
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The effect of manganese oxidation state on antiferromagnetic order in SrMn1−xSbxO3 (0 < x < 0.5) perovskite solid solutions†
Gennady V.
Bazuev
a,
Alexander P.
Tyutyunnik
 a,
Alexander V.
Korolev
b,
Emmanuelle
Suard
c,
Cheuk-Wai
Tai
d and
Nadezda V.
Tarakina
*ef
a,
Alexander V.
Korolev
b,
Emmanuelle
Suard
c,
Cheuk-Wai
Tai
d and
Nadezda V.
Tarakina
*ef
aInstitute of Solid State Chemistry, Ural Branch of the Russian Academy of Sciences, 91 Pervomayskaya, Ekaterinburg, Russia
bM.N. Mikheev Institute of Metal Physics of the Ural Branch of the Russian Academy of Sciences, Ekaterinburg, Russia
cInstitut Laue-Langevin, BP 156, 6, rue Jules Horowitz, Grenoble Cedex 9, France
dDepartment of Materials and Environmental Chemistry, Arrhenius Laboratory, Stockholm University, S-106 91 Stockholm, Sweden
eMax Planck Institute of Colloids and Interfaces, Am Mühlenberg 1, 14476 Potsdam, Germany. E-mail: nadja.tarakina@mpikg.mpg.de
fSchool of Engineering and Materials Science, Queen Mary University of London, Mile End, London E1 4NS, UK
First published on 17th January 2019
Abstract
The mixed-valence manganese (Mn3+/Mn4+) solid solution, SrMn1−xSbxO3, was prepared for the first time. Two ranges of solid solutions were found: (1) SrMn1−xSbxO3 (0.025 ≤ x ≤ 0.09) with monoclinically distorted 6H-SrMnO3 polytype (sp. gr. C/2c) and (2) SrMn1−xSbxO3 (0.17 ≤ x ≤ 0.50) with a tetragonal unit cell (sp. gr. I4/mcm). Crystal structure refinement using X-ray and neutron powder diffraction data showed that the structure of the monoclinic solid solution consists of corner-sharing octahedra around sites occupied by manganese and antimony ions and face-sharing octahedra around sites occupied by manganese ions only, while the tetragonal solid solution has a random distribution of B-site cations. The presence of long-range antiferromagnetic order with a Néel temperature of about 148 K for SrMn0.80Sb0.20O3 and about 280 K for SrMn0.925Sb0.075O3 was found from the results of DC and AC susceptibility and neutron diffraction experiments at 5 K and 80 K.
Introduction
Manganites with perovskite-like structures attract considerable attention due to their unique electronic, magnetoelectrical and magnetic properties. Spontaneous electric polarization and magnetic ordering in the Sr1−xBaxMnO3 solid solution at x ≥ 0.45,1 ferromagnetic coupling and metallic conductivity in La1−xCaxMn4−xMn4+xO3,2 strong magnetoelectric effects near room temperature in single-crystal BaMnO2.99 and its derivative BaMn0.97M0.03O3 (M = Li or K),3 soft ferromagnetic behaviour in La0.75K0.25AMnTiO6 (A = Sr or Ba) at low-temperature,4 collinear-magnetism-driven ferroelectricity in the Ising chains in Ca3CoMnO6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 5,6 are just a few examples. Such variety of properties results from the interplay between charge, exchange and phonon interactions,7 which can be adjusted by substituting either A-site cations or Mn with other cations (usually cations of different sizes/charges). The latter influences the ratio of Mn cations in different oxidation states in the final compound and can also lead to the formation of different structural modifications within the perovskite type structure.
5,6 are just a few examples. Such variety of properties results from the interplay between charge, exchange and phonon interactions,7 which can be adjusted by substituting either A-site cations or Mn with other cations (usually cations of different sizes/charges). The latter influences the ratio of Mn cations in different oxidation states in the final compound and can also lead to the formation of different structural modifications within the perovskite type structure.
In this work we studied structural and magnetic properties of SrMn4+O3–Sr2Mn3+SbO6 solid solutions. In order to explain why we chose to study this system, let's first look at the structure and properties of the two parent compounds SrMnO3 and Sr2MnSbO6. They differ considerably in chemical composition, oxidation state of manganese, physical properties and their crystal structure.
Strontium manganite SrMnO3 can be synthesised with three different modifications. SrMnO3 produced at high temperatures and oxidized at 350 °C in air crystallises in an ideal cubic structure (sp. gr. Pm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m, a = 3.8041(3) Å, Z = 18), with Sr2+ cations occupying the 12-fold coordination site between corner-sharing Mn4+O6 octahedra. At moderate temperatures, SrMnO3 forms a hexagonal 4H structure (sp. gr. P63/mmc, a = 5.454(1) Å, c = 9.092(2) Å, Z = 4),9 and at high pressures and high temperatures (6 GPa and 1773 K) a 6H structure (sp. gr. P63/mmc, a = 5.42892(1) Å, c = 13.4025(3) Å, V = 342.0929(9) Å3, Z = 6) is formed.8,10 The 4H- and 6H-variants of SrMnO3 form face-sharing octahedral dimers and pairs of corner-sharing MnO6 octahedra in the periodic sequence of hchc and cchcch, respectively, where c and h denote cubic and hexagonal types of layer stacking, respectively.
m, a = 3.8041(3) Å, Z = 18), with Sr2+ cations occupying the 12-fold coordination site between corner-sharing Mn4+O6 octahedra. At moderate temperatures, SrMnO3 forms a hexagonal 4H structure (sp. gr. P63/mmc, a = 5.454(1) Å, c = 9.092(2) Å, Z = 4),9 and at high pressures and high temperatures (6 GPa and 1773 K) a 6H structure (sp. gr. P63/mmc, a = 5.42892(1) Å, c = 13.4025(3) Å, V = 342.0929(9) Å3, Z = 6) is formed.8,10 The 4H- and 6H-variants of SrMnO3 form face-sharing octahedral dimers and pairs of corner-sharing MnO6 octahedra in the periodic sequence of hchc and cchcch, respectively, where c and h denote cubic and hexagonal types of layer stacking, respectively.
The hexagonal 6H structure of SrMnO3 can be stabilized at normal pressures by partial replacement of Sr or Mn by other elements. During the synthesis of the 15-layered rhombohedral (15R) perovskite, SrMn0.9Fe0.1MnO3−x,12,13 a 6H-layered perovskite of the same composition was found to be an intermediate phase. The latter phase was obtained in pure form at 1350 °C. In the Sr1−xLaxMnO3 solid solutions, a 6H-layered perovskite was also found to form at x = 0.1.14
The crystal structure and magnetic properties of the double perovskite Sr2MnSbO6 have been extensively studied.15–19 Depending on the synthesis method, different crystal structure modifications of Sr2MnSbO6 can be formed. Samples prepared at low temperatures have a random distribution of Mn3+ and Sb5+ cations at the octahedral positions, resulting in the space group I4/mcm.15 In ref. 17 and 18, the structure of Sr2MnSbO6 was described in the space group I4/m with an ordered distribution of cations at the octahedral positions. The presence of the Jahn–Teller cation Mn3+ distorts the coordination environment and contributes considerably to octahedral tilting. Accordingly,19 this structure of Sr2MnSbO6 is stable in the temperature range of 2–750 K. Above 750 K, a cubic modification with space group Fmm is formed.
The magnetic properties of SrMn4+O3 and Sr2Mn3+SbO6 differ considerably because the latter has half of the octahedral positions occupied by diamagnetic Sb5+ ions. Cubic SrMnO3 is antiferromagnetic with the Néel temperature TN = 240 K.8 4H-SrMnO3 also possesses antiferromagnetic properties with TN = 280 K,20 and short-range antiferromagnetic interactions are retained in the paramagnetic region. The magnetic properties of 6H-SrMnO3 are expected to be defined by Mn–Mn inter- and intradimer interactions, and the temperature dependence of the magnetic susceptibility by the dimeric behaviour. However, in ref. 8 it was established that the temperature dependence of the magnetic susceptibility χ(T) for 6H-SrMnO3 is typical of a three-dimensional antiferromagnetic compound with a maximum at TN = 235 K. The magnetic susceptibility of 6H-SrMnO3 above TN follows the Curie–Weiss law with a Curie–Weiss constant θ of −743(3) K and an effective magnetic moment μeff of 4.099(6) μB (the theoretical value μtheor for the Mn4+ cation is 3.873 μB). The ratio θ/TN = 3.2 is indicative of the presence of frustration. The magnetization vs. magnetic field curves are linear up to 10 kOe, and there are upturn deviations from the linear behaviour for higher magnetic fields.
The magnetic susceptibility of Sr2MnSbO6 exhibits a divergence between field cooling (FC) and zero field cooling (ZFC) curves at 25 K18 and nonequilibrium dynamics and memory at low temperatures.19 No long-range magnetic ordering was found at 2 K. It is believed that Sr2MnSbO6 reaches an unconventional spin-glass state at low temperatures. The magnetization vs. magnetic field dependence at 5 K displays a Brillouin-like curvature with a hysteresis loop. The Curie–Weiss law χ = C/(T − θ) is fulfilled only in the temperature range of 300–400 K with a positive Weiss constant. The experimental value of μeff (5.68 μB) is significantly higher than the calculated value μcal = 4.90 μB for Mn3+. These results show that the magnetic behaviour of Sr2MnSbO6 is determined by weak cluster-type ferromagnetic interactions.18 The reasons for the lack of magnetic ordering in Sr2MnSbO6 are not yet completely clear. However, magnetic ordering of B-site cations in Sr2MnSbO6-based compounds is possible to realise through heterovalent substitution of Sr2+ by La3+.21 The then obtained SrLaMnSbO6 has a monoclinically distorted double-perovskite structure with ordered Mn2+ and Sb5+ cations and shows long-range antiferromagnetic (AFM) ordering below TN = 10 K and significant ferromagnetic correlation below 90 K. At the same time, it has been shown that the magnetic properties of the rhombohedral perovskite La0.67Ba0.33Mn1−xSbxO3 (x = 0.01, 0.03 and 0.07) can be modified by varying the Sb doping.22 A small increase of the Sb content changes the Mn3+/Mn4+ ratio and thus influences the double exchange interactions and leads to a decrease in the Curie temperature (TC) from 326 to 296 K.22
The above data motivated us to examine systems based on SrMn4+O3 and Sr2Mn3+SbO6 manganites to identify the structural and magnetic properties of compounds and/or solid solutions with mixed manganese oxidation states Mn3+/Mn4+. The wide variety of properties of manganites is determined not only by the differences in chemical composition, but also by the differences in their structure, in particular the disordered or ordered arrangement of B-site cations (Mn and Sb) at octahedral positions. The aim of this study was to find out how the structure of substituted manganites SrMn1−xSbxO3 with a mixed oxidation state of manganese, Mn4+/Mn3+, affects their magnetic characteristics.
Experimental
SrCO3 (99.95%), Mn2O3 (99.95%) and Sb2O3 (99.95%) were used as the initial compounds for the synthesis of SrMnO3–Sr2MnSbO6 solid solutions. Mixtures of initial reagents were pressed into pellets and calcined in air at 950 °C for 30 h and at 1150 °C, 1250 °C and 1350 °C for 12 h with intermediate cooling and regrinding in an agate mortar.The purity of the synthesized product was checked using X-ray powder diffraction (XRPD). XRPD patterns were collected at room temperature on a XRD Shimadzu 7000S diffractometer using Cu Kα radiation. The possible impurity phases were checked by comparing their XRPD patterns with those in the PDF2 database (ICDD, USA, Release 2016). For the crystal structure refinements, XRPD patterns were collected on a STADI-P (Stoe) diffractometer in transmission geometry with a linear mini-PSD detector, using Cu Kα1 radiation in the 2θ range 5° to 120° with a step of 0.02°. Polycrystalline silicon (a = 5.43075(5) Å) was used as an external standard.
The microstructure and chemical composition of obtained samples were analysed using a JEOL JSM-5900 LV microscope equipped with a JEOL energy-dispersive X-ray detector (EDX).
Neutron powder diffraction patterns (NPD) were collected at the ILL facility at Grenoble (France) using the D2B high-resolution two-axis diffractometer (λ = 1.594 Å) equipped with a cryofurnace, which enables to perform measurements at temperatures in the range of 2–500 K. For all measurements, the samples were placed in a vanadium cylinder with a diameter of 8 mm.
The crystal structure refinement was carried out with the GSAS23,24 program suite using the NPD data for low and high temperatures and a combination of XRPD and NPD data for room temperature. The peak profiles were fitted with a pseudo-Voigt function, I(2θ) = x × L(2θ) + (1 − x) × G(2θ) (where L and G are the Lorentzian and Gaussian part, respectively). The angular dependence of the peak width was described by the relation (FWHM)2 = Utg2θ + Vtgθ + W, where FWHM is the full line width at half maximum. The background level was described by a combination of thirty-sixth-order Chebyshev polynomials. The absorption correction function for a flat plate sample in transmission geometry has been applied. Since neutron scattering lengths for Mn and Sb are −3.73 and 5.57 fm, respectively, the resulting scattering length of the mixed sites with Mn/Sb ratios equal to 0.667/0.333 would be −3.73 × 2/3 + 5.57 × 1/3 = −0.63 fm, as close to zero as the value of −0.3824 fm tabulated for vanadium which is considered as transparent for low energy neutrons and used as a material for the capillary for neutron diffraction data collection. In this regard, we have fixed the Mn/Sb ratios to 0.667/0.333 and set the values of the thermal motions to 0.025 for this site for all full-profile refinements of the SrMn0.665Sb0.335O3 structure based on neutron data only, namely for the temperatures of 5 K and 500 K. In the analysis of the low-temperature NPD data, a two-phase refinement for every temperature was performed. In each case, the crystal structure was refined taking as starting parameters those obtained at room temperature. The magnetic structure was refined as an independent phase with the P1 space group for which only magnetic atoms were defined. The scale, atomic coordinates and displacement parameters were constrained for both the nuclear and magnetic structures. The bond valence sums (BVS) were calculated using the VaList program by Wills25 according to equations given by Brown and Altermatt26 and the bond-valence parameters, r0 and b, tabulated by Brese and O’Keeffe.27
A scanning transmission electron microscopy (STEM) study was performed using a FEI (S)TEM Titan 80-300 microscope, operated at 300 kV. For the STEM study an ethanol suspension of the sample was prepared and kept in an ultrasonic bath for 5 minutes; then a drop of this suspension was put onto a holey carbon film supported on a Cu grid. High-temperature transmission electron microscopy investigations were performed using a double-tilt heating holder (Gatan model 652) on a JEOL 2100 transmission electron microscope. The selected-area electron diffraction (SAED) patterns were recorded on a Gatan Orius 200D camera.
Magnetic measurements were performed on a SQUID magnetometer MPMS-XL-5 produced by QUANTUM DESIGN.30 The temperature was varied from 5 K to 400 K. The controlled magnetic field strength H was set to 0.5 kOe. Measurements were carried out when a specimen was cooled in zero and controlled (ZFC and FC) magnetic field. A specimen was first cooled in the zero field to 5 K, after which the magnetization (M) on heating up to 400 K (ZFC mode) was measured, followed by measurements when cooling from 400 K to 5 K (FC mode). From these measurements we found the static magnetic susceptibility χ = M/H, whereas the AC susceptibility measurement technique was used to determine the real χ′ and imaginary χ′′ components of the dynamic susceptibility for an amplitude value of the variable magnetic field of 4 Oe and a frequency of 80 Hz.
Results and discussion
In order to obtain comprehensive information about the SrMnO3–Sr2MnSbO6 system, we have synthesized 11 samples of the general formula SrMn1−xSbxO3 with x = 0.025, 0.05, 0.075, 0.10, 0.125, 0.165, 0.20, 0.25, 0.335 and 0.415. This integrated formula was used for a more convenient comparison of the compositions of the solid solutions formed on the basis of SrMnO3 and Sr2MnSbO6.A preliminary analysis of the XRPD patterns of the obtained solid solutions SrMn1−xSbxO3 (x = 0.025–0.415) collected at room temperature indicates a morphotropic transition with a variation in the antimony content. Fig. 1 and Table S1 (ESI†) show three regions: (1) x = 0.025–0.09, where the monoclinically distorted 6H-perovskite structure8,29 is stable, (2) x = 0.17–∼0.45, where the solid solution adopts a tetragonal double perovskite structure,15–19 and the (1 + 2) two-phase region, where the limiting compositions of each modification coexist, with a change in Sb-content only the mass fractions of each modification change.
 | ||
| Fig. 1 Dependence of formula unit volume V/Z on antimony content for SrMn1−xSbxO3 (x = 0.025–0.415). Three regions can be distinguished: (1) x = 0.025–0.09 where the monoclinic modification (C2/c) is stable and (2) x = 0.17–0.415, tetragonal modification (I4/mcm), and (1 + 2) where mixtures with various ratios of these two modifications exist. | ||
The unit cell parameters and volumes for both monoclinic and tetragonal modifications obey Vegard's law. For a detailed study of the crystal and magnetic structures, low and high temperature NPD data have been collected for four single phases and well-crystallized samples of SrMn1−xSbxO3 with x = 0.075, 0.20, 0.335 and 0.415.
Crystal structures of SrMn1−xSbxO3 solid solutions
| T = 1.5 K NPD | T = 80 K NPD | T = 298 K XRPD + NPD | T = 500 K NPD | ||||
|---|---|---|---|---|---|---|---|
| a R-Factors obtained without taking into account the magnetic ordering. b Thermal parameters of Mn/Sb(1) and Mn(2) atoms were constrained as single variable. | |||||||
| Space group, #, Z | C2/c, 15, 12 | C2/c, 15, 12 | C2/c, 15, 12 | C2/c, 15, 12 | |||
| Cell constants | |||||||
| a, Å | 5.4602(2) | 5.4612(2) | 5.46826(7) | 5.4829(3) | |||
| b, Å | 9.4339(2) | 9.4353(2) | 9.4629(1) | 9.4902(5) | |||
| c, Å | 13.4951(4) | 13.4971(4) | 13.5140(2) | 13.5423(6) | |||
| β, ° | 90.773(2) | 90.764(2) | 90.6545(9) | 90.555(3) | |||
| V, Å3 | 695.08(4) | 695.42(3) | 699.24(2) | 704.64(6) | |||
| Sr(1)-4e (0, y, 1/4) | Y | −0.0025(8) | −0.0009(9) | −0.0015(4) | 0.0027(17) | ||
| U i/Ue × 100 | 1.46(9) | 1.4(1) | 1.11(7) | 2.34(13) | |||
| Sr(2)-8f (x, y, z) | X | 0.0071(7) | 0.0063(7) | 0.0056(3) | 0.0049(10) | ||
| Y | 0.3324(7) | 0.3333(7) | 0.3334(4) | 0.3341(12) | |||
| Z | 0.0900(3) | 0.0902(3) | 0.09109(9) | 0.0903(3) | |||
| U i/Ue × 100 | 1.71(8) | 1.67(7) | 1.26(5) | 2.65(10) | |||
| Mn/Sb(1)-4a (0, 0, 0) | U i/Ue × 100 | 1.20(6)b | 1.16(6)b | 0.66(9) | 1.71(9)b | ||
| Fraction | 0.775/0.225 | 0.775/0.225 | 0.775/0.225 | 0.775/0.225 | |||
| Mn(2)-8f (x, y, z) | X | 0.9905(9) | 0.9905(9) | −0.0060(4) | −0.0040(10) | ||
| Y | 0.3334(11) | 0.3345(11) | 0.3307(6) | 0.3290(18) | |||
| Z | 0.8422(3) | 0.8429(3) | 0.8427(1) | 0.8426(4) | |||
| U i/Ue × 100 | 1.20(7)b | 1.16(6)b | 1.55(7) | 1.71(9)b | |||
| O(1)-4e (0, y, 1/4) | Y | 0.5140(8) | 0.5125(9) | 0.5121(8) | 0.5145(19) | ||
| U i/Ue × 100 | 1.7(2) | 1.7(2) | 0.6(2) | 2.2(4) | |||
| O(2)-8f (x, y, z) | X | 0.2691(11) | 0.2688(11) | 0.2680(12) | 0.2687(20) | ||
| Y | 0.2428(6) | 0.2420(6) | 0.2387(6) | 0.2413(9) | |||
| Z | 0.2436(4) | 0.2436(4) | 0.2437(4) | 0.2468(5) | |||
| U i/Ue × 100 | 1.7(1) | 1.7(1) | 1.7(1) | 2.2(2) | |||
| O(3)-8f (x, y, z) | X | 0.0303(14) | 0.0298(14) | 0.0272(15) | 0.0146(33) | ||
| Y | 0.8340(7) | 0.8348(7) | 0.8350(7) | 0.8347(11) | |||
| Z | 0.0824(5) | 0.0821(5) | 0.0825(5) | 0.0819(9) | |||
| U i/Ue × 100 | 2.1(2) | 2.0(2) | 1.0(2) | 2.7(3) | |||
| O(4)-8f (x, y, z) | X | 0.2719(9) | 0.2731(10) | 0.2702(12) | 0.2603(24) | ||
| Y | 0.0833(7) | 0.0836(7) | 0.0844(8) | 0.0841(15) | |||
| Z | 0.0716(3) | 0.0718(4) | 0.0742(4) | 0.0761(7) | |||
| U i/Ue × 100 | 1.7(1) | 1.8(1) | 1.5(1) | 3.1(2) | |||
| O(5)-8f (x, y, z) | X | 0.7761(11) | 0.7768(11) | 0.7688(14) | 0.7690(21) | ||
| Y | 0.0834(8) | 0.0832(8) | 0.0818(8) | 0.0835(14) | |||
| Z | 0.0913(4) | 0.0916(4) | 0.0893(4) | 0.0886(6) | |||
| U i/Ue × 100 | 1.4(1) | 1.4(1) | 1.5(2) | 2.6(2) | |||
| wRp, X-ray/neutron, % | —/4.46 | —/4.72a | —/4.45 | —/4.55a | 1.15/4.53 | —/4.33 | |
| R p, X-ray/neutron, % | —/3.41 | —/3.60a | —/3.39 | —/3.46a | 0.84/3.54 | —/3.43 | |
| χ 2 | —/1.698 | —/1.916a | —/1.691 | —/1.783a | 1.925 | 1.579 | |
| R(F2), X-ray/neutron, % | —/2.20 | —/2.43a | —/2.31 | —/2.41a | 2.75/2.92 | —/2.76 | |
| Thermal expansion | |||||||
| a a, 10−6 K−1 | 2.333 | 5.922 | 13.218 | ||||
| a b, 10−6 K−1 | 1.890 | 13.379 | 14.241 | ||||
| a c, 10−6 K−1 | 1.888 | 5.737 | 10.345 | ||||
| a V, 10−6 K−1 | 6.228 | 25.060 | 37.938 | ||||
 | ||
| Fig. 2 Temperature dependence of the unit cell parameters of SrMn1−xSbxO3 (a) for x = 0.075 and (b) for x = 0.20 (filled marks) and 0.335 (unfilled marks). For clarity, in the tetragonal cell the a value has been divided by √2, the c value by 2 and V by 4 to yield a cubic cell. The estimated errors do not exceed the sizes of the marks. | ||
 | ||
| Fig. 3 Crystal structure of the monoclinically distorted 6H-perovskite SrMn0.925Sb0.075O3, (a and c) and the arrangement of magnetic moments in monoclinic (b) and parent hexagonal lattices (d). Red, yellow, green, light blue and blue spheres indicate Sr(1), Sr(2), Mn/Sb(1), Mn(2) and O atoms, respectively. Black and red dashed lines indicate monoclinic and hexagonal unit cells. Red and blue planes in (b) indicate up and down ferromagnetically ordered planes, which alternate antiferromagnetically along the c axis. | ||
Strontium ions in sites 4e and 8f, respectively, center two polyhedra 12-fold coordinated by oxygen ions. Linear and volume thermal expansion coefficients (Table 1) are expressed by
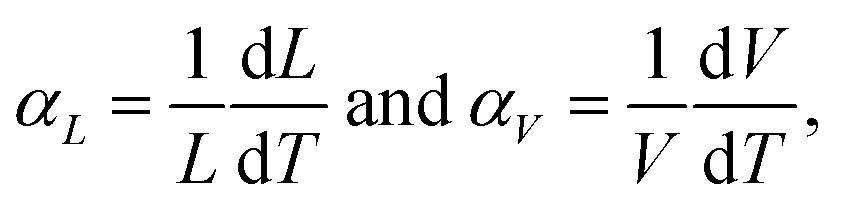 | (1) |
The magnitude of the thermal expansion is small, comparable with those of iron and quartz, and anisotropy is clearly seen for the b axis at around room temperature, which is most probably a result of the rotation of polyhedra into configuration corresponding to the ideal hexagonal lattice. The oxidation state of manganese for this composition is +3.92. The calculated bond valence sum (BVS) values (Table S2, ESI†) were in good agreement with expected oxidation states, with the exception of the BVSs of Mn(1) and Sb(1), which were found to be 3.722 and 6.202, respectively, not really reflecting the expected +4 and +5 that could be explained by mixed occupation of this site.
Analysis of the selected-area electron diffraction (SAED) patterns confirmed that SrMn0.925Sb0.075O3 crystalizes in a monoclinic unit cell with the space group C2/c at room temperature.
Both HAADF-STEM images taken along the 100 direction and SAED patterns confirm the absence of planar defects or local cation ordering in the structure (Fig. 4 and Fig. S1, ESI†).
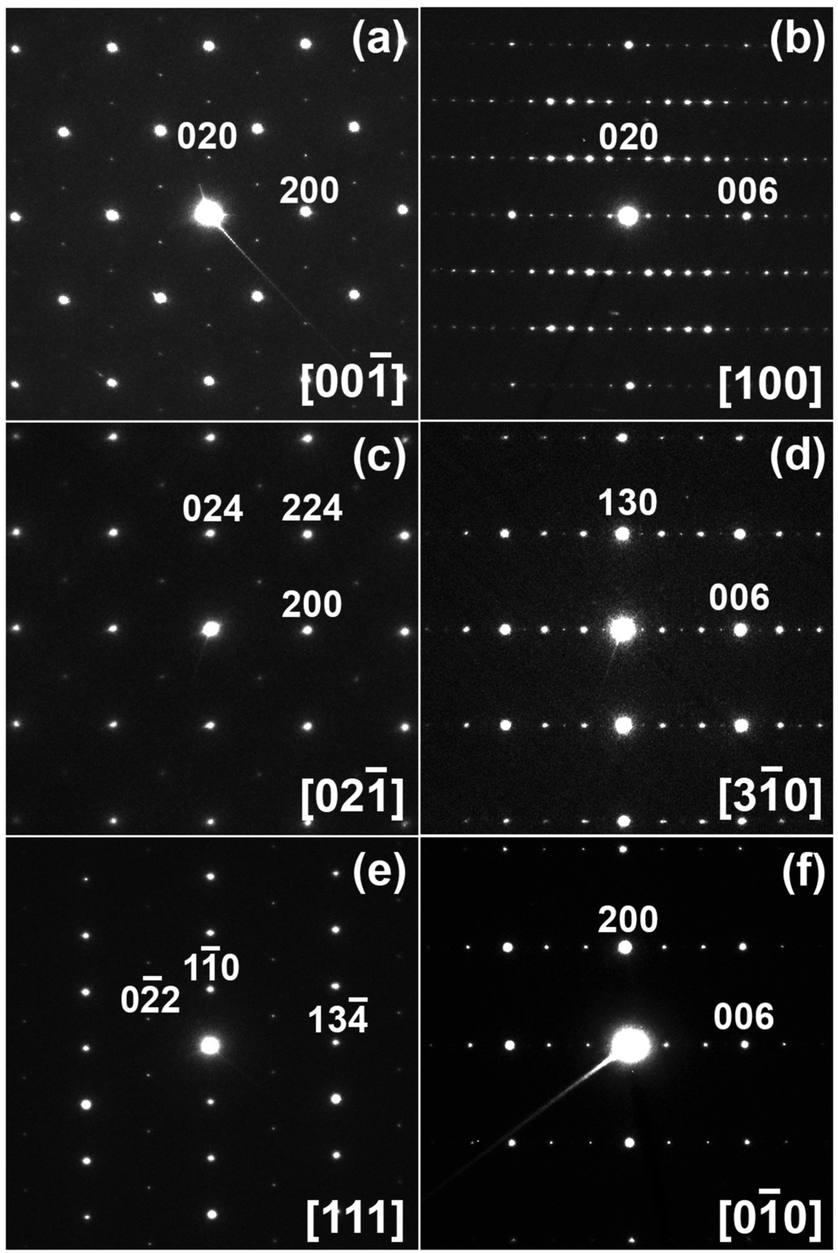 | ||
| Fig. 4 SAED patterns of SrMn0.925Sb0.075O3 taken along the main zone axes at room temperature. | ||
Linear and volume thermal expansion coefficients (Table S3, ESI†) calculated according to expression (1) exceed those for the monoclinic solid solutions, and the sign of the coefficient along the c axis is negative in the whole temperature range. The crystal structure of SrMn1−xSbxO3 with x = 0.20, 0.335 and 0.415 is described as a double perovskite with lattice parameters related to the cubic perovskite as follows: at ∼ ac√2 and ct ∼ 2ac and results from tilting of the octahedra (I4/mcm – a0a0c−) (Fig. 5a).
 | ||
| Fig. 5 Crystal structure (a) and arrangement of magnetic moments (b) of SrMn0.80Sb0.20O3 at 5 K, and (c) cubic polymorph at 500 K. Red, green and blue spheres indicate Sr, Mn/Sb and O atoms, respectively. AFM planes ferromagnetically stacking along the c axis are show in green. Black and red lines in (c) indicate primitive and face-centered cells. | ||
Since there is a large amount of discussion on the ordering of manganese and antimony among octahedral positions15–19 we tested all the proposed models and found that our synthetic route produces materials with random distribution of cations, having the highly symmetrical space group I4/mcm. The high-temperature modification of SrMn0.80Sb0.20O3 observed at 500 K is also described with the highest symmetry as a cubic perovskite (Fig. 5c) with the space group Pmm (Pmm – a0a0a0) and unit cell parameter a = 3.87998(2) Å, unlike the structure published for Sr2MnSbO6 at 900 K in ref. 19.
One should note that all full-profile refinements for all temperatures give the same R-values and quality of fit for models with and without refinement of ordering of Mn and Sb and oxygen shifts. No displacements from special sites of oxygen atoms, no ordering of manganese and antimony atoms exceeding experimental uncertainties have been detected, and the corresponding standard deviations for such parameters were much higher than those for other similar variables. These observations, along with the instability of the refinement for space groups I4/m and Fmm for the high-temperature modification usually arising from strong correlations indicate an excess in degrees of freedom and a wrong or too-low symmetry of the model.
So, we performed final refinements (Table S2, ESI†) with space groups I4/mcm for SrMn1−xSbxO3 compounds with x = 0.20, 0.335 and 0.415 and Pmm for the high-temperature modification with x = 0.20. The oxidation states of manganese for x = 0.20, 0.335 and 0.415 are +3.75, +3.5 and +3.29, respectively. The calculated BVS values were in good agreement with the expected oxidation states (Table S2, ESI†), except for those of Mn(1) and Sb(1) because manganese is present in two oxidation sates, +3 and +4, in addition to pentavalent antimony.
Average interatomic distances (Table S4, ESI†) are in good agreement with the sums of the crystal radii according to Shannon.28
Mean values of Sr–O bond length vary from 2.738 Å to 2.783 Å for all solid solutions, similar to 2.729–2.738 Å in 6H-SrMnO38 and 2.701 Å in Sr2MnSbO6.19 The sequential Mn/Sb–O distance increase of 1.930 Å, 1.939 Å, 1.964 Å and 1.973 Å for x = 0.075, 0.20, 0.335 and 0.415, respectively, reflects the growing content of Sb5+, with a crystal radius of 0.74 Å, which is larger than 0.67 Å for Mn4+ and 0.72 Å for Mn3+. The distance Mn(2)–Mn(2) in SrMn0.925Sb0.075O3 in face-sharing octahedra is short and are 2.491(1) Å, 2.510(9) Å, 2.508(3) Å and 2.508(9) Å at 1.5 K, 80 K, 298 K and 500 K, respectively, but agrees well with 2.511(2) Å reported for the undistorted 6H-SrMnO3 polymorph.8
Small substitution of larger antimony for manganese in corner-sharing octahedra sites stabilizes the monoclinically distorted high-pressure modification of SrMnO3. While further increase of antimony content results in a morphotropic transition, and the phase diagram contains two solid solubility regions based on monoclinic and tetragonal structures and a miscibility gap in the region from x = 0.09 to 0.17, where both modifications coexist without a change of unit cell parameters, only their mass fractions are changed.
 | ||
| Fig. 6 SAED patterns of SrMn0.80Sb0.20O3 taken along the same direction in the crystal at room temperature and at 513 K; h0l spots (h + l = 2n) on the [010]tetr diffraction pattern vanish upon heating. | ||
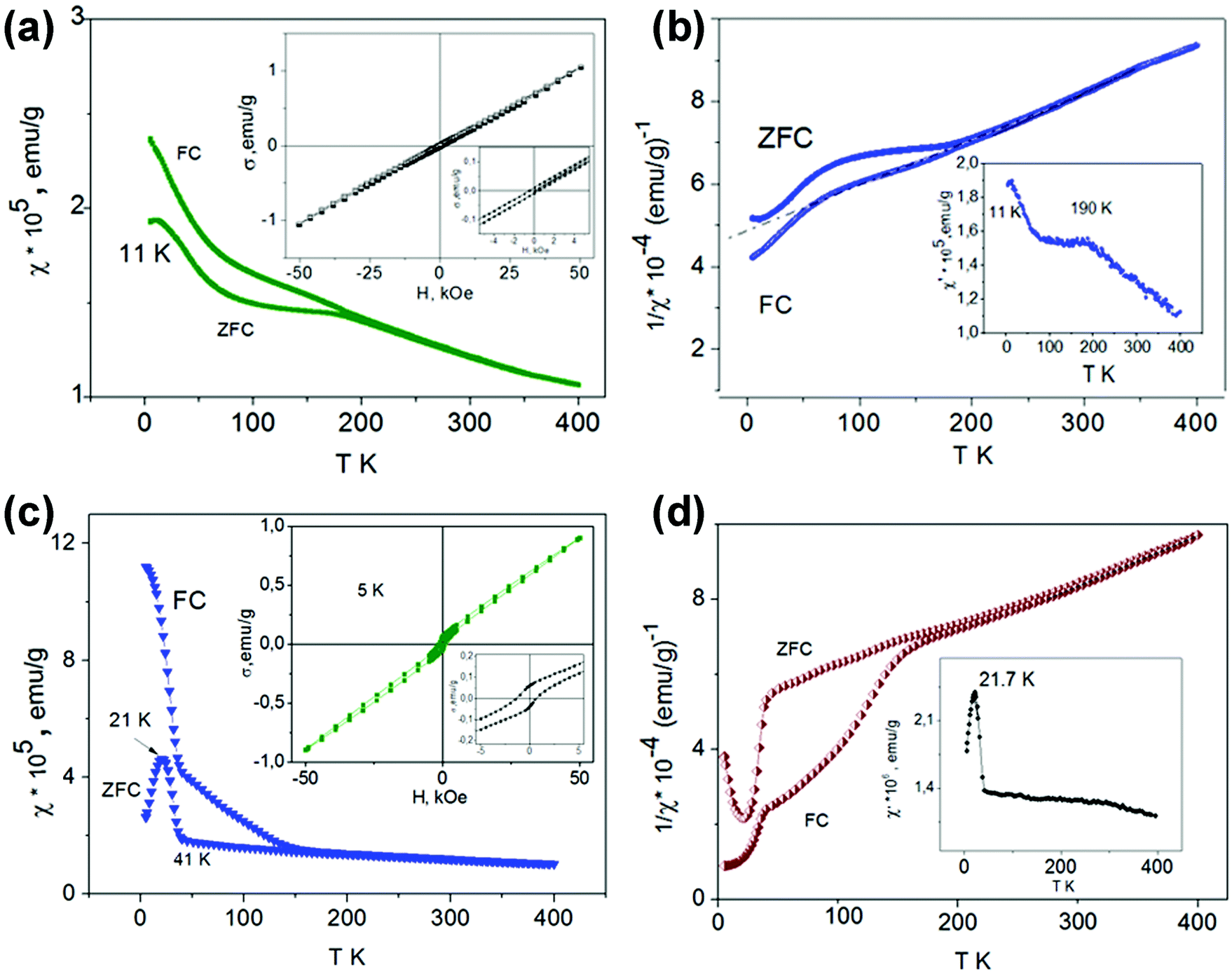 | ||
| Fig. 7 Temperature dependence of the ZFC, and FC susceptibility in an external field of 50 kOe and field dependence of σ = f(H) (inset) for SrMn0.95Sb0.05O3 (a) and SrMn0.925Sb0.075O3 (c); temperature dependence of 1/χ and AC susceptibility χ′(T) (inset) for SrMn0.95Sb0.05O3 (b) and SrMn0.925Sb0.075O3 (d). | ||
At high temperatures, both solid solutions are paramagnetic, and the magnetic susceptibility in the temperature range of ∼250–400 K follows the Curie–Weiss law χ = C/(T − Θ) (Fig. 7b and d) with negative Θ values (−500 K for x = 0.05 and −368 K for x = 0.075) and μexp = 3.866 and 3.902 μB, respectively. These μexp values are close to μtheor for the Mn4+ cation (electron configuration d3, μ = 3.872 μB). The presence of Mn3+ cations with electron configuration d4 (μ = 4.90 μB) led to an overestimated value of μexp for the solid solution with x = 0.075. The negative sign and the large Curie–Weiss temperatures Θ are indicative of the presence of very strong antiferromagnetic interactions and magnetic frustration appearing due to the structural peculiarities of these perovskites. The crystal structure of SrMn1−xSbxO3 with x = 0.05 and 0.075—a monoclinically distorted 6H-perovskite—can be considered as a two-dimensional triangular magnet,29 in which the Mn sublattice is divided by double layers occupied by manganese and antimony ions. The presence of magnetic disorder, due to the implantation of diamagnetic cations Sb5+, promotes a decrease in TN compared with 6H-SrMnO38 and an abrupt growth of susceptibility when the temperature lowers (Fig. 7a and c). Similar to 4H-SrMnO3,20 for the monoclinic solid solution SrMn1−xSbxO3 (x = 0.05 and 0.075), the short-range antiferromagnetic interactions persist in the paramagnetic region.
The χ(T) curves for the samples with x = 0.05 and 0.075 cooled in the ZFC mode and in measured field (FC mode) diverge at 190 K and ∼280 K, respectively. For SrMn0.925Sb0.075O3, a considerable increase in χ measured in the FC mode is observed at 160 K (Fig. 7c). During further cooling of the sample, another abrupt increase in χ is observed at 41 K and a maximum at 21 K appears on the ZFC curve. The effect on the AC susceptibility at 21 K (Fig. 7d) may be an indication that at low temperature not all of the manganese cations are involved in long-range antiferromagnetic order and can form clusters that form a spin-glass state. These data suggest that the temperature vs. susceptibility dependence is determined by dimeric behavior for the monoclinic solid solution SrMn1−xSbxO3 (x = 0.05 and 0.075); in contrast to 6H-SrMnO3.8
The insets in Fig. 7c display the magnetization measurement results as a function of the magnetic field at 5 K. The measurements were performed during a reduction of the magnetic field from 50 kOe on samples cooled under ZFC conditions. The fact that the σ vs. H curves are nonlinear and that the magnetization does not reach saturation at 50 kOe suggest the presence of frustrated spins in the magnetic system. These results, together with the χ = f(T) data (Fig. 7a–d), may be indicative of a rather complicated model of antiferromagnetic ordering of monoclinic SrMn1−xSbxO3 solid solutions below the temperatures at which the divergence between FC and ZFC magnetic susceptibility data is observed (190 K and about 280 K for x = 0.05 and 0.075, respectively). Distinct from 6H-SrMnO3 produced at 6 GPa,11 the χ(T) curve which displays a sharp maximum near TN = 235 K, there is only a flat maximum near 190 K on the ZFC curve (Fig. 7a) and the χ′ = f(T) curve (Fig. 7b) of the solid solution with x = 0.05.
![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) ) at d ≈ 4.47 Å and (11
) at d ≈ 4.47 Å and (11![[2 with combining macron]](https://www.rsc.org/images/entities/char_0032_0304.gif) ) at d ≈ 3.88 Å (Fig. 8).
) at d ≈ 3.88 Å (Fig. 8).
 | ||
| Fig. 8 Neutron powder diffraction pattern of SrMn0.925Sb0.075O3 at 1.5 K without magnetic ordering (top) and with magnetic ordering (bottom). The red crosses are experimental points, the solid black line is the calculated profile and the vertical marks correspond to the positions of the Bragg reflections for the crystallographic (purple, first row) and magnetic (blue, second row) structures. The difference curve is plotted at the bottom of the figure. | ||
These reflections of magnetic origin can be indexed with a magnetic cell of the same dimensions and space group as the structural one, which means that we need the single propagation vector k = (0, 0, 0). The magnetic symmetry analysis was performed using the program BasIreps in the FullProf suite.31 Full details on the determination of the magnetic structure are given in the ESI† (including Tables S5–S7) and ref. 32. The relations between the cells and the arrangement of magnetic moments in monoclinic and parent hexagonal lattices are shown in Fig. 3; the difference in the fit of the NPD pattern of SrMn0.925Sb0.075O3 at 1.5 K without taking into account the magnetic ordering and with long-range magnetic ordering is presented in Fig. 8. In general, Fourier coefficients are (u, v and w) and (−u, v and −w) and both magnetic subsystems might have antiferromagnetic spin canting in the direction of the a axis and ferromagnetic in the direction of the b axis, which means that spins are slightly tilted rather than being parallel, and a nonzero net moment is possible. In first approximation, the magnetic structure is well described as A-type antiferromagnetic, where spins are parallel to the c axis. In-plane, the coupling is ferromagnetic, while the inter-plane coupling is antiferromagnetic (Fig. 3b). The refined magnetic moments for SrMn0.925Sb0.075O3 at 1.5 K and 80 K are presented in Table S8 (ESI†). The magnitude of the saturated moments at 1.5 K, 1.85(9) μB for 4a and 1.13(6) μB for 8f sites, respectively, is much lower than the predicted value of 2.6 μB for Mn4+ with a degree of covalency.33 The combined populations on both sites of 92.5% of S = 3/2 Mn4+ and 7.5% of S = 2 Mn3+ ions mainly leads to antiferromagnetic interactions, although local orbital ordering of Mn3+ could create some ferromagnetic couplings causing frustration. Similar long-range magnetic ordering has been reported for the 9R-type perovskite, BaRu0.2Mn0.8O334 and hematite, α-Fe2O3,35 below the Morin temperature. More recently, the same magnetic ordering has been published for undistorted 6H perovskite Ba3Fe2TeO9.36
 | ||
| Fig. 9 (a) Temperature dependence of 1/χ′, AC susceptibility χ′(T) (inset) of SrMn0.665Sb0.335O3. (b) ZFC and FC magnetic susceptibility of SrMn0.665Sb0.335O3 measured at 0.5 kOe. The inset shows the dependence of σ = f(H) on different magnetic fields. (c) ZFC and FC magnetic susceptibility for SrMn0.80Sb0.20O3 measured at 0.5 kOe. The inset shows the dependence of χ′ = f(T). | ||
The increase in the Mn content and in the average oxidation state to +3.75 in tetragonal SrMn0.80Sb0.20O3 qualitatively changes the temperature dependence of the magnetic susceptibility (Fig. 9c). The appearance of a maximum on the χ = f(T) dependence at 248 K and a very narrow region of validity of the Curie–Weiss law on the 1/χ = f(T) dependence (in the range of 250–350 K, Θ = 1300 K, not shown) allow us to suppose a long-range magnetic ordering of this solid solution, with TN = 248 K. The presence of a maximum at this temperature is also confirmed by the results of AC susceptibility measurements (inset on Fig. 9c). A small anomaly in the ZFC-susceptibility at 43 K may be the evidence of the magnetic transition in Mn3O4, which was not found by X-ray diffraction. The magnetization-field dependence does not show significant hysteresis (not shown). The transformation of this solid solution into the cubic form is also seen on the χ = f(T) curve at about 350 K.
 | ||
| Fig. 10 Neutron diffraction patterns of SrMn0.8Sb0.2O3 at 500 K (a), 298 K (b) and 5 K (c). The red crosses are the experimental points, the solid black line is the calculated profile and the vertical marks correspond to the positions of the Bragg reflections for the crystallographic (purple, first row) and magnetic (blue, second row) structures. The difference curve is plotted at the bottom of the figure. | ||
These reflections can be indexed with a magnetic cell of the same dimensions as the structural one, but the crystallographic unit cell is I-centered, whereas the magnetic unit cell is primitive, which means that we need the single propagation vector k = (1, 0, 0). Full details on the determination of the magnetic structure is given in the ESI† (including Table S9). The refinement showed that the Γ7 model perfectly describes the extra peaks (Fig. 10c). The magnetic structure is described as C-type antiferromagnetic, where spins are parallel to the c axis with antiferromagnetic intraplanar and ferromagnetic interplanar coupling (Fig. 5b). The refined saturated magnetic moments at 5 K, 2.79(2) μB and 2.00(4) μB for SrMn1−xSbxO3 with x = 0.20 and 0.335, respectively (Table S10, ESI†), fit well to the predicted value for Mn4+, but are lower than those for Mn3+.
Conclusions
Solid solutions of the SrMn1−xSbxO3 system with a perovskite structure and mixed-manganese valence Mn4+/Mn3+ were prepared for the first time by conventional solid-state synthesis. Two ranges of solid solutions were found: (1) SrMn1−xSbxO3 (0.025 ≤ x ≤ 0.09) with a monoclinically distorted 6H-SrMnO3 polytype (sp. gr. C/2c) and (2) SrMn1−xSbxO3 (0.17 ≤ x ≤ 0.5) with a tetragonal unit cell (sp. gr. I4/mcm). The crystal structure refinement was carried out using the NPD data for low and high temperatures and a combination of XRPD and NPD data for room temperature. The crystal structure of the monoclinic solid solution consists of corner-sharing octahedra around sites 4a randomly occupied by manganese and antimony ions and face-sharing octahedra around sites 8f occupied by manganese ions only. The tetragonal solid solution SrMn1−xSbxO3 has a random distribution of cations. The high-temperature modification observed at 500 K has been described with the highest symmetry: a cubic perovskite with a space group Pmm and a unit cell parameter a = 3.87998(2) Å (x = 0.20).
Magnetic susceptibility measurements and low-temperature NPD data show that the solid solutions adopt antiferromagnetic ordered structures. The magnetic structure of the tetragonal solid solutions SrMn1−xSbxO3 (x = 0.20 and 0.335) is described as C-type AFM, where spins are parallel to the c axis with AFM intraplanar and FM interplanar coupling. The refined saturated magnetic moments at 5 K, 2.79(2) μB and 2.00(4) μB for SrMn1−xSbxO3 for x = 0.20 and 0.335, respectively, fit well to the predicted value for Mn4+, but are lower than those for Mn3+. In first approximation, the magnetic structure of the monoclinic solid solution is well described as an A-type AFM, where spins are parallel to the c axis. Inside the plane, coupling is ferromagnetic while inter-plane coupling is antiferromagnetic.
Our results clearly demonstrate that antiferromagnetic order can be induced in both perovskite structures of the SrMn1−xSbxO3 system through chemical substitution. This result contradicts earlier published data for the SrMn0.5Sb0.5O3 tetragonal perovskite in which magnetic order was not found.18,19 The magnetic behavior of SrMn0.5Sb0.5O3 was described by a spin-glass state with a weak ferromagnetism at low temperatures and a positive Weiss constant in the paramagnetic area indicating the presence of ferromagnetic clusters; there was no convincing explanation for the lack of a magnetic ordered state in Sr2MnSbO6 given so far. However, similar magnetic properties of Sr2MnTaO6 at low temperature37 were explained by competing double-exchange (ferromagnetism) and super-exchange (antiferromagnetism) interactions. This conclusion was based on the assumption that Mn in this compound has a mixed oxidation state between +3 and +4. At the same time, in isostructural manganites (monoclinic Sr2FeSbO6 and Sr2FeTaO6) but with sufficient structural disorder, a partial cation ordering in the structure leads to the coexistence of a magnetically-ordered spin state and a spin-glass state.38
It is important that in our work, as a result of the replacement of 33% of Sb5+ cations by manganese in SrMn0.5Sb0.5O3, the Weiss constant in SrMn0.67Sb0.33O3 began to have a negative value thus pointing towards domination of AFM interactions and the formation of antiferromagnetic frustrated-spin clusters. Considering that according to our NPD data the oxygen content in the solid solutions corresponds precisely to the theoretical values, we suggest that the mean oxidation states of manganese in the solid solutions with x = 0.33 and 0.20 are +3.5 and +3.75, respectively. Consequently, the emergence of antiferromagnetic ordering in these solid solutions can be explained by the formation of mixed-valence manganese Mn3+/Mn4+ states due to decrease of the diamagnetic Sb5+ ion content in antiferromagnetic frustrated-spin clusters.
Further substitution of Mn cations by Sb5+ is also a reason for the formation of the monoclinic structure with mixed-valence Mn3+/Mn4+ states. Thus, a change in composition provokes a change in structure and magnetic properties, inducing antiferromagnetic ordering in the tetragonal (x = 0.20 and 0.33) and the monoclinic (at x = 0.025 and 0.075) solid solution based on SrMn1−xSbxO3.
This conclusion can be further supported by the analysis of results of the other works already mentioned in the introduction, in which possibilities of modification of the crystal structures of manganites and their magnetic properties by chemical replacements are clearly shown.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work was carried out in accordance with the state assignment for the Institute of Solid State Chemistry of the Ural Branch of the Russian Academy of Sciences (RAS). The X-ray powder diffraction study was carried out at the multi-access center for X-ray structure analysis at the Institute of Solid State Chemistry (Ekaterinburg) in the frame of the Comprehensive Program of Basic Scientific Research of the Ural Branch of the RAS (theme No. 18-10-3-32). The Knut and Alice Wallenberg (KAW) Foundation is acknowledged for funding the electron microscopy facilities at Stockholm University, financial support for C. W. T. under the Project No. 3DEM-NATUR. Open Access funding provided by the Max Planck Society.Notes and references
- H. Sakai, J. Fujioka, T. Fukuda, D. Okuyama, D. Hashizume, F. Kagawa, H. Nakao, Y. Murakami, T. Arima, A. Q. R. Baron, Y. Taguchi and Y. Tokura, Phys. Rev. Lett., 2011, 107, 137601 CrossRef CAS PubMed.
- J. M. D. Coey, M. Viret and S. von Molnar, Adv. Phys., 1999, 48, 167 CrossRef CAS.
- O. Korneta, T. F. Qi, M. Ge, M. S. Parkin, L. E. De Long, P. Schlottmann and G. Cao, J. Phys.: Condens. Matter, 2011, 23, 435901 CrossRef CAS PubMed.
- G. Zhang, H. Chen, Z. Gu, P. Zhang, T. Zeng and F. Huang, Inorg. Chem., 2017, 56, 10404 CrossRef CAS PubMed.
- V. G. Zubkov, G. V. Bazuev, A. P. Tyutyunnik and I. F. Berger, J. Solid State Chem., 2001, 160, 293 CrossRef CAS.
- Y. J. Choi, H. T. Yi, S. Lee, Q. Huang, V. Kiryukhin and S.-W. Cheong, Phys. Rev. Lett., 2008, 100, 047601 CrossRef CAS PubMed.
- Y. Tokura and N. Nagaosa, Science, 2000, 288, 462 CrossRef CAS PubMed.
- A. A. Belik, Y. Matsushita, Y. Katsuya, M. Tanaka, T. Kolodiazhnyi, M. Isobe and E. Takayama-Muromachi, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 84, 094438 CrossRef.
- B. L. Chamberland, A. W. Sleight and J. F. Weiher, J. Solid State Chem., 1970, 1, 506 CrossRef.
- E. Cussen, J. Sloan, J. F. Vente, P. D. Battle and T. C. Gibb, Inorg. Chem., 1998, 37, 6071 CrossRef CAS PubMed.
- A. Kumar, A. S. Verma and S. R. Bhardwaj, Open Appl. Phys. J., 2008, 1, 11 CrossRef CAS.
- P. Lightfoot and P. D. Battle, J. Solid State Chem., 1990, 89, 174 CrossRef CAS.
- H. Kozuka, H. Yamada, T. Hishida, K. Ohbayashi and K. Koumoto, J. Mater. Chem. A, 2013, 1, 3249 RSC.
- G. Blasse, J. Inorg. Nucl. Chem., 1965, 27, 993 CrossRef CAS.
- M. W. Lufaso, P. M. Woodward and J. Goldberger, J. Solid State Chem., 2004, 177, 1651 CrossRef CAS.
- E. D. Politova, G. M. Kaleva, I. N. Danilenko, V. F. Chuprakov, S. A. Ivanov and N. Y. Venevtsev, Izv. Akad. Nauk SSSR, Neorg. Mater., 1990, 26, 2352 CAS.
- M. Cheah, P. A. Saines and B. J. Kennedy, J. Solid State Chem., 2006, 179, 1775 CrossRef CAS.
- K. Mandal, V. V. Poltavets, M. Croft and M. Greenblatt, J. Solid State Chem., 2008, 181, 2325 CrossRef.
- S. A. Ivanov, P. Nordblad, R. Tellgren and A. Hewat, Mater. Res. Bull., 2009, 44, 822 CrossRef CAS.
- P. Battle, T. C. Gibb and C. W. Jones, J. Solid State Chem., 1988, 74, 60 CrossRef CAS.
- T. K. Mandal, A. M. Abakumov, M. V. Lobanov, M. Croft, V. V. Poltavets and M. Greenblatt, Chem. Mater., 2008, 20, 4653 CrossRef CAS.
- A. Ben Hassine, A. Dhahri, L. Bouazizi, M. Oumezzine and E. K. Hlil, Solid State Commun., 2016, 233, 6 CrossRef CAS.
- B. H. Toby, J. Appl. Crystallogr., 2001, 34, 210 CrossRef CAS.
- A. C. Larson and R. B. Von Dreele, “General Structure Analysis System (GSAS)”, Los Alamos National Laboratory Report LAUR 2004, 86-748.
- A. S. Wills, VaList, Program available from http://www.ccp14.ac.uk.
- I. D. Brown and D. Altermatt, Acta Crystallogr., Sect. B: Struct. Sci., 1985, 41, 244 CrossRef.
- N. E. Brese and M. O’Keeffe, Acta Crystallogr., Sect. B: Struct. Sci., 1991, B47, 192 CrossRef CAS.
- G. V. Bazuev and A. V. Korolyov, J. Magn. Magn. Mater., 2008, 320, 2262 CrossRef CAS.
- Y. Doi, Y. Hinatsu and K. Ohoyama, J. Phys.: Condens. Matter, 2009, 16, 8923 CrossRef.
- R. D. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751 CrossRef.
- FullProf, http://www.ill.eu/sites/fullprof/.
- A. S. Wills, J. Phys. IV France, 2001, 11, 133 CAS.
- C. Yin, G. Li, W. A. Kockelmann, J. Lin and J. P. Attfield, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 80, 094420 CrossRef.
- C. G. Shull, W. A. Strauser and E. O. Wollan, Phys. Rev., 1951, 83, 333 CrossRef CAS.
- F. J. Morin, Phys. Rev., 1950, 78, 819 CrossRef CAS.
- Y. Tang, R. P. Sena, M. Avdeev, P. D. Battle, J. M. Cadogan, J. Hadermann and E. C. Hunter, J. Solid State Chem., 2017, 253, 347 CrossRef CAS.
- M. Valldor, S. Esmaeilzadeh, M. Andersson and A. Morawski, J. Magn. Magn. Mater., 2006, 299, 161 CrossRef CAS.
- E. J. Cussen, J. F. Vente, P. D. Battle and T. C. Gibb, J. Mater. Chem., 1997, 7, 459–463 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8tc05717f |
| This journal is © The Royal Society of Chemistry 2019 |