Diazaspirocycles: novel platforms for efficient phosphorescent organic light-emitting diodes†
Received
30th October 2018
, Accepted 24th December 2018
First published on 25th December 2018
Abstract
Azafluorenone was discovered decades ago and its derivatives have received widespread attention in various research fields. However, the application of azafluorenone and its derivatives in organic light-emitting diode (OLED) materials is rarely reported and the related device performance is still not satisfactory. In this work, 1,4-diazafluorenone and its two analogues were prepared as precursors and three novel spiro compounds, SAIP, SAIQ, and SABIQ, were synthesized. The basic physical and chemical properties of SAIP, SAIQ, and SABIQ were studied in detail. From SAIP to SAIQ and SABIQ, the triplet energies (ETs) and optical band gaps (Egs) decrease gradually with the extension of the intramolecular conjugation length. In addition, blue/green/red and green/red phosphorescent OLEDs (PHOLEDs) were fabricated with SAIP and SAIQ as host materials, respectively. It is worth noting that all green and red devices based on SAIP or SAIQ showed a relatively high device performance with maximum external quantum efficiencies (EQEs) of 17.8% and 21.4%, respectively. These results further show the high potential of azafluorenone and its analogues in the building of efficient host materials.
Introduction
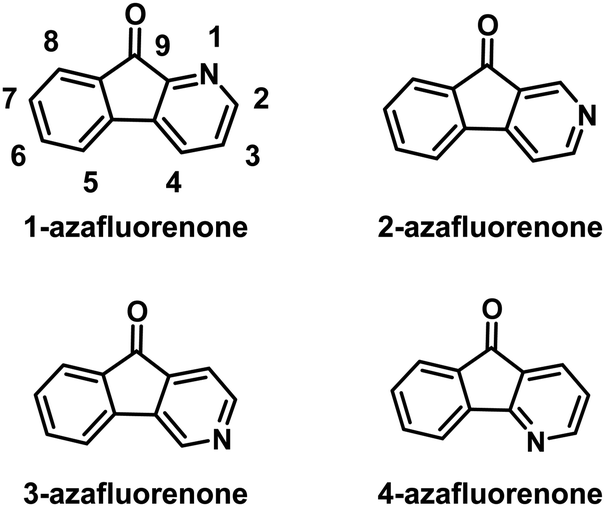
The chemistry of azafluorenones (Fig. 1) has been well studied in the early literature,1 and there has been significant development in the synthetic methods for these fused ring systems over the last four decades. A survey of the preparation of azafluorenones shows that there are three typical synthetic routes. The first one is to make use of oxidative ring contractions of nitrogen-containing fused compounds. For example, azafluorenone derivatives can be obtained by oxidation of the corresponding substituted azaphenanthrenes with potassium permanganate (KMnO4).1b,2 The second method is to increase the number of fused rings by annelation of the heterocycle. For instance, the condensation of the indane–1,3-dione-aminocrotonate–aldehyde three-component system is the most efficient and therefore widely used for the synthesis of 1,4-dihydro-4-azafluorenes,3 which can be easily aromatized with oxidants or under UV irradiation to obtain the corresponding 4-azafluorenones.4 The third method is intramolecular cyclocondensation of aryl- or aroyl pyridines. The availability of diverse pyridine derivatives (phenylpyridinecarboxylic acids,5 and respective acid chlorides,6 nitriles, esters5e,f,7 and acid amides8) has offered broad possibilities for the synthesis of all isomeric azafluorenones. The corresponding cyclisation often requires sulfuric9 and polyphosphoric acids (PPAs),7d,10 aluminum chloride (AlCl3),11 and some other compounds5e,f,8,12 as condensing agents. Other specific methods such as the photochemical Pschorr cyclization of 2-diazoniodiaryl ketones,13 radical Pschorr cyclization of organoboronic acids and trifluoroborates,14 gas-phase thermolysis of diazo compounds,1b intramolecular Heck reactions of 3-aroylpyridines,15 intramolecular carbonylation via oxidative C–H functionalization of the methyl group,16 and C–H functionalization of the methyl/hydroxymethyl/aldehyde group17 have also been reported.
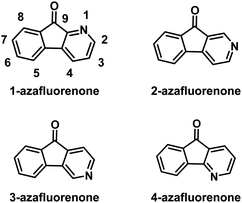 |
| | Fig. 1 The structures of isomeric azafluorenones. | |
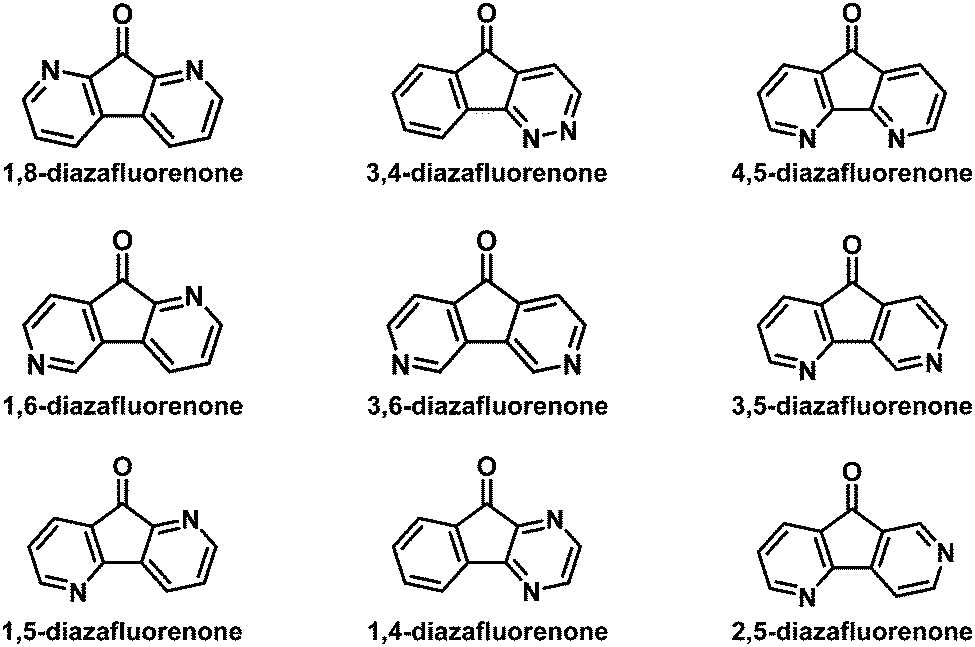
Recently, various nitrogenated rigid molecular building blocks18 such as carbazole,19 dibenzo[b,d]furan (DBF),20 dibenzo[b,d]thiophene (DBT),21 and 9,9′-spirobifluorene (SBF)22 have been extensively used as functional materials in organic light-emitting diodes (OLEDs).19b–g,20,21,22a–e Many research studies have illustrated that the introduction of an sp2 nitrogen atom into the backbone of organic conjugated materials can improve their electron transport ability.18 For this purpose, azafluorenones (Fig. 2) and diazafluorenones (Fig. 3) are good starting materials to construct the corresponding aza-SBF derivatives.23–25 It is worth noting that 4,5-diaza-SBF derivatives have been more extensively studied22c–f,23 relative to other diaza-SBF compounds. There are several reported ways to synthesize diazafluorenones, including the ring contraction reactions of phenanthrolinchinones,2d,26 the condensation reaction of ninhydrin/aryl methyl ketone and hydrazine hydrate,23,27 the condensation reaction of 1-(N,N-dimethylaminomethyliden)indan-2-one and benzamidine hydrochloride hydrate,28 and the cyclocondensation reaction of ninhydrin and ethylenediamine.29
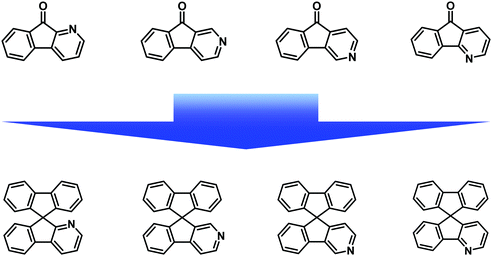 |
| | Fig. 2 Reported structures of azafluorenones and the corresponding azaspirobifluorenes. | |
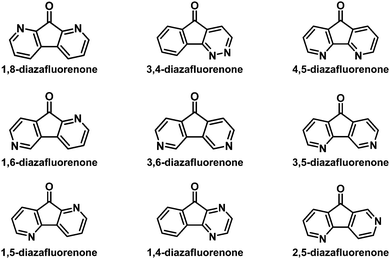 |
| | Fig. 3 Reported structures of diazafluorenones. | |
In OLEDs, phosphorescent emitters are normally doped into suitable host materials to suppress the competitive decay process such as triplet–triplet annihilation and triplet–polaron annihilation.30 Therefore, the chemical and physical properties of the host materials such as good thermal stability, suitable triplet energy, and balance charge transfer properties are important for fabricating efficient phosphorescent OLEDs (PHOLEDs).31 Very recently, plenty of work has been reported on the development of highly efficient host materials for blue, green, and red PHOLEDs,32 as well as the optimization of the device structure.33 For instance, Liao and co-workers reported a novel thermally activated delayed fluorescence (TADF) material as the host material for a red PHOLED with an external quantum efficiency (EQE) over 31%.32e So far, many novel molecular platforms such as dispiro,32a propellane32a and 9-silafluorene32b have been used for the construction of highly efficient host materials. On the other hand, high performance PHOLEDs have also been realized via device optimization such as applying a co-host,33a,b a double emission layer,33c and a novel electron-transporting material.33d,e
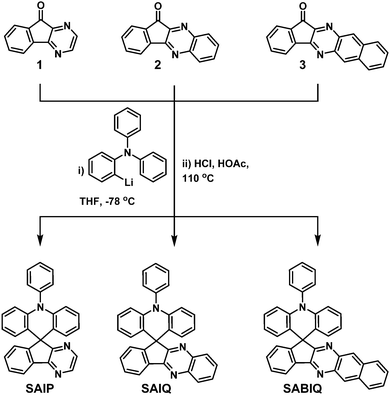
To develop high performance host materials, several empirical design strategies that would improve the charge transport capability of aza-SBFs have been reported in recent years, such as introduction of aromatic amine substituents (diphenylamine, carbazole, etc.) to aza-SBF backbones34 and the replacement of fluorene with a triphenylamine or diphenylsulfane unit.35 In this article, three novel spirocycles, 10-phenyl-10H-spiro[acridine-9,9′-indeno[1,2-b]pyrazine] (SAIP), 10-phenyl-10H-spiro[acridine-9,11′-indeno[1,2-b]quinoxaline] (SAIQ) and 10-phenyl-10H-spiro[acridine-9,13′-benzo[g]indeno[1,2-b]quinoxaline] (SABIQ), are constructed. Basic properties such as the thermal and photophysical properties and the electrochemistry behaviors of SAIP, SAIQ, and SABIQ were fully characterized. The introduction of different electron acceptor segments has a great influence on the basic physical and chemistry properties of these spirocycles, but has a minor effect on their electroluminescence performance. Blue, green, and red PHOLEDs were fabricated with bis(4,6-(difluorophenyl)pyridinato-N,C2′)picolinate iridium(III) (FIrpic), bis(2-phenylpyridine)(acetylacetonate)iridium(III) (Ir(ppy)2(acac)), and bis(2-methyldibenzo-[f,h]-quinoxaline)iridium(III) (acetylacetonate) (Ir(MDQ)2(acac)) as dopants, respectively. To our great delight, the green and red devices demonstrated moderate device performance with external quantum efficiencies above 17% and 21%, respectively.
Experimental section
Materials synthesis
The precursors 1,4-diazafluorenone (1),29 11H-indeno[1,2-b]quinoxalin-11-one (2),36 and 13H-benzo[g]indeno[1,2-b]quinoxalin-13-one (3)37 were prepared in accordance with literature methods.
1,4-Diazafluorenone (1).
1H NMR (400 MHz, chloroform-d) δ 8.56–8.52 (m, 1H), 8.50–8.47 (m, 1H), 7.89 (d, J = 7.5 Hz, 1H), 7.82 (d, J = 7.4 Hz, 1H), 7.68 (t, J = 7.6 Hz, 1H), 7.53 (t, J = 7.5 Hz, 1H) ppm.
11H-Indeno[1,2-b]quinoxalin-11-one (2).
1H NMR (400 MHz, chloroform-d) δ 8.24 (d, J = 8.3 Hz, 1H), 8.14–8.08 (m, 2H), 7.93 (d, J = 7.5 Hz, 1H), 7.87–7.71 (m, 3H), 7.61 (t, J = 7.5 Hz, 1H) ppm.
13H-Benzo[g]indeno[1,2-b]quinoxalin-13-one (3).
1H NMR (400 MHz, chloroform-d) δ 8.79 (s, 1H), 8.62 (s, 1H), 8.20 (d, J = 7.6 Hz, 1H), 8.09 (t, J = 8.7 Hz, 2H), 7.98 (d, J = 7.6 Hz, 1H), 7.82 (t, J = 7.5 Hz, 1H), 7.69–7.55 (m, 3H) ppm.
10-Phenyl-10H-spiro[acridine-9,9′-indeno[1,2-b]pyrazine] (SAIP).
2-Bromo-N,N-diphenylaniline (5 g, 15.4 mmol) was dissolved in THF (80 mL) at −78 °C, and n-butyl lithium (11.6 mL, 18.5 mmol, 1.6 M in n-hexane) was added dropwise using a syringe. After stirring for 1 hour at −78 °C, 1 (2.8 g, 15.4 mmol) in THF (50 mL) was added slowly over a period of 30 min. The resulting mixture was stirred for 2 hours at −78 °C, and the mixture was gradually warmed to room temperature overnight. The reaction was quenched with water, and the product was extracted with EtOAc (3 × 50 mL). Then the organic layer was separated and dried over sodium sulfate (Na2SO4). The removal of the solvent under reduced pressure afforded the crude product as a light yellow oil, which was directly used in the next reaction without further purification.
The crude product was dissolved in acetic acid (100 mL) at 110 °C and 10 mL of hydrochloric acid was added dropwise. The mixture was heated to reflux overnight. Then the reaction system was cooled down to room temperature. After the removal of the solvent, the raw product was purified by column chromatography using petroleum ether/EtOAc (3/1, v/v) as an eluent to give SAIP as a pale-yellow powder (3.8 g, ∼60%). 1H NMR (600 MHz, chloroform-d) δ 8.45 (d, J = 2.6 Hz, 1H), 8.29 (d, J = 2.6 Hz, 1H), 8.19–8.10 (m, 1H), 7.68 (t, J = 7.6 Hz, 2H), 7.60–7.47 (m, 6H), 6.94 (ddd, J = 8.4, 7.1, 1.5 Hz, 2H), 6.57 (t, J = 7.5 Hz, 2H), 6.40 (d, J = 8.4 Hz, 2H), 6.31 (dd, J = 7.9, 1.5 Hz, 2H) ppm. 13C NMR (151 MHz, chloroform-d) δ 168.45, 154.67, 152.18, 143.36, 142.76, 141.79, 141.02, 136.86, 131.62, 131.27, 130.94, 128.58, 128.43, 127.83, 127.23, 126.55, 121.77, 121.44, 120.41, 115.17, 55.34 ppm. MALDI-TOF (m/z), calculated for: 409.492, found: 407.994. Anal. calcd for: C29H19N3 (%): C, 85.06; H, 4.68; N, 10.26; found: C, 85.09; H, 4.72; N, 10.18.
10-Phenyl-10H-spiro[acridine-9,11′-indeno[1,2-b]quinoxaline] (SAIQ) and 10-phenyl-10H-spiro[acridine-9,13′-benzo[g]indeno[1,2-b]quinoxaline] (SABIQ) were prepared by a similar method to that used for SAIP.
SAIQ
.
Bright yellow solid (68%). 1H NMR (600 MHz, chloroform-d) δ 8.34–8.29 (m, 1H), 8.14 (d, J = 8.2 Hz, 1H), 7.99 (d, J = 8.3 Hz, 1H), 7.75–7.67 (m, 3H), 7.67–7.55 (m, 7H), 6.95 (t, J = 8.5 Hz, 2H), 6.56 (t, J = 8.1 Hz, 2H), 6.45 (d, J = 8.4 Hz, 2H), 6.37 (d, J = 7.8 Hz, 2H) ppm. 13C NMR (151 MHz, chloroform-d) δ 167.72, 154.61, 153.28, 142.62, 142.09, 141.77, 141.12, 136.64, 132.73, 131.31, 130.92, 129.77, 129.43, 128.95, 128.83, 128.63, 128.44, 127.76, 127.42, 126.80, 123.08, 122.06, 120.40, 115.14, 55.16 ppm. MALDI-TOF (m/z), calculated for: 459.552, found: 458.343. Anal. calcd for: C33H21N3 (%): C, 86.25; H, 4.61; N, 9.14; found: C, 86.30; H, 4.75; N, 9.10.
SABIQ
.
Orange-yellow solid (62%). 1H NMR (600 MHz, chloroform-d) δ 8.69 (s, 1H), 8.53 (s, 1H), 8.39–8.36 (m, 1H), 8.08 (d, J = 8.2 Hz, 1H), 8.00 (d, J = 8.1 Hz, 1H), 7.75 (t, J = 7.7 Hz, 2H), 7.71–7.67 (m, 2H), 7.66–7.57 (m, 4H), 7.55–7.49 (m, 2H), 6.96 (t, J = 8.5 Hz, 2H), 6.58 (t, J = 8.0 Hz, 2H), 6.47 (d, J = 8.5 Hz, 2H), 6.43 (d, J = 7.9 Hz, 2H) ppm. 13C NMR (151 MHz, chloroform-d) δ 168.16, 154.57, 154.36, 141.66, 141.12, 139.29, 138.84, 136.62, 133.65, 133.37, 133.16, 131.35, 130.96, 128.99, 128.48, 128.28 (d, J = 6.3 Hz), 127.98, 127.76 (d, J = 13.5 Hz), 127.02 (d, J = 7.0 Hz), 126.51, 126.37, 123.33, 122.27, 120.45, 115.15, 55.06 ppm. MALDI-TOF (m/z), calculated for: 509.612, found: 509.169. Anal. calcd for: C37H23N3 (%): C, 87.20; H, 4.55; N, 8.25; found: C, 87.34; H, 4.61; N, 8.13.
Results and discussion
Preparation and characterization
The synthesis routes to SAIP, SAIQ, and SABIQ are outlined in Scheme 1. The key precursors 1,292,36 and 337 were synthesized according to literature methods that provided good yields. Then the ketones underwent nucleophilic addition with 2-lithium-N,N-diphenylaniline to give the corresponding tertiary alcohols. The subsequent acid catalyzed cyclization produced the target products SAIP, SAIQ, and SABIQ in moderate yields. In addition, 1H and 13C nuclear magnetic resonance (1H/13C NMR), mass spectrometry (MS) and elemental analysis (EA) were utilized to further validate the structures of SAIP, SAIQ, and SABIQ.
 |
| | Scheme 1 Synthetic routes to SAIP, SAIQ, and SABIQ. | |
Thermal properties
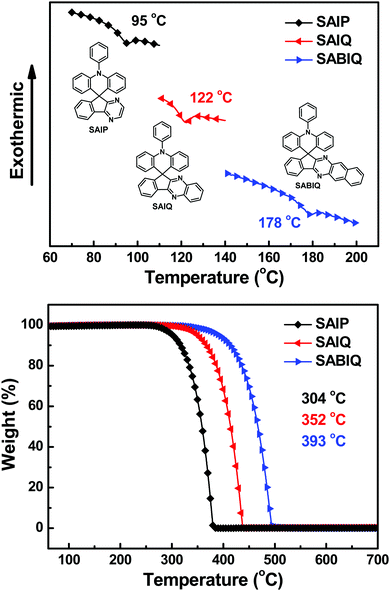
The thermal properties of SAIP, SAIQ, and SABIQ were investigated by differential scanning calorimetry (DSC) and thermogravimetric analysis (TGA) in nitrogen. The experimental results are shown in Fig. 4 and the detailed data are listed in Table 1. The glass transition temperatures (Tgs) and decomposition temperatures (Tds, 5 wt% weight loss) were found to be 95, 122 and 178 °C and 304, 352 and 393 °C for SAIP, SAIQ, and SABIQ, respectively. The good thermal properties of these three materials might be due to the rigid and bulky molecular platforms of the spiro-structures. It is worth noting that the nearly 100% weight loss at 381 °C, 439 °C, and 495 °C for SAIP, SAIQ, and SABIQ, respectively, could be due to the sublimation of these compounds.
 |
| | Fig. 4 DSC and TGA curves of SAIP, SAIQ, and SABIQ. | |
Table 1 Summary of the physical properties of SAIP, SAIQ, and SABIQ
| Molecule |
Abs λmaxa |
PL λmaxa |
T
g
/Tdc |
E
g
|
E
T
|
HOMOf |
LUMOg |
| [nm] |
[nm] |
[°C] |
[eV] |
[eV] |
[eV] |
[eV] |
|
Measured in dichloromethane solution at room temperature.
T
g: glass transition temperature.
T
d: decomposition temperature.
E
g: optical band gap energies were calculated from the corresponding absorption onset in dichloromethane solution.
E
T: measured in a toluene glass matrix at 77 K.
HOMO levels were calculated from CV data.
LUMO levels were calculated from the HOMOs and Egs.
|
|
SAIP
|
321 |
478 |
95/304 |
3.58 |
2.71 |
−5.33 |
−1.75 |
|
SAIQ
|
348, 366 |
539 |
122/352 |
3.30 |
2.47 |
−5.33 |
−2.03 |
|
SABIQ
|
374, 394 |
570 |
178/393 |
2.92 |
2.38 |
−5.34 |
−2.42 |
Photophysical properties
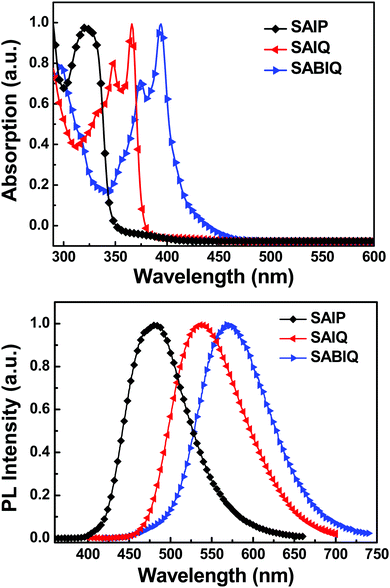
The UV-Vis absorption and photoluminescence (PL) spectra of SAIP, SAIQ, and SABIQ were recorded at room temperature in a dichloromethane (DCM) solution, and phosphorescence (Phos) spectra were recorded at 77 K in a toluene glass matrix (Fig. 5 and Fig. S1, ESI†). Their photophysical results are summarized in Table 1. According to the UV-Vis absorption spectra, the maximum absorption peaks were observed at 321 nm, 348/366 nm, and 374/394 nm for SAIP, SAIQ, and SABIQ, respectively. In addition, the energy gaps (Egs) are calculated based on the absorption edge, which are 3.58 eV, 3.30 eV, and 2.92 eV for SAIP, SAIQ, and SABIQ, respectively. On the other hand, we can clearly see that the PL spectra of SAIP, SAIQ, and SABIQ are broader and structureless, and their emission peaks were at 478 nm, 539 nm, and 570 nm, respectively. In contrast, the Phos spectra of SAIP, SAIQ, and SABIQ are well-defined (Fig. S1, ESI†), and the corresponding triplet energies (ETs) were determined from the first emission peaks in the Phos spectra to be 2.71 eV for SAIP, 2.47 eV for SAIQ, and 2.38 eV for SABIQ. It is noteworthy that from pyrazine to quinoxaline to benzo[g]quinoxaline, the π-conjugation systems are extending gradually, which make a direct contribution to the red-shift of their absorption and emission spectra, and the narrowing of their Egs. In addition, the molecular configuration of SAIP resembles that of 10-phenyl-10H-spiro[acridine-9,9′-fluorene] (SAF),38 but the triplet energy of SAIP is not as high as that of SAF (2.87 eV).38c The replacement of benzene with pyrazine could be the reason for the reduction in the triplet state energy,39 but the introduction of the electron-withdrawing group might be beneficial for achieving a more balanced charge transport characteristic, thus resulting in an efficient PHOLED.35
 |
| | Fig. 5 UV-Vis absorption and PL spectra of SAIP, SAIQ, and SABIQ. | |
In an attempt to understand the electronic structures of the excited states of SAIP, SAIQ, and SABIQ, natural transition orbital (NTO) analysis was used to study the nature of singlet (S1) and triplet (T1) excitations of these molecules.40 As shown in Fig. S2 (ESI†), the holes of the S1 states of SAIP, SAIQ, and SABIQ were mainly localized at the acridine unit. However, the particles of these three molecules were confined to the electron acceptors, 9H-indeno[1,2-b]pyrazine, 11H-indeno[1,2-b]quinoxaline, and 13H-benzo[g]indeno[1,2-b]quinoxaline, which indicate the charge transfer (CT) characteristics of the S1 states of these compounds. The solvation effect of SAIP, SAIQ, and SABIQ could support their intramolecular charge transfer (ICT) features (Fig. S3, ESI†).41 Furthermore, for the T1 state, the holes and particles of SAIP and SAIQ were similar to those of S1, but the holes were partially delocalized over pyrazine to a certain extent, which exhibited CT and locally excited (LE) hybrid state characteristics. However, the holes and particles of SABIQ were mainly localized at the benzo[g]quinoxaline motif, which demonstrated an LE state characteristic.
Electrochemical properties and theoretical calculations
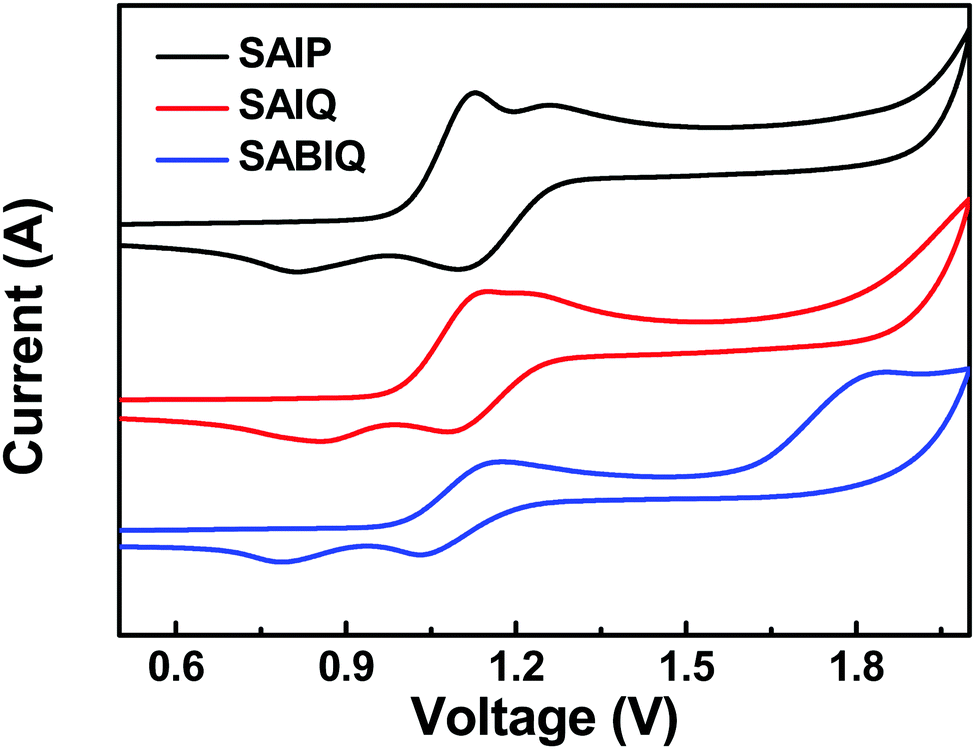
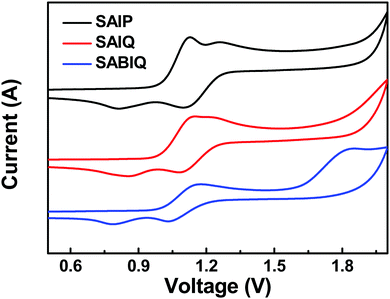
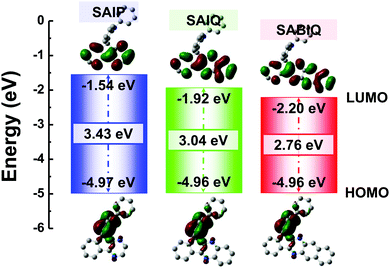
The HOMO and LUMO energy levels of a host play an important role in device fabrication and optimization. Therefore, we investigated the electrochemical behaviors of SAIP, SAIQ, and SABIQ by cyclic voltammetry (CV). The HOMO energy levels can be estimated from the onset of oxidation waves. As shown in Fig. 6, the electrochemical behaviors of SAIP, SAIQ, and SABIQ were alike at the onset of the oxidation waves around 1.0 V. Therefore, SAIP, SAIQ, and SABIQ exhibited very similar HOMO energy levels. On the other hand, the LUMO energy levels of SAIP (−1.75 eV), SAIQ (−2.03 eV), and SABIQ (−2.42 eV) can be calculated by subtracting the optical energy gaps (Egs) from the corresponding HOMO energy levels. Furthermore, we also studied the frontier molecular orbital (FMO) electronic distributions and the energy levels of SAIP, SAIQ, and SABIQ using density function theory (DFT) calculations at the B3LYP/6-31G(d) level. As illustrated in Fig. 7, the HOMOs were mainly distributed on the electron rich acridine motifs, which led to SAIP, SAIQ, and SABIQ having almost the same HOMO energy level. The LUMOs reside over the diaza-containing motifs in these compounds. From SAIP to SAIQ and to SABIQ, the Egs decrease gradually with the extension of molecular π-conjugation, and the LUMO energy levels decrease as well.
 |
| | Fig. 6 Cyclic voltammograms of SAIP, SAIQ, and SABIQ. | |
 |
| | Fig. 7 Frontier molecular orbital distributions and energy levels of SAIP, SAIQ, and SABIQ. | |
Electroluminescence properties
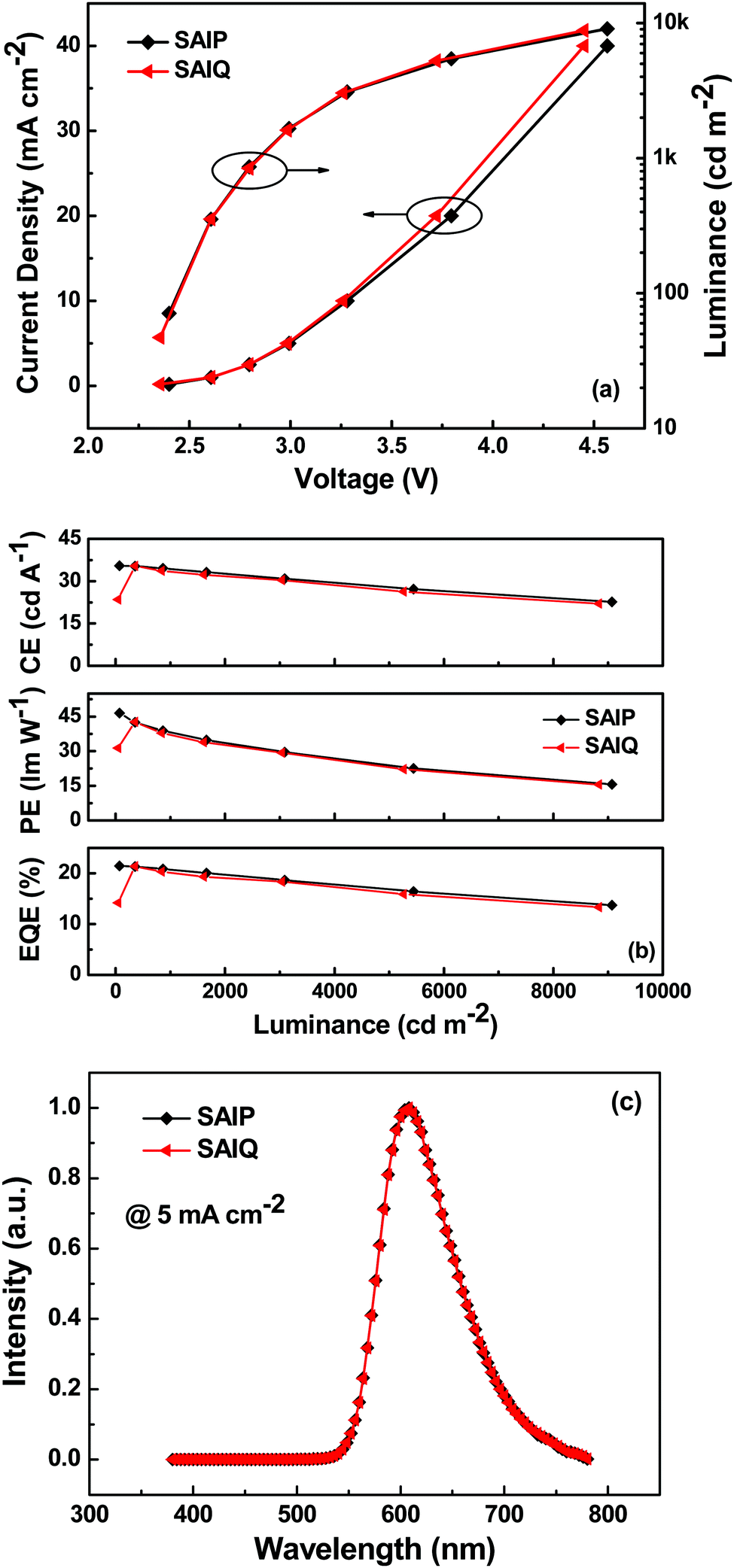
In order to investigate the electroluminescence (EL) properties of these novel spirocycles, blue, green and red PHOLEDs were fabricated with the following device configurations: ITO/HAT-CN (10 nm)/TAPC (55 nm)/TCTA (10 nm)/SAIP: 10 wt% FIrpic (20 nm)/B4PyMPM (45 nm)/Liq (2 nm)/Al (120 nm) (blue device, denoted as B1), ITO/HAT-CN (10 nm)/TAPC (55 nm)/SAIP or SAIQ: 10 wt% Ir(ppy)2(acac) (20 nm)/B4PyMPM (40 nm)/Liq (2 nm)/Al (120 nm) (green device, denoted as G1 or G2), and ITO/HAT-CN (10 nm)/TAPC (45 nm)/TCTA (10 nm)/SAIP or SAIQ: 6 wt% Ir(MDQ)2(acac) (20 nm)/TmPyPB (45 nm)/Liq (2 nm)/Al (120 nm) (red device, denoted as R1 or R2). Fig. 8, 9 and Fig. S5 (ESI†) show the current density–voltage–luminance (J–V–L) characteristics, current efficiency (CE)/power efficiency (PE)/external quantum efficiency (EQE)–L curves, and EL spectra of the corresponding PHOLEDs. These device parameters are listed in Table 2.
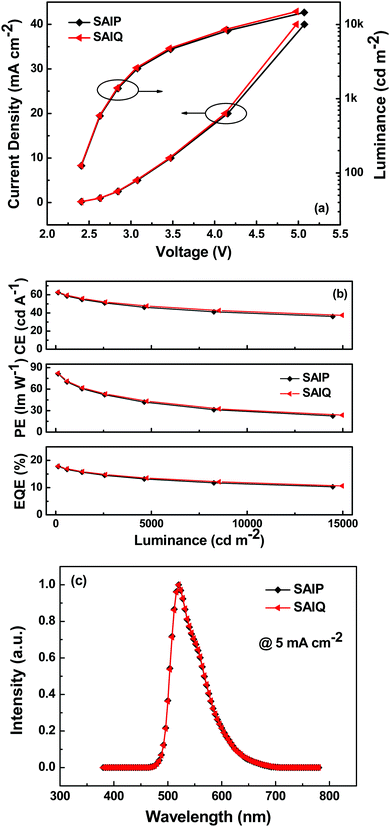 |
| | Fig. 8 (a) J–V–L characteristics, (b) CE–, PE–, and EQE–L curves, and (c) EL spectra of the green devices. | |
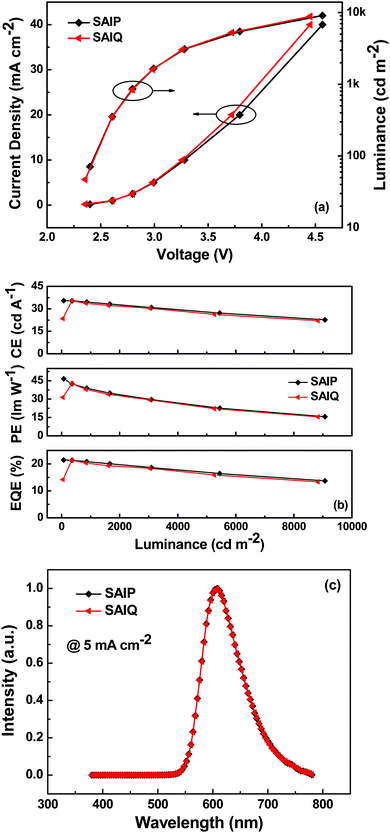 |
| | Fig. 9 (a) J–V–L characteristics, (b) CE–, PE–, and EQE–L curves, and (c) EL spectra of the red devices. | |
Table 2 Electroluminescence characteristics of the blue, green, and red devices
| Devicea |
Host |
V
|
η
CE
|
η
PE
|
EQEc |
CIEd |
| [V] |
[cd A−1] |
[lm W−1] |
[%] |
[x, y] |
|
Device configuration: B1: ITO/HAT-CN (10 nm)/TAPC (55 nm)/TCTA (10 nm)/SAIP: 10 wt% FIrpic (20 nm)/B4PyMPM (45 nm)/Liq (2 nm)/Al (120 nm); G1/G2: ITO/HAT-CN (10 nm)/TAPC (55 nm)/SAIP or SAIQ: 10 wt% Ir(ppy)2(acac) (20 nm)/B4PyMPM (40 nm)/Liq (2 nm)/Al (120 nm); and R1/R2: ITO/HAT-CN (10 nm)/TAPC (45 nm)/TCTA (10 nm)/SAIP or SAIQ: 6 wt% Ir(MDQ)2(acac) (20 nm)/TmPyPB (45 nm)/Liq (2 nm)/Al (120 nm).
Voltages at 200 cd m−2.
Efficiencies in the order of the maxima and at 500 cd m−2.
Commission Internationale de l’Eclairage coordinates measured at 5 mA cm−2.
|
| B1 |
SAIP
|
2.6 |
22.0, 18.7 |
28.0, 20.9 |
11.0, 9.4 |
0.16, 0.33 |
| G1 |
SAIP
|
2.5 |
62.5, 59.7 |
81.5, 71.8 |
17.8, 16.7 |
0.32, 0.63 |
| G2 |
SAIQ
|
2.5 |
63.0, 60.6 |
82.3, 73.6 |
17.9, 17.4 |
0.32, 0.63 |
| R1 |
SAIP
|
2.5 |
35.5, 35.1 |
46.5, 41.7 |
21.5, 21.2 |
0.61, 0.39 |
| R2 |
SAIQ
|
2.5 |
35.4, 35.0 |
42.6, 41.2 |
21.4, 21.0 |
0.61, 0.39 |
The green and red devices based on SAIP and SAIQ exhibited very similar device performances. More specifically, the SAIP and SAIQ based green and red devices showed relatively low driving voltages (2.5 V) at a brightness of 200 cd m−2. Furthermore, the green and red devices also demonstrated relatively high device efficiencies. The maximum CE/PE/EQE achieved were 62.5 cd A−1/81.5 lm W−1/17.8% and 63.0 cd A−1/82.3 lm W−1/17.9% for the SAIP and SAIQ hosted green devices, respectively. The maximum CE, PE, and EQE could reach 35.5/35.4 cd A−1, 46.5/42.6 lm W−1, and 21.5%/21.4% for the SAIP/SAIQ based red devices, respectively. The Commission Internationale de l’Eclairage (CIE) coordinates of the SAIP and SAIQ based green and red devices are (0.32, 0.63) and (0.61, 0.39), respectively, which are consistent with those of the reported Ir(ppy)2(acac)42 and Ir(MDQ)2(acac)43 doped devices. For the SAIP and SAIQ hosted devices, there are some factors that can account for their similar device performance. The high ETs of SAIP and SAIQ would result in an efficient energy transfer process from the host to the dopant, and prevent reverse energy transfer from the dopant to the host.44 Moreover, suitable HOMO/LUMO energy levels of SAIP and SAIQ would reduce the hole and electron injection barrier, thus lowering the device driving voltages and achieving a high PE. In addition, the good thermal stabilities of SAIP and SAIQ will be favorable for the device operation stability. In addition, the PLQYs of Ir(PPy)2(acac) (10 wt%) and Ir(MDQ)2(acac) (6 wt%) in SAIP/SAIQ were measured to be 66%/69% and 75%/74%, respectively, which were consistent with the observed EQEs for the SAIP (green, 17.8%; red, 21.5%) and SAIQ (green, 17.9%; red, 21.4%) hosted devices. These results further suggest that SAIP and SAIQ could effectively confine the triplet energies of green and red dopants within devices. On the other hand, hole-only and electron-only devices were fabricated to study the charge transport properties of SAIP and SAIQ with the following device structures: hole-only devices, ITO/MoO3 (10 nm)/SAIP or SAIQ (100 nm)/MoO3 (10 nm)/Al (100 nm); electron-only: ITO/TmPyPB (20 nm)/SAIP or SAIQ (100 nm)/TmPyPB (20 nm)/Liq (2 nm)/Al (100 nm) (Fig. 10). Both SAIP and SAIQ demonstrated the bipolar transport character with similar hole and electron mobilities, which could account in part for their similar device performance. In addition, the hole- and electron-mobility of SAIP and SAIQ were estimated according to the literature method45 to be μSAIP,h = 6.2 × 10−5 cm V−1 s−1, μSAIP,e = 1.3 × 10−7 cm V−1 s−1; μSAIQ,h = 5.7 × 10−5 cm V−1 s−1, and μSAIQ,e = 3.2 × 10−7 cm V−1 s−1. Thus, detailed studies of the relationships between molecular structures, physical/chemical properties, and device performance are important. As shown in Fig. S5 (ESI†), the performance of the SAIP hosted blue device was relatively low compared with those of the corresponding green and red devices. The maximum CE, PE, and EQE were 22.0 cd A−1, 28.0 lm W−1, and 11.0%, respectively. This result is not satisfactory, probably due to the small ET difference between SAIP (2.71 eV) and the blue dopant FIrpic (2.62 eV),31b which could favor the reverse energy transfer from the dopant to the host, although the emission from the host was not observed in the EL spectrum.
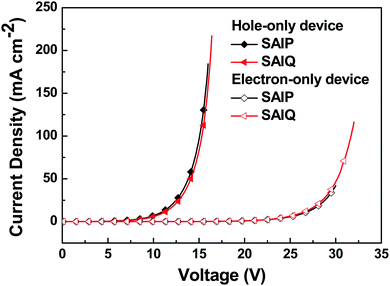 |
| | Fig. 10 Hole- and electron-only devices based on SAIP and SAIQ. | |
Conclusions
In conclusion, three novel spirocycle compounds (SAIP, SAIQ, and SABIQ) containing an electron donating acridine unit and an electron withdrawing pyrazine segment were designed and synthesized. The different π-conjugation lengths of the acceptor units led to the different photophysical properties of SAIP, SAIQ, and SABIQ. Our results show that the green and red PHOLEDs exhibit a similar and relatively high device performance with those of SAIP and SAIQ as host materials. The maximum EQE of the green and red devices could exceed 17% and 21%, respectively. Correspondingly, we hope our study will expand the application of 1,4-diazafluorenone and its analogues in the field of OLED materials and shed new light on the development of efficient host materials.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We acknowledge financial support from the National Key R&D Program of China (2016YFB0400703), the National Natural Science Foundation of China (21472135, 61307036), the Natural Science Foundation of Jiangsu Province of China (BK20151216), and the 111 Project. This project is also funded by the Collaborative Innovation Center of Suzhou Nano Science and Technology (CIC-Nano), Soochow University and by the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD).
References
-
(a) N. S. Prostakov, Usp. Khim., 1969, 38, 1710 (
Russ. Chem. Rev.
, 1969
, 38
, 774
) CrossRef CAS;
(b) C. Mayor and C. Wentrup, J. Am. Chem. Soc., 1975, 97, 7464 CrossRef;
(c) N. S. Prostakov, A. T. Soldatenkov, N. M. Kolyadina and A. A. Obynochnyi, Russ. Chem. Rev., 1997, 66, 121 CrossRef.
-
(a) O. Doebner and P. Kuntze, Ann. Chem., 1988, 249, 109 CrossRef;
(b) O. Doebner and J. Peters, Ber. Dtsch. Chem. Ges., 1890, 23, 1228 CrossRef;
(c) R. Oberkobusch, Ber., 1953, 86, 975 CrossRef CAS;
(d) K. Kloc, J. Mlochowski and Z. Szulc, J. Prakt. Chem., 1977, 319, 959 CrossRef CAS.
-
(a) V. Petrov, J. Saper and B. Sturgeon, J. Chem. Soc., 1949, 2134 RSC;
(b) J. N. Chatterjea, S. C. Shaw and S. N. Singh, Indian J. Chem., 1978, 55, 149 CAS;
(c) E. Y. Ozola and G. Y. Vanag, Khim. Geterotsikl. Soedin., 1969, 5, 103 (
Chem. Heterocycl. Compd.
, 1969
, 5
, 82
) Search PubMed;
(d) Y. E. Pelcher, A. K. Aren, Z. A. Bomika and G. Y. Vanag, Khim. Geterotsikl. Soedin., 1969, 5, 305 (
Chem. Heterocycl. Compd.
, 1969
, 5
, 232
) Search PubMed.
-
(a) B. A. Vigante, Y. Ozols, E. M. Belash and Y. I. Beilis, Khim. Geterotsikl. Soedin., 1984, 2, 210 (
Chem. Heterocycl. Compd.
, 1984
, 20
, 170
) Search PubMed;
(b) Y. Ozols, B. A. Vigante and G. Y. Dubur, Geterotsikl. Soedin., 1994, 1603 (
Chem. Heterocycl. Compd.
, 1994
, 20
, 1386
) CAS;
(c) R. Ulrich, J. Heterocycl. Chem., 1990, 27, 237 CrossRef.
-
(a) J. Beger and W. Triebs, Ann. Chem., 1962, 652, 204 CrossRef CAS;
(b) N. S. Prostakov, L. M. Kirillova, D. Pkhal'gumani, L. A. Shakhparonova and V. P. Zvolinskii, Khim. Geterotsikl. Soedin., 1967, 3, 1068 (
Chem. Heterocycl. Compd.
, 1967
, 3
, 832
) Search PubMed;
(c) N. S. Prostakov, O. I. Sorokin and A. Y. Ismailov, Khim. Geterotsikl. Soedin., 1967, 3, 674 (
Chem. Heterocycl. Compd.
, 1967
, 3
, 541
) Search PubMed;
(d) M.-J. Shiao, C.-J. Peng, J.-S. Wang and Y.-T. Ma, J. Chin. Chem. Soc., 1992, 39, 173 CrossRef CAS;
(e) A.-S. Rebstock, F. Mongin, F. Trécourt and G. Quéguiner, Tetrahedron, 2003, 59, 4973 CrossRef CAS;
(f) D. Tilly, A.-S. Castanet and J. Mortier, Tetrahedron Lett., 2006, 47, 1121 CrossRef CAS.
- J. T. Plati, A. K. Ingberman and W. Wenner, J. Org. Chem., 1957, 22, 2615 CrossRef.
-
(a) B. F. Bowden, K. Picker, R. Ritchie and W. C. Taylor, Aust. J. Chem., 1975, 28, 2681 CrossRef CAS;
(b) J. C. Powers and I. Ponticello, J. Am. Chem. Soc., 1968, 90, 7102 CrossRef CAS PubMed;
(c) M. J. Shiao, K. H. Liu and P. Y. Lin, Heterocycles, 1993, 36, 507 CrossRef CAS;
(d) M. T. Du Priest, C. L. Schmidt, D. Kuzmich and S. B. Willams, J. Org. Chem., 1986, 51, 2021 CrossRef CAS.
- M. Alessi, A. L. Larkin, K. A. Ogilvie, L. A. Green, S. Lai, S. Lopez and V. Snieckus, J. Org. Chem., 2007, 72, 1588 CrossRef CAS PubMed.
- W. H. Mills, W. H. Palmer and M. G. Tomkinson, J. Chem. Soc., 1924, 2364 Search PubMed.
- R. H. Prager, C. Tsopelas and T. Heisler, Aust. J. Chem., 1991, 44, 227 Search PubMed.
- L. M. Yagupolskii, K. I. Petko, A. N. Retchitsky and I. I. Maletina, J. Fluorine Chem., 1994, 67, 5 CrossRef CAS.
- R. C. Fuson and J. J. Miller, J. Am. Chem. Soc., 1957, 79, 3477 CrossRef CAS.
- E. P. Kyba, S.-T. Liu, K. Chockalingam and B. R. Reddy, J. Org. Chem., 1988, 53, 3513 CrossRef CAS.
- J. W. Lockner, D. D. Dixon, R. Risgaard and P. S. Baran, Org. Lett., 2011, 13, 5628 CrossRef CAS PubMed.
-
(a) D. Mueller, R. A. Davis, S. Duffy, V. M. Avery, D. Camp and R. J. Quinn, J. Nat. Prod., 2009, 72, 1538 CrossRef CAS PubMed;
(b) S. Dhara, A. Ahmed, S. Nandi, S. Baitalik and J. K. Ray, Tetrahedron Lett., 2013, 54, 63 CrossRef CAS;
(c) N. Marquise, V. Dorcet, F. Chevallier and F. Mongin, Org. Biomol. Chem., 2014, 12, 8138 RSC;
(d) N. Marquise, P. J. Harford, F. Chevallier, T. Roisnel, A. E. H. Wheatley, P. C. Gros and F. Mongin, Tetrahedron Lett., 2013, 54, 3154 CrossRef CAS;
(e) N. Marquise, P. J. Harford, F. Chevallier, T. Roisnel, V. Dorcet, A.-L. Gagez, S. Sable, L. Picot, V. Thiery, A. E. H. Wheatley, P. C. Gros and F. Mongin, Tetrahedron, 2013, 69, 10123 CrossRef CAS.
- J. K. Laha, K. P. Jethava and S. Patel, Org. Lett., 2015, 17, 5890 CrossRef CAS PubMed.
- J. K. Laha, K. P. Jethava, S. Patel and K. V. Patel, J. Org. Chem., 2017, 82, 76 CrossRef CAS PubMed.
-
(a) S.-J. Su, C. Cai and J. Kido, Chem. Mater., 2011, 23, 274 CrossRef CAS;
(b) S.-J. Su, C. Cai and J. Kido, J. Mater. Chem., 2012, 22, 3447 RSC;
(c) D. Chen, S.-J. Su and Y. Cao, J. Mater. Chem. C, 2014, 2, 9565 RSC.
-
(a) Q. Yan, E. Gin, M. G. Banwell, A. C. Willis and P. D. Carr, J. Org. Chem., 2017, 82, 4328 CrossRef CAS PubMed;
(b) Y. Im and J. Y. Lee, Chem. Commun., 2013, 49, 5948 RSC;
(c) T. Motoyama, H. Sasabe, Y. Seino, J. Takamatsu and J. Kido, Chem. Lett., 2011, 40, 306 CrossRef CAS;
(d) S. J. Kim, Y. J. Kim, Y. H. Son, J. A. Hur, H. A. Um, J. Shin, T. W. Lee, M. J. Cho, J. K. Kim, S. Joo, J. H. Yang, G. S. Chae, K. Choi, J. H. Kwon and D. H. Choi, Chem. Commun., 2013, 49, 6788 RSC;
(e) E. Varathan, D. Vijay and V. Subramanian, J. Phys. Chem. C, 2014, 118, 21741 CrossRef CAS;
(f) Z. Zhang, J. Xie, Z. Wang, B. Shen, H. Wang, M. Li, J. Zhang and J. Cao, J. Mater. Chem. C, 2016, 4, 4226 RSC;
(g) J. S. Moon, D. H. Ahn, S. W. Kim, S. Y. Lee, J. Y. Lee and J. H. Kwon, RSC Adv., 2018, 8, 17025 RSC.
-
(a) C. W. Lee, J.-K. Kim, S. H. Joo and J. Y. Lee, ACS Appl. Mater. Interfaces, 2013, 5, 2169 CrossRef CAS PubMed;
(b) C. W. Lee and J. Y. Lee, Adv. Mater., 2013, 25, 596 CrossRef CAS PubMed;
(c) M.-M. Xue, C.-C. Huang, Y. Yuan, Y.-X. Zhang, M.-K. Fung and L.-S. Liao, Chem. Commun., 2017, 53, 263 RSC;
(d) J. H. Kim, D. R. Lee, S. H. Han and J. Y. Lee, J. Mater. Chem. C, 2018, 6, 5363 RSC.
- C. W. Lee and J. Y. Lee, Chem. Commun., 2013, 49, 1446 RSC.
-
(a) P. Wu, J. Zhu, Z. Zhang, D. Dou, H. Wang, B. Wei and Z. Wang, Dyes Pigm., 2018, 156, 185 CrossRef CAS;
(b) S. Jeong, C. Kim, S. H. Kim, D. Y. Kim, S. E. Lee, Y. K. Kim and S. S. Yoon, Mol. Cryst. Liq. Cryst., 2018, 660, 24 CrossRef CAS;
(c) H.-F. Chen, W.-Y. Hung, S.-W. Chen, T.-C. Wang, S.-W. Lin, S.-H. Chou, C.-T. Liao, H.-C. Su, H.-A. Pan, P.-T. Chou, Y.-H. Liu and K.-T. Wong, Inorg. Chem., 2012, 51, 12114 CrossRef CAS PubMed;
(d) K.-T. Wong, T.-Y. Hwu, A. Balaiah, T.-C. Chao, F.-C. Fang, C.-T. Lee and Y.-C. Peng, Org. Lett., 2006, 8, 1415 CrossRef CAS PubMed;
(e) M. L. Keshtova, S. I. Pozinb, D. V. Marochkinc, V. P. Perevalovc, P. V. Petrovskiia, I. V. Blagodatskikha and A. A. R. Khokhlova, Dokl. Chem., 2012, 442, 50 CrossRef;
(f) K.-T. Wong, R.-T. Chen, F.-C. Fang, C.-c. Wu and Y.-T. Lin, Org. Lett., 2005, 7, 1979 CrossRef CAS PubMed;
(g) H.-C. Liao, C.-H. Lee, Y.-C. Ho, M.-H. Jao, C.-M. Tsai, C.-M. Chuang, J.-J. Shyue, Y.-F. Chen and W.-F. Su, J. Mater. Chem., 2012, 22, 10589 RSC;
(h) K.-T. Wong, H.-F. Chen and F.-C. Fang, Org. Lett., 2006, 8, 3501 CrossRef CAS PubMed;
(i) Z. Jiang, H. Yao, Z. Zhang, C. Yang, Z. Liu, Y. Tao, J. Qin and D. Ma, Org. Lett., 2009, 11, 2607 CrossRef CAS PubMed;
(j) C. Fan, Y. Chen, P. Gan, C. Yang, C. Zhong, J. Qin and D. Ma, Org. Lett., 2010, 12, 5648 CrossRef CAS PubMed.
- H.-C. Su, F.-C. Fang, T.-Y. Hwu, H.-H. Hsieh, H.-F. Chen, G.-H. Lee, S.-M. Peng, K.-T. Wong and C.-C. Wu, Adv. Funct. Mater., 2007, 17, 1019 CrossRef CAS.
- L.-L. Yang and Q.-C. Liu, Chin. J. Synth. Chem., 2011, 19, 73 CAS.
- A. Mustafa, N. M. Mikhailova, N. I. Golovtsov and N. S. Prostakov, Chem. Heterocycl. Compd., 1992, 28, 1155 CrossRef.
-
(a) J. Druey and P. Schmidt, Helv. Chim. Acta, 1950, 33, 1080 CrossRef CAS;
(b) Y.-Z. Li and G. B. Schuster, J. Org. Chem., 1987, 52, 3975 CrossRef CAS.
-
(a) R. Frédérick, W. Dumont, F. Ooms, L. Aschenbach, C. J. Van der Schyf, N. Castagnoli, J. Wouters and A. Krief, J. Med. Chem., 2006, 49, 3743 CrossRef PubMed;
(b) S. Kneubühler, U. Thull, C. Altomare, V. Carta, P. Gaillard, P.-A. Carrupt, A. Carotti and B. Testa, J. Med. Chem., 1995, 38, 3874 CrossRef;
(c) S. Kneubühler, V. Carta, C. Altomare, A. Carotti and B. Testa, Helv. Chim. Acta, 1993, 76, 1956 CrossRef.
- C. Altomare, S. Cellamare, L. Summo, M. Catto and A. Carotti, J. Med. Chem., 1998, 41, 3812 CrossRef CAS PubMed.
- A. A. C. Takaizumi, F. R. dos Santos, M. T. Silva and J. C. Netto-Ferreira, Quim. Nova, 2009, 32, 1799 CrossRef CAS.
-
(a) Y. Tao, C. Yang and J. Qin, Chem. Soc. Rev., 2011, 40, 2943 RSC;
(b) R. C. Kwong, S. Sibley, T. Dubovoy, M. Baldo, S. R. Forrest and M. E. Thompson, Chem. Mater., 1999, 11, 3709 CrossRef CAS;
(c) C. Adachi, M. A. Baldo, S. R. Forrest, S. Lamansky, M. E. Thompson and R. C. Kwong, Appl. Phys. Lett., 2001, 78, 1622 CrossRef CAS;
(d) C. Adachi, M. A. Baldo, M. E. Thompson and S. R. Forrest, J. Appl. Phys., 2001, 90, 5048 CrossRef CAS;
(e) M. A. Baldo, S. Lamansky, P. E. Burrows, M. E. Thompson and S. R. Forrest, Appl. Phys. Lett., 1999, 75, 4 CrossRef CAS;
(f) S.-J. Su, H. Sasabe, T. I. Takeda and J. Kido, Chem. Mater., 2008, 20, 1691 CrossRef CAS.
-
(a) L.-X. Xiao, Z.-J. Chen, B. Qu, J.-X. Luo, S. Kong, Q.-H. Gong and J. Kido, Adv. Mater., 2011, 23, 926 CrossRef CAS PubMed;
(b) C. Adachi, R. C. Kwong, P. Djurovich, V. Adamovich, M. A. Baldo, M. E. Thompson and S. R. Forrest, Appl. Phys. Lett., 2001, 79, 2082 CrossRef CAS;
(c) R. J. Holmes, S. R. Forrest, Y. J. Tung, R. C. Kwong, J. J. Brown, S. Garon and M. E. Thompson, Appl. Phys. Lett., 2003, 82, 2422 CrossRef CAS.
-
(a) X.-Y. Liu, X. Tang, Y. Zhao, D. Zhao, J. Fan and L.-S. Liao, ACS Appl. Mater. Interfaces, 2018, 10, 1925 CrossRef CAS PubMed;
(b) X.-Y. Liu, X. Tang, Y. Zhao, D. Zhao, J. Fan and L.-S. Liao, J. Mater. Chem. C, 2018, 6, 1023 RSC;
(c) W.-C. Chen, Y. Yuan, Z.-L. Zhu, Z.-Q. Jiang, S.-J. Su, L.-S. Liao and C.-S. Lee, Chem. Sci., 2018, 9, 4062 RSC;
(d) Y.-K. Wang, S.-H. Li, S.-F. Wu, C.-C. Huang, S. Kumar, Z.-Q. Jiang, M.-K. Fung and L.-S. Liao, Adv. Funct. Mater., 2018, 28, 1706228 CrossRef;
(e) D. Liu, F. Wang and R. Yao, J. Mater. Chem. C, 2018, 6, 7839 RSC.
-
(a) K.-H. Kim, S. Lee, C.-K. Moon, S.-Y. Kim, Y.-S. Park, J.-H. Lee, J. W. Lee, J. Huh, Y. You and J.-J. Kim, Nat. Commun., 2014, 5, 4769 CrossRef CAS PubMed;
(b) C.-J. Shih, C.-C. Lee, T.-H. Yeh, S. Biring, K. K. Kesavan, N. R. Al Amin, M.-H. Chen, W.-C. Tang, S.-W. Liu and K.-T. Wong, ACS Appl. Mater. Interfaces, 2018, 10, 24090 CrossRef CAS PubMed;
(c) Y. Wang, W. Wang, Z. Huang, H. Wang, J. Zhao, J. Yu and D. Ma, J. Mater. Chem. C, 2018, 6, 7042 RSC;
(d) D. Zhang, J. Qiao, D. Zhang and L. Duan, Adv. Mater., 2017, 1702847 CrossRef PubMed;
(e) M. Bian, D. Zhang, Y. Wang, Y.-H. Chung, Y. Liu, H. Ting, L. Duan, Z. Chen, Z. Bian, Z. Liu and L. Xiao, Adv. Funct. Mater., 2018, 1800429 CrossRef.
-
(a) W.-Y. Hung, T.-C. Wang, H.-C. Chiu, H.-F. Chen and K.-T. Wong, Phys. Chem. Chem. Phys., 2010, 12, 10685 RSC;
(b) H.-F. Chen, T.-C. Wang, W.-Y. Hung, H.-C. Chiu, C. Yun and K.-T. Wong, J. Mater. Chem., 2012, 22, 9658 RSC.
-
(a) M. Romain, D. Tondelier, O. Jeannin, B. Geffroy, J. Rault-Berthelot and C. Poriel, J. Mater. Chem. C, 2015, 3, 9701 RSC;
(b) S. Thiery, D. Tondelier, B. Geffroy, O. Jeannin, J. Rault-Berthelot and C. Poriel, Chem. – Eur. J., 2016, 22, 10136 CrossRef CAS PubMed.
-
(a) S. Ruhemann, J. Chem. Soc., 1910, 97, 1438 RSC;
(b) B. D. Pearson, R. A. Mitsch and N. H. Cromwell, J. Org. Chem., 1962, 27, 1674 CrossRef CAS;
(c) F. D. Popp, J. Heterocycl. Chem., 1972, 9, 1399 CrossRef CAS;
(d) L. W. Deady, J. Desneves and A. C. Ross, Tetrahedron, 1993, 49, 9823 CrossRef CAS.
- C. Zhang, S. Li, L. Ji, S. Liu, Z. Li, S. Li and X. Meng, Bioorg. Med. Chem. Lett., 2015, 25, 4693 CrossRef CAS PubMed.
-
(a) C.-J. Lin, H.-L. Huang, M.-R. Tseng and C.-H. Cheng, J. Disp. Technol., 2009, 5, 236 CAS;
(b) C.-Y. Chan, Y.-C. Wong, M.-Y. Chan, S.-H. Cheung, S.-K. So and V. W.-W. Yam, ACS Appl. Mater. Interfaces, 2016, 8, 24782 CrossRef CAS PubMed;
(c) W.-C. Chen, Y. Yuan, Z.-L. Zhu, S.-F. Ni, Z.-Q. Jiang, L.-S. Liao, F.-L. Wong and C.-S. Lee, Chem. Commun., 2018, 54, 4541 RSC;
(d) W. Sun, N. Zhou, Y. Xiao, S. Wang and X. Li, Chem. – Asian J., 2017, 12, 3069 CrossRef CAS PubMed.
-
(a) X.-Y. Liu, F. Liang, Y. Yuan, Z.-Q. Jiang and L.-S. Liao, J. Mater. Chem. C, 2016, 4, 7869 RSC;
(b) X.-Y. Liu, F. Liang, L.-S. Cui, X.-D. Yuan, Z.-Q. Jiang and L.-S. Liao, Chem. – Asian J., 2015, 10, 1402 CrossRef CAS PubMed.
-
(a) Z. Chen, Z. Wu, F. Ni, C. Zhong, W. Zeng, D. Wei, K. An, D. Ma and C. Yang, J. Mater. Chem. C, 2018, 6, 6543 RSC;
(b) C. Li, X. Fan, C. Han and H. Xu, J. Mater. Chem. C, 2018, 6, 6747 RSC.
-
(a) N. Mataga, Y. Kaifu and M. Kaizumi, Bull. Chem. Soc. Jpn., 1956, 29, 465 CrossRef CAS;
(b) N. G. Bakhshiev, M. I. Knyazhanskii, V. I. Minkin, O. A. Osipov and G. V. Saidov, Russ. Chem. Rev., 1969, 38, 740 CrossRef;
(c) Y. Zhang, K. Wang, G. Zhuang, Z. Xie, C. Zhang, F. Cao, G. Pan, H. Chen, B. Zou and Y. Ma, Chem. – Eur. J., 2015, 21, 2474 CrossRef CAS PubMed.
- S. Lamansky, P. Djurovich, D. Murphy, F. Abdel-Razzaq, H. E. Lee, C. Adachi, E. Paul, P. E. Burrows, S. R. Forrest and M. E. Thompson, J. Am. Chem. Soc., 2001, 123, 4304 CrossRef CAS PubMed.
- J. P. Duan, P. P. Sun and C. H. Cheng, Adv. Mater., 2003, 15, 224 CrossRef CAS.
- J. Zhao, G.-H. Xie, C.-R. Yin, L.-H. Xie, C.-M. Han, R.-F. Chen, H. Xu, M.-D. Yi, Z.-P. Deng, S.-F. Chen, Y. Zhao, S.-Y. Liu and W. Huang, Chem. Mater., 2011, 23, 5331 CrossRef CAS.
-
(a) B. Wang, X. Lv, J. Tan, Q. Zhang, Z. Huang, W. Yi and L. Wang, J. Mater. Chem. C, 2016, 4, 8473 RSC;
(b) J. Tan, B. Wang, Z. Huang, X. Lv, W. Yi, S. Zhuang and L. Wang, J. Mater. Chem. C, 2016, 4, 5222 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8tc05462b |
| ‡ These two authors contributed equally to this paper. |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 * and
Jian
Fan
* and
Jian
Fan