) (Fig. S2, ESI†). | ||
| Scheme 1 Synthesis of SAzTrz. | ||
2.2 Photophysical properties
Density functional theory (DFT) and time-dependent DFT (TD-DFT) calculations were used to predict the geometries, HOMO–LUMO distribution, natural transition orbitals (NTOs), and transition energies of SAzTrz. Calculations of the ground state geometry and excited state geometries were performed with Gaussian09, using the B3LYP functional for the ground state and the CAM-B3LYP functional for excited states. Single point calculations for transition energies and NTOs were performed with Jaguar software (Schrödinger, USA), using the LC-wPBE functional with modified parameter ω = 0.1. The 6-31G(d,p) basis set was used for both geometry optimization and single point calculations. The dihedral angles of silafluorene/phenazasiline and phenazasiline/triphenyltriazine were ∼90° in both the ground and excited states. Due to an orthogonal donor and acceptor conformation, the HOMO and LUMO were localized at the donor and acceptor, respectively. NTOs of the lowest singlet vertical emission were almost identical to the HOMO–LUMO distribution, indicating absorption and emission from a charge transfer state (Fig. S3, ESI†). The calculated S1 vertical emission energy of 2.925 eV matched well with the onset energy of experimental SAzTrz spectra, but the T1 energy of 2.912 eV was an overestimate compared to the experimental value of 2.79 eV. This was due to overestimation of the locally excited triplet state energy by a long-range-corrected functional, leading to a calculation result indicating that the charge transfer state is the lowest triplet state. The experimentally measured T1 energy of SAzTrz and the related delayed fluorescence will be discussed below.The photophysical properties of SAzTrz in various media are shown in Fig. 2, 3 and Fig. S4 (ESI†) and are summarized in Table 1. The solution PL of SAzTrz was measured in cyclohexane (ε = 2.02), toluene (ε = 2.38), chlorobenzene (ε = 5.62), tetrahydrofuran (ε = 7.58), and acetonitrile (ε = 37.5) (Fig. S4, ESI†). SAzTrz exhibited strong solvatochromism with a red-shift of its PL peak from 410 nm in cyclohexane to 585 nm in acetonitrile, indicating charge transfer-type emission as predicted from TD-DFT calculations. Fig. 2a depicts the absorbance, fluorescence, and phosphorescence spectra of 10−5 M SAzTrz in toluene. The intense and structured absorption in the range of 310–350 nm could be assigned to the absorption of spiro-silafluorene–phenazasiline, while the relatively weak and broad absorption from 330 nm to 430 nm could be assigned to the intramolecular charge transfer excitation. The phosphorescence of SAzTrz in frozen toluene (cooled with liquid N2, 77 K) was measured by integrating its delayed luminescence between 100 and 500 ms after 325 nm ultraviolet (UV) excitation. The phosphorescence spectrum of SAzTrz was identical to that of spiro-silafluorene–phenazasiline and the lowest triplet state of SAzTrz could be assigned to the locally excited triplet state of the donor (3LED). The 3CT energy could not be assigned directly from the phosphorescence spectrum since it decays nonradiatively through internal conversion to the lowest triplet (3LED). However, the estimated 1CT–3CT energy gap of SAzTrz obtained from TD-DFT calculations is very small (0.013 eV), due to well-separated S1 → S0 and T1 → S0 transition orbitals (Fig. S3, ESI†). Thus, we may infer that the 3CT state of SAzTrz lies very close to the 1CT state. Recently, Monkman et al. showed that the RISC of TADF emitters occurs from second-order transitions, including the coupling of 3CT–3LE and 3LE–1CT, and that the rate of RISC is maximized when the 1CT–3LE gap is minimized.44 Hence, the RISC efficiency (ΦRISC) of SAzTrz will be strongly affected by the 1CT energy, which is determined by the polarity of the host matrix.
 | ||
| Fig. 2 Photophysical properties of SAzTrz. (a) Absorbance and PL spectra of a toluene solution. Fluorescence: PL at 300 K. Phosphorescence: PL of a frozen toluene solution at 77 K, 100 ms delay. (b) PL spectra and (c) transient PL decay of 10 wt% SAzTrz doped in various host materials. | ||
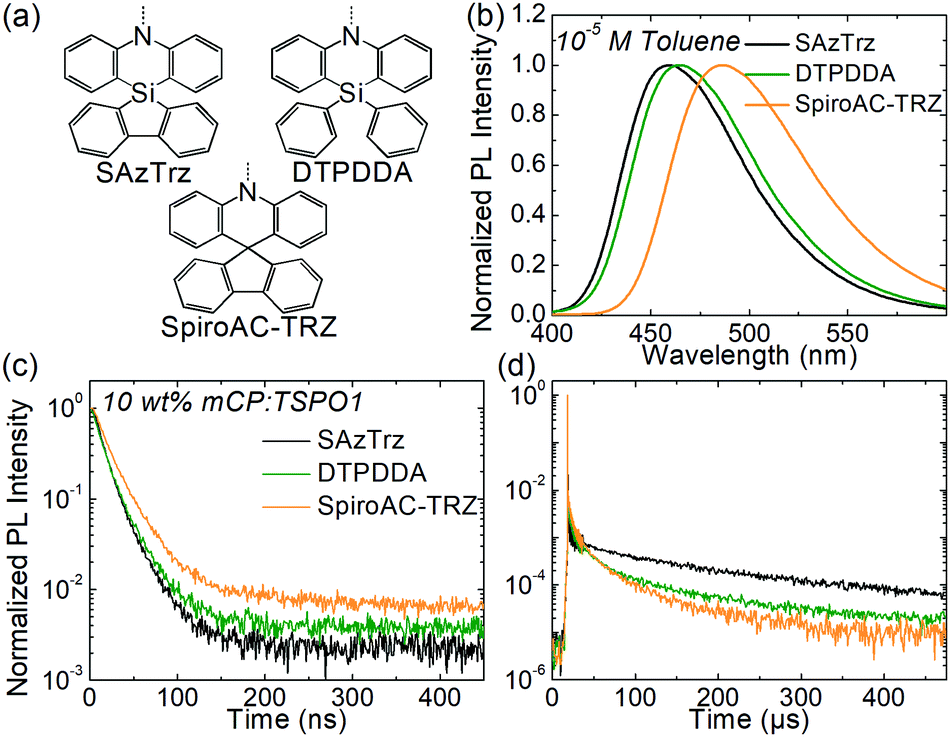 | ||
| Fig. 3 Comparison of the photophysical properties of SAzTrz and blue TADF emitters based on similar donor moieties. (a) Donor structure of SAzTrz, DTPDDA, and SpiroAC-TRZ. (b) PL spectra in toluene. (c and d) Transient PL decay in mCP:TSPO1. | ||
| Solution/film | λ PL (nm) | ΔESTb (eV) | PLQYc (%) | Φ p (%) | Φ d (%) | τ p (ns) | τ d (μs) | Θ (%) |
|---|---|---|---|---|---|---|---|---|
| a Photoluminescence (PL) peak. b S1–T1 gap. c PL quantum yield. d Prompt portion of total emission. e Delayed portion of total emission. f Prompt decay lifetime. g Delayed decay lifetime. h Horizontal emitting dipole ratio. | ||||||||
| Toluene | 458 | 0.20 | — | — | — | — | — | — |
| mCP | 457 | 0.35 | 40 | 23 | 77 | 15.1 | 103 | — |
| mCP-CN | 462 | 0.31 | 43 | 24 | 76 | 15.1 | 93 | — |
| mCBP-CN | 459 | 0.33 | 35 | 35 | 65 | 15.2 | 72 | — |
| mCP:TSPO1 | 465 | 0.25 | 65 | 11 | 89 | 15.6 | 173 | 70 |
| DPEPO | 469 | 0.20 | 68 | 10 | 90 | 16.8 | 165 | 79 |
To evaluate the performance and emission properties of SAzTrz as the emitter for electroluminescence devices, the photophysical properties of doped films were investigated prior to OLED fabrication. For host materials, mCP (T1 = 3.02 eV), mCP-CN (T1 = 3.0 eV), mCBP-CN (T1 = 2.81 eV),45 TSPO1 (T1 = 3.36 eV),46 and DPEPO (T1 = 3.00 eV)47 were chosen to confine triplet excitons within the TADF emitter. The emission peak of SAzTrz red-shifted from 457 nm (mCP) to 469 nm (DPEPO) with increasing polarity of the host material (Fig. 2b). We also determined the singlet–triplet energy gap (ΔEST) of SAzTrz in various host materials from the difference in emission onset energy between fluorescence at 300 K in a doped film and phosphorescence at 77 K in a toluene solution. ΔEST ranged from 0.35 eV in mCP to 0.20 eV in DPEPO. ΔEST is the barrier for reverse intersystem crossing (RISC) from a triplet exciton to a singlet exciton, and the effect of ΔEST on delayed fluorescence can be observed from the transient PL decay (Fig. 2(c)). The delayed fluorescence of SAzTrz in mCP:TSPO1 and DPEPO persisted for over 10 ms and the delayed portion of the total emission (Φd) was as high as 90%. However, in mCP, mCP-CN, and mCBP-CN, the delayed fluorescence of SAzTrz ended at around 4 ms with a delayed portion of Φd = 65–77%, indicating decreased RISC efficiency compared to that of SAzTrz in the mCP:TSPO1 and DPEPO hosts. The longer lifetimes of SAzTrz delayed fluorescence in mCP:TSPO1 (173 μs) and DPEPO (165 μs) compared to that of SAzTrz in other host materials (72–103 μs) can be attributed to an enhanced ISC/RISC cycle by small ΔEST values. The delayed PL lifetime of SAzTrz was especially short in the mCBP-CN host. Here we may infer that the triplet exciton is not perfectly confined to SAzTrz, due to the similar triplet energies of mCBP-CN (2.81 eV) and SAzTrz (2.79 eV).
The emitting dipole orientations of SAzTrz in the mCP:TSPO1 and DPEPO hosts were determined by angle-dependent PL measurements in doped films, and by optical simulations (Fig. S5, ESI†). The proportion of horizontally oriented emitting dipoles (Θ) of SAzTrz was 70% for SAzTrz in mCP:TSPO1 and 79% in DPEPO (note that Θ = 66.7% indicates an isotropic orientation). Therefore, SAzTrz exhibited a preferred horizontal orientation in both host materials. This is due to a parallel alignment between the emitting dipole moment and the long-axis of the SAzTrz molecule, and a coplanar arrangement between the silafluorene and triphenyltriazine moieties.
The photophysical properties of SAzTrz and blue TADF emitters based on similar donor moieties were compared and are shown in Fig. 3, Fig. S6, S7 and Table S1 (ESI†). These three blue TADF emitters, SAzTrz, DTPDDA,43 and SpiroAC-TRZ,8 all incorporate triphenyltriazine as an acceptor but incorporate spiro-silafluorene–phenazasiline, diphenyl-phenazasiline, and spiro-fluorene–acridine as donors, respectively. Among these TADF emitters, SAzTrz shows the deepest PL emission for both the toluene solution (Fig. 3(b)) and doped film in mCP:TSPO1 (Fig. S6, ESI†). The bond length of Si–C is 1.86 Å, which is longer than the C–C bond (1.53 Å), and it results in a larger C–N–C bond angle (127.1°) of diphenylamine in SAzTrz compared to that of 121.5° for SpiroAC-TRZ (Fig. S7, ESI†). Hence, the conjugation within the donor moiety of SAzTrz is weaker than that of SpiroAC-TRZ, leading to a larger HOMO–LUMO gap and higher S1 energy of SAzTrz. Although it is not as significant as the difference of the PL spectra of SAzTrz and SpiroAC-TRZ, SAzTrz also shows a blue-shifted PL spectrum compared to DTPDDA. Formation of spiro-silicon (linking two phenyls into silafluorene) slightly increases the C–N–C bond angle of the diphenylamine and results in a blueshifted PL spectrum. Formation of spiro-silafluorene also effects the molecular orientation of the emitter doped in host materials. As the silafluorene and triphenyltriazine of SAzTrz are co-planar, it exhibits a higher Θ of 70% than DTPDDA (66%).
Modification of the donor structure gives rise to a difference in the PL decay curves. The prompt decay rate is the fastest for SAzTrz (Fig. 3(c)) and we may infer from the PLQY and prompt portion of the total emission (Φp) (Table S1, ESI†) that the nonradiative decay rate of the singlet state is the largest for SAzTrz. We expected that linking two freely rotating phenyls (DTPDDA) into silafluorene (SAzTrz) would suppress nonradiative relaxation but this was not the result. In the delayed emission region, the decay rate of DTPDDA is faster than the decay rate of SAzTrz until 200 μs and then becomes similar. The larger change in the delayed decay rate of DTPDDA implies that it has a broader distribution of the molecular configuration than SAzTrz. The difference in delayed decay tendency of SAzTrz and DTPDDA might also originate from different RISC mechanisms. As shown in Fig. S6 (ESI†), DTPDDA exhibits broad CT-like phosphorescence whereas the phosphorescence of SAzTrz is from the donor. As the lowest triplet state of DTPDDA is 3CT and the locally excited triplet of the donor and acceptor is much higher than 1CT and 3CT, 3CT would be upconverted to 1CT by direct RISC. On the other hand, the lowest triplet of SAzTrz is 3LE, and 3CT lies between 1CT and 3LE. Therefore, the main RISC mechanism of SAzTrz would be population of 3CT from 3LE by vibronic coupling followed by second-order coupling of 1CT and 3CT induced by spin–orbit coupling of 1CT–3LE and vibronic coupling of 3LE–3CT.44
The device performances of TADF OLEDs based on SAzTrz, DTPDDA, and SpiroAC-TRZ are summarized in Table S2 (ESI†). The RISC efficiencies of the emitters estimated by optical simulations based on the PLQY and Θ are compared.
2.3 Electroluminescent properties
Several EL devices were fabricated to investigate the EL properties of SAzTrz and their dependence on the host material and device structure (Fig. 4, 5, and Table 2). The structures of the fabricated OLEDs were as follows: (a) ITO (70 nm)/6 wt% ReO3:mCP (45 nm)/mCP (10 nm)/10 wt% SAzTrz-mCP![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :TSPO1 (1:1) (30 nm)/TSPO1 (10 nm)/6 wt% Rb2CO3:TSPO1 (45 nm)/LiF (0.7 nm)/Al (100 nm), (b) ITO (70 nm)/MoO3 (1 nm)/H2 (45 nm)/mCBP (5 nm)/10,20 wt% SAzTrz:mCBP-CN (15 nm)/CN-T2T (50 nm)/LiF (0.7 nm)/Al (100 nm), and (c) ITO (70 nm)/MoO3 (1 nm)/H2 (45 nm)/mCP (5 nm)/10 wt% SAzTrz:mCP, mCP-CN, or DPEPO (15 nm)/DPEPO (5 nm)/TPBi (45 nm)/LiF (0.7 nm)/Al (100 nm).
:TSPO1 (1:1) (30 nm)/TSPO1 (10 nm)/6 wt% Rb2CO3:TSPO1 (45 nm)/LiF (0.7 nm)/Al (100 nm), (b) ITO (70 nm)/MoO3 (1 nm)/H2 (45 nm)/mCBP (5 nm)/10,20 wt% SAzTrz:mCBP-CN (15 nm)/CN-T2T (50 nm)/LiF (0.7 nm)/Al (100 nm), and (c) ITO (70 nm)/MoO3 (1 nm)/H2 (45 nm)/mCP (5 nm)/10 wt% SAzTrz:mCP, mCP-CN, or DPEPO (15 nm)/DPEPO (5 nm)/TPBi (45 nm)/LiF (0.7 nm)/Al (100 nm).
 | ||
| Fig. 4 Device structures of organic light-emitting diodes (OLEDs) based on SAzTrz. (a) mCP:TSPO1 mixed host structure. (b) mCBP-CN:CN-T2T interface exciplex structure. (c) Single host device structures based on mCP, mCP-CN, and DPEPO hosts. | ||
 | ||
| Fig. 5 Electroluminescent characteristics of OLEDs based on SAzTrz. (a) Current density–voltage–luminance curves. (b) Current density–power efficiency/external quantum efficiency (EQE) curves. (c) EL spectrum and CIE color coordinates (inset) at 200 cd m−2. | ||
| Device | EQEmaxa (%) | EQE100b (%) | PEmaxc (lm W−1) | λ EL (nm) | CIEe | Turn onf (V) | L max (cd m−2) |
|---|---|---|---|---|---|---|---|
| a Maximum external quantum efficiency (EQE). b EQE at 100 cd m−2. c Maximum power efficiency (PE). d Electroluminescence (EL) peak. e Commission Internationale de L’Eclairage (CIE) 1931 coordinates at 200 cd m−2. f Turn on voltage. g Maximum luminance. | |||||||
| mCP:TSPO1 | 8.2 | 3.9 | 12.4 | 468 | (0.157, 0.206) | 3.4 | 2240 |
| mCBP-CN:CN-T2T (10 wt%) | 1.6 | 1.6 | 1.9 | 452 | (0.150, 0.120) | 2.8 | 4500 |
| mCBP-CN:CN-T2T (20 wt%) | 2.3 | 2.3 | 3.1 | 456 | (0.150, 0.134) | 2.8 | 6930 |
| mCP | 0.6 | 0.3 | 0.5 | 456 | (0.160, 0.139) | 4.0 | 210 |
| mCP-CN | 4.6 | 2.3 | 4.6 | 456 | (0.151, 0.133) | 3.8 | 400 |
| DPEPO | 20.6 | 7.6 | 20.7 | 468 | (0.150, 0.184) | 4.1 | 440 |
Device (a) employs a mixed host of mCP:TSPO1, a thick (30 nm) emitting layer (EML), a doped charge transport layer to attain charge balance in the EML and a wide recombination zone. Device (b) utilizes an interface exciplex (Fig. S8, ESI†) composed of mCBP-CN:CN-T2T to form excitons on the emitter by energy transfer from the interface exciplex rather than direct charge trapping on the emitter. Device (c) is a single-host structured device incorporating mCP, mCP-CN, and DPEPO as host materials with hole/electron blocking layers.
As expected from the photophysical properties of SAzTrz in various host materials, the DPEPO device exhibited the highest external quantum efficiency (EQE) (20.6%), followed by an EQE of 8.2% with the mCP:TSPO1 device. Theoretically achievable EQEs were calculated as a function of the thickness of the transport layers by optical simulations based on the PLQY and Θ of the emitter, assuming that the RISC efficiency (ΦRISC) is 100% (Fig. S9, ESI†). The EQE of the DPEPO device was close to the calculated maximum achievable EQE of 20.8%, implying that the RISC efficiency was nearly 100%. Conversely, the EQE of the mCP:TSPO1 device was much smaller than the calculated EQE of 20.2%, implying a relatively lower amount of harvested triplets. Other devices, including those based on the interface exciplex structure and single-host structures with mCP and mCP-CN hosts, showed poor efficiency with EQEs under 5%, indicating that the majority of the emission from these devices originated from singlet excitons instead of upconverted triplets.
The maximum luminance of the fabricated devices did not trend with efficiency performances with regard to host materials. Devices employing an interface exciplex based on mCBP-CN:CN-T2T exhibited the highest luminance up to 6930 nit, while the luminance values of mixed- and single-host devices were 2240 nit and 210–440 nit, respectively. The large differences in maximum luminance observed among the devices can be attributed to the triplet lifetime of the emitter. For the case of the interface exciplex device, most of the excitons are formed at the interface between mCBP-CN and CN-T2T, and only singlet exciplexes are transferred to the emitter via Förster energy transfer. In addition, the triplet lifetime of SAzTrz (T1 = 2.79 eV) was shortest in mCBP-CN (T1 = 2.81 eV) due to their similar triplet energies. Hence, the population of “dark” triplet states in SAzTrz was smallest in the interfacial exciplex device, resulting in the highest luminance. With a single-host device, the majority of excitons are formed by charge trapping at the emitter. Triplets are confined to the SAzTrz molecule in the mCP and mCP-CN hosts, which leads to a large population of dark triplet states and low brightness. Although the RISC process of SAzTrz is efficient in a DPEPO host, the triplet lifetime is the longest due to a prolonged ISC/RISC cycle and the efficiency decreases rapidly at high current densities, resulting in a low maximum luminance. The exciton dynamics of SAzTrz in mCP:TSPO1 are similar to those of SAzTrz in DPEPO, but the wider exciton recombination zone and thicker EML of the former result in higher luminance.
3. Conclusion
In summary, we designed and synthesized the first TADF emitter containing a spiro-silicon scaffold in its molecular structure. Spiro-silicon-connected silafluorene–phenazasiline and triphenyltriazine were used as the donor and acceptor moieties, respectively, resulting in a D–π–A structured TADF molecule, SAzTrz. The lowest triplet energy of silafluorene–phenazasiline was 2.79 eV, which was high enough to yield a small singlet–triplet energy gap (ΔEST = 0.20 eV) with efficient triplet upconversion (ΦRISC = 1). The photophysical properties of SAzTrz were investigated in various host materials and OLEDs were fabricated with mixed-host, single-host, and interface exciplex structures. Highly efficient blue OLEDs were realized with DPEPO as a host material, attaining an EQE value of 20.6% with CIE coordinates of (0.150, 184).4. Experimental section
4.1 Fabrication of organic thin films and OLEDs
Organic thin films for photoluminescence (PL) measurements were fabricated by thermal deposition onto pre-cleaned fused silica substrates at a base pressure of <5 × 10−7 Torr. OLEDs were fabricated by thermal deposition onto 70 nm-thick patterned ITO glass substrates at a base pressure of <5 × 10−7 Torr. Before deposition of the organic layers, the ITO substrates were pre-cleaned with acetone and isopropyl alcohol and then exposed to UV-ozone for 10 minutes. Samples for photoluminescence and electroluminescence measurements were encapsulated in a nitrogen-filled glove box.4.2 Measurements of absorption, photoluminescence, and electroluminescence
The absorbance spectrum of 10−5 M SAzTrz dissolved in toluene was measured with a Varian Cary 5000 ultraviolet-visible near-infrared (UV-vis-NIR) spectrometer. The PLQY was measured using a monochromator-attached photomultiplier tube (PMT) with a PL sample in an integrating sphere (LabSphere, USA) using a continuous wave 325 nm He/Cd laser (Kimmon Koha, Japan) for excitation. Photoluminescence was measured with a CCD spectrometer (Maya 2000; Ocean Optics, USA) using a He/Cd laser (325 nm) for excitation. Phosphorescence of the toluene solution was measured by freezing the solution in liquid nitrogen at 77 K. For angle-dependent PL measurements, p-polarized light emitted from PL samples was measured by attaching the film substrate to a half-cylinder lens with index-matching oil and changing the angle between the sample and the detector from −90° to 90° using a motorized rotational stage. Transient PL was measured with a streak camera (Hamamatsu Photonics, Japan) using a nitrogen laser (337 nm; Usho Optical Systems, Japan) as the excitation source. The J–V–L curves of the OLEDs were measured using a programmable source meter (Keithley 2400; Keithley Instruments, USA) and a spectrophotometer (Spectrascan PR650; Photo Research, USA).4.3 Thermogravimetric analysis and cyclic voltammetry
Thermal analyses were performed with a TGA-2100 thermogravimetric analyzer in a nitrogen atmosphere at a heating rate of 10 °C min−1. Cyclic voltammetry (CV) was performed using a CH instruments electrochemical analyzer. A three-electrode system composed of a glassy carbon working electrode, a platinum wire counter electrode, and a Ag/AgCl reference electrode was used. A 0.1 M solution of tetrabutylammonium hexafluorophosphate (TBAP) in chloroform was used as the electrolyte. The ferrocenium/ferrocene (4.43 eV) redox couple was used as an internal standard. Cyclic voltammograms were obtained at a scan rate of 100 mV s−1. The HOMO level was obtained from the CV oxidation onset and the LUMO level was obtained by adding the optical bandgap energy to the HOMO level.4.4 Synthesis
A detailed synthesis procedure and characterization of SAzTrz are described in the ESI.†Author contributions
Y. K., Y.-H. K., and S.-K. K. designed and synthesized the emitter compound. S.-J. W. designed and carried out the photoluminescence and electroluminescence experiments. S.-J. W. and J.-J. Kim wrote the manuscript. S.-J. W. and Y. K. equally contributed to this work.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Industrial Strategic Technology Development Program [10048317] funded by Korea Evaluation Institute of Industrial Technology (KEIT) and Ministy of Trade, Industry and Energy (MOTIE).References
- A. Endo, K. Sato, K. Yoshimura, T. Kai, A. Kawada, H. Miyazaki and C. Adachi, Appl. Phys. Lett., 2011, 98, 083302 CrossRef.
- H. Uoyama, K. Goushi, K. Shizu, H. Nomura and C. Adachi, Nature, 2012, 492, 234–238 CrossRef CAS PubMed.
- Z. Yang, Z. Mao, Z. Xie, Y. Zhang, S. Liu, J. Zhao, J. Xu, Z. Chi and M. P. Aldred, Chem. Soc. Rev., 2017, 46, 915–1016 RSC.
- M. Y. Wong and E. Zysman-Colman, Adv. Mater., 2017, 29, 1605444 CrossRef PubMed.
- T. Nakagawa, S. Y. Ku, K. T. Wong and C. Adachi, Chem. Commun., 2012, 48, 9580–9582 RSC.
- Q. Zhang, J. Li, K. Shizu, S. Huang, S. Hirata, H. Miyazaki and C. Adachi, J. Am. Chem. Soc., 2012, 134, 14706–14709 CrossRef CAS PubMed.
- M. Kim, S. K. Jeon, S. H. Hwang and J. Y. Lee, Adv. Mater., 2015, 27, 2515–2520 CrossRef CAS PubMed.
- T. A. Lin, T. Chatterjee, W. L. Tsai, W. K. Lee, M. J. Wu, M. Jiao, K. C. Pan, C. L. Yi, C. L. Chung, K. T. Wong and C. C. Wu, Adv. Mater., 2016, 28, 6976–6983 CrossRef CAS PubMed.
- K.-C. Pan, S.-W. Li, Y.-Y. Ho, Y.-J. Shiu, W.-L. Tsai, M. Jiao, W.-K. Lee, C.-C. Wu, C.-L. Chung, T. Chatterjee, Y.-S. Li, K.-T. Wong, H.-C. Hu, C.-C. Chen and M.-T. Lee, Adv. Funct. Mater., 2016, 26, 7560–7571 CrossRef CAS.
- I. S. Park, H. Komiyama and T. Yasuda, Chem. Sci., 2017, 8, 953–960 RSC.
- Q. Zhang, B. Li, S. Huang, H. Nomura, H. Tanaka and C. Adachi, Nat. Photonics, 2014, 8, 326–332 CrossRef CAS.
- M. Y. Wong, S. Krotkus, G. Copley, W. Li, C. Murawski, D. Hall, G. J. Hedley, M. Jaricot, D. B. Cordes, A. M. Z. Slawin, Y. Olivier, D. Beljonne, L. Muccioli, M. Moral, J. C. Sancho-Garcia, M. C. Gather, I. D. W. Samuel and E. Zysman-Colman, ACS Appl. Mater. Interfaces, 2018, 10, 33360–33372 CrossRef CAS PubMed.
- C.-Y. Chan, L.-S. Cui, J. U. Kim, H. Nakanotani and C. Adachi, Adv. Funct. Mater., 2018, 28, 1706023 CrossRef.
- H. Noda, H. Nakanotani and C. Adachi, Sci. Adv., 2018, 4, eaao6910 CrossRef PubMed.
- S. Y. Lee, T. Yasuda, Y. S. Yang, Q. Zhang and C. Adachi, Angew. Chem., Int. Ed., 2014, 53, 6402–6406 CrossRef CAS PubMed.
- J. W. Sun, J. Y. Baek, K.-H. Kim, J.-S. Huh, S.-K. Kwon, Y.-H. Kim and J.-J. Kim, J. Mater. Chem. C, 2017, 5, 1027–1032 RSC.
- C. Duan, J. Li, C. Han, D. Ding, H. Yang, Y. Wei and H. Xu, Chem. Mater., 2016, 28, 5667–5679 CrossRef CAS.
- M. Numata, T. Yasuda and C. Adachi, Chem. Commun., 2015, 51, 9443–9446 RSC.
- Y. H. Lee, S. Park, J. Oh, J. W. Shin, J. Jung, S. Yoo and M. H. Lee, ACS Appl. Mater. Interfaces, 2017, 9, 24035–24042 CrossRef CAS PubMed.
- J. A. Seo, Y. Im, S. H. Han, C. W. Lee and J. Y. Lee, ACS Appl. Mater. Interfaces, 2017, 9, 37864–37872 CrossRef CAS PubMed.
- Y. Im, S. H. Han and J. Y. Lee, J. Mater. Chem. C, 2018, 6, 5012–5017 RSC.
- Y. J. Kang and J. Y. Lee, Dyes Pigm., 2017, 138, 176–181 CrossRef CAS.
- Y. Wada, S. Kubo and H. Kaji, Adv. Mater., 2018, 30, 1705641 CrossRef PubMed.
- P. Rajamalli, N. Senthilkumar, P. Y. Huang, C. C. Ren-Wu, H. W. Lin and C. H. Cheng, J. Am. Chem. Soc., 2017, 139, 10948–10951 CrossRef CAS PubMed.
- I. Lee and J. Y. Lee, Org. Electron., 2016, 29, 160–164 CrossRef CAS.
- W. L. Tsai, M. H. Huang, W. K. Lee, Y. J. Hsu, K. C. Pan, Y. H. Huang, H. C. Ting, M. Sarma, Y. Y. Ho, H. C. Hu, C. C. Chen, M. T. Lee, K. T. Wong and C. C. Wu, Chem. Commun., 2015, 51, 13662–13665 RSC.
- D. R. Lee, M. Kim, S. K. Jeon, S. H. Hwang, C. W. Lee and J. Y. Lee, Adv. Mater., 2015, 27, 5861–5867 CrossRef CAS PubMed.
- Y. H. Lee, S. Park, J. Oh, S.-J. Woo, A. Kumar, J.-J. Kim, J. Jung, S. Yoo and M. H. Lee, Adv. Opt. Mater., 2018, 6, 1800385 CrossRef.
- D. Zhang, M. Cai, Y. Zhang, D. Zhang and L. Duan, Mater. Horiz., 2016, 3, 145–151 RSC.
- L. S. Cui, H. Nomura, Y. Geng, J. U. Kim, H. Nakanotani and C. Adachi, Angew. Chem., 2017, 56, 1571–1575 CrossRef CAS PubMed.
- X. Ren, J. Li, R. J. Holmes, P. I. Djurovich, S. R. Forrest and M. E. Thompson, Chem. Mater., 2004, 16, 4743–4747 CrossRef CAS.
- W.-S. Han, H.-J. Son, K.-R. Wee, K.-T. Min, S. Kwon, I.-H. Suh, S.-H. Choi, D. H. Jung and S. O. Kang, J. Phys. Chem. C, 2009, 113, 19686–19693 CrossRef CAS.
- T.-H. Han, M.-R. Choi, C.-W. Jeon, Y.-H. Kim, S.-K. Kwon and T.-W. Lee, Sci. Adv., 2016, 2, e1601428 CrossRef PubMed.
- E. Wang, C. Li, Y. Mo, Y. Zhang, G. Ma, W. Shi, J. Peng, W. Yang and Y. Cao, J. Mater. Chem. C, 2006, 16, 4133–4140 RSC.
- M. Shimizu, K. Mochida, M. Katoh and T. Hiyama, J. Phys. Chem. C, 2010, 114, 10004–10014 CrossRef CAS.
- S. F. Chen, Y. Tian, J. Peng, H. Zhang, X. J. Feng, H. Zhang, X. Xu, L. Li and J. Gao, J. Mater. Chem. C, 2015, 3, 6822–6830 RSC.
- X. Xu, K. Bai, J. Ding and L. Wang, Org. Electron., 2018, 59, 77–83 CrossRef CAS.
- X. Xu, X. Li, S. Wang, J. Ding and L. Wang, J. Mater. Chem. C, 2018, 6, 9599–9606 RSC.
- Y. Cai, A. Qin and B. Z. Tang, J. Mater. Chem. C, 2017, 5, 7375–7389 RSC.
- X.-Y. Liu, X. Tang, Y. Zhao, D. Zhao, J. Fan and L.-S. Liao, J. Mater. Chem. C, 2018, 6, 1023–1030 RSC.
- H. Li, Y. Wang, K. Yuan, Y. Tao, R. Chen, C. Zheng, X. Zhou, J. Li and W. Huang, Chem. Commun., 2014, 50, 15760–15763 RSC.
- S. Xu, H. Li, R. Chen, Z. Chen, L. Xu, Y. Tang and W. Huang, Adv. Opt. Mater., 2018, 6, 1701105 CrossRef.
- J. W. Sun, J. Y. Baek, K.-H. Kim, C.-K. Moon, J.-H. Lee, S.-K. Kwon, Y.-H. Kim and J.-J. Kim, Chem. Mater., 2015, 27, 6675–6681 CrossRef CAS.
- M. K. Etherington, J. Gibson, H. F. Higginbotham, T. J. Penfold and A. P. Monkman, Nat. Commun., 2016, 7, 13680 CrossRef CAS PubMed.
- S. G. Ihn, N. Lee, S. O. Jeon, M. Sim, H. Kang, Y. Jung, D. H. Huh, Y. M. Son, S. Y. Lee, M. Numata, H. Miyazaki, R. Gomez-Bombarelli, J. Aguilera-Iparraguirre, T. Hirzel, A. Aspuru-Guzik, S. Kim and S. Lee, Adv. Sci., 2017, 4, 1600502 CrossRef PubMed.
- S. O. Jeon, S. E. Jang, H. S. Son and J. Y. Lee, Adv. Mater., 2011, 23, 1436–1441 CrossRef CAS PubMed.
- C. Han, Y. Zhao, H. Xu, J. Chen, Z. Deng, D. Ma, Q. Li and P. Yan, Chem. – Eur. J., 2011, 17, 5800–5803 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthesis and characterization of the materials, experimental methods, and supporting tables and figures. See DOI: 10.1039/c9tc00193j |
| ‡ Seung-Je Woo and Youheon Kim equally contributed to this work. |
| This journal is © The Royal Society of Chemistry 2019 |