
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhysical vapour deposition of cyanine salts and their first application in organic electronic devices†
Donatas
Gesevičius
ab,
Antonia
Neels
 c,
Léo
Duchêne
d,
Erwin
Hack
e,
Jakob
Heier
*a and
Frank
Nüesch
af
c,
Léo
Duchêne
d,
Erwin
Hack
e,
Jakob
Heier
*a and
Frank
Nüesch
af
aLaboratory for Functional Polymers, Swiss Federal Laboratories for Materials Science and Technology, Empa, Überlandstrasse 129, Dübendorf, Switzerland. E-mail: jakob.heier@empa.ch
bInstitute of Chemical Sciences and Engineering, ISIC, Ecole Polytechnique Fédérale de Lausanne, EPFL, Station 6, CH-1015 Lausanne, Switzerland
cCenter for X-ray Analytics, Swiss Federal Laboratories for Materials Science and Technology, Empa, Überlandstrasse 129, Dübendorf, Switzerland
dLaboratory Materials for Energy Conversion, Swiss Federal Laboratories for Materials Science and Technology, Empa, Überlandstrasse 129, Dübendorf, Switzerland
eLaboratory for Transport at Nanoscale Interfaces, Swiss Federal Laboratories for Materials Science and Technology, Empa, Überlandstrasse 129, Dübendorf, Switzerland
fInstitut des Matériaux, Ecole Polytechnique Fédérale de Lausanne, EPFL, Station 6, CH-1015 Lausanne, Switzerland
First published on 18th December 2018
Abstract
A symmetrical trimethine indocyanine dye chromophore was modified with the bistriflylimide anion to study the changes in Coulomb interactions, thermal stability and the volatility of the organic salt. The bulky bistriflylimide anion minimizes electrostatic interactions and increases the vapour pressure of the compound resulting in an increased volatility of the organic salt. First examples of entirely vacuum-processed bulk heterojunction organic photovoltaic devices show the proof of concept. We give evidence that the well dispersed negative charge of the bistriflylimide introduces quasi gas phase conditions to the cyanine chromophore. This overcomes the existing restriction of thin film formation via spincasting in cyanine dye based organic electronics and expands the thin film fabrication process choices towards physical vapour deposition.
1. Introduction
Organic electronic devices have an over 60 year long history of intense research.1,2 Besides the fundamental understanding of electronic processes in organic semiconductor devices, proper layer formation by suitable deposition techniques plays a crucial role in obtaining a successfully working device. In academic research wet film formation processes3–8 are most popular and especially spincasting9 is established as a fast technique to form thin films of small area. Despite its popularity, spincasting has several drawbacks such as little control over the desired film geometry. In addition, the resulting film quality strongly depends on substrate surface wetting of the solution. Organic electronic devices in research often consist of several thin layers with similar solubility, making fabrication of a multistack device via spincasting a challenging task. However, industry requires large area homogenous thin film layers, high precision in geometry control and long term stability, which are usually achieved by vacuum deposition techniques.8,10–15The majority of small molecule organic semiconductors are neutral.16 However, indolenium–cyanine dyes belong to the class of organic salts, with the characteristic presence of an electron accepting quaternary ammonia group (antiauxochrome) as well as an electron donating ternary amino moiety (auxochrome) which are connected via a polymethine chain. Such a geometry creates a fully delocalised charge over the polymethine backbone within the cyanine limit and enables many of their attractive optical properties, such as large extinction coefficients.17–19 Since their discovery in 185620 cyanine dyes were used in various applications, according to the scientific interest in each decade starting from photography,20 CD-R based data storage,21–24 life science,25,26 non-linear optics27,28 and organic photovoltaics (OPV) or photodiodes.17,29–39
While neutral organic semiconductors can be easily evaporated mainly due to the weak intermolecular van der Waals forces, ionic organic functional materials such as cyanine dyes suffer from strong Coulomb interactions which results in high melting points and decomposition at ambient temperatures.40,41 Heteroatoms in the polymethine chain or in aromatic moieties as well as high molecular weight of the cyanine salts and counterions with localised charge induce additional decomposition pathways when thermal stress is applied.24 These drawbacks prevented successful implementation of physical vapour deposition techniques in cyanine dye based semiconductor research.
Weakly coordinating anions as the key to improve both chromophore stability in the excited state and organic electronic device performance are emerging topics.37,42 The influence of a weakly coordinating anion on the volatility of the cyanine salts was not reported in the literature so far. Our work presents guidelines for a successful design of evaporable cyanine organic salts with the aim to open future research possibilities regarding the synthesis of evaporable cyanine salts, organic electronic device fabrication and optimisation as well as theoretical calculations on new emerging thermodynamic properties. Besides the fundamental interest in overcoming present limitations and making a cyanine salt thermally stable but volatile enough to be evaporated before decomposition occurs, we demonstrate first applications in organic electronic devices where thin film formation had taken place by a physical vapour deposition based fabrication method.
2. Results and discussion
2.1. From material synthesis to optical and electrochemical properties
The bistriflylimide (TFSI) anion was introduced into 1-ethyl-2-[3-(1-ethyl-1,3-dihydro-3,3-dimethyl-2H-indol-2-ylidene)-1-propen-1-yl]-3,3-dimethyl-3H-indolium iodide (Cy3I), exchanging the partially redox active iodide anion from the starting material by using an Amberlyst® A26 (OH− form) wet anion exchange resin. The detailed working principles and experimental setup were reported elsewhere.42 For simplicity the dye shall be denoted as Cy3TFSI (Fig. 1 and Fig. S1, ESI†) in the following. | ||
| Fig. 1 On the left: Cartoon depicting the molecular structure of the Cy3TFSI salt with the corresponding energy level alignment respective possible device fabrication. On the right: Qualitative UV-vis spectra of evaporated thin films on different substrates. The films have different average thicknesses. | ||
Four different substrates were chosen and compared to the solution spectrum to investigate the UV-vis properties of vapour deposited Cy3TFSI films with respect to further application in organic electronic devices (Fig. 1 and Fig. S2, ESI†). Cy3TFSI has an absorbance maximum in acetonitrile solution attributed to the 0 → 0 transition at 544 nm with a shoulder shifted by 1182 cm−1 towards shorter wavelengths at 512 nm due to a dominant vibronic mode resulting from the 0 → 1 or 0 → 2 transition. The absorbance peak spans a range between 455 and 567 nm. With a relative molar extinction coefficient of 2.40 × 105 L mol−1 cm−1 in acetonitrile solution the dye manifests strong light absorption which is most beneficial to light harvesting in thin films as required for OPV devices (Fig. S3 and Table S1, ESI†). Depending on the substrate used the solid state absorbance broadens up to 1302 cm−1 for glass, 2269 cm−1 for MoO3, 3401 cm−1 for PEDOT:PSS and 4371 cm−1 for mesoporous TiO2. The blue shifted shoulder grows and becomes a dominant species on TiO2 (1366 cm−1) and MoO3 (1496 cm−1) substrates with a measured ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.91 and 1:0.87 respectively. The opposite effect is observed on glass with a shift of 1531 cm−1 and PEDOT:PSS (1405 cm−1) showing a ratio between the shoulder and the main peak of 0.92:1 and 0.7:1, respectively. Such differences in peak/shoulder ratios, absorbance peak shapes and shoulder shifts indicate differences in the thin film morphology. A film with large thickness variations could cause the observed peak broadening in the UV-vis spectrum due to geometrical reasons and points either towards dewetted or rough surfaces. Dimerization and aggregation effects induced by solid state packing of the molecules could contribute as well to shoulder formation at shorter wavelengths. In general the optical properties of vapour deposited molecules are similar to those of spin cast films.43
:0.91 and 1:0.87 respectively. The opposite effect is observed on glass with a shift of 1531 cm−1 and PEDOT:PSS (1405 cm−1) showing a ratio between the shoulder and the main peak of 0.92:1 and 0.7:1, respectively. Such differences in peak/shoulder ratios, absorbance peak shapes and shoulder shifts indicate differences in the thin film morphology. A film with large thickness variations could cause the observed peak broadening in the UV-vis spectrum due to geometrical reasons and points either towards dewetted or rough surfaces. Dimerization and aggregation effects induced by solid state packing of the molecules could contribute as well to shoulder formation at shorter wavelengths. In general the optical properties of vapour deposited molecules are similar to those of spin cast films.43
The Cy3TFSI dye belongs to the class of intermediate band gap semiconductors possessing an electrochemical energy gap of 1.86 eV with intermediate electron affinity and high positive oxidation potential (Fig. S4 and Table S4, ESI†). The TFSI anion is not redox active within the electrochemical window of the chromophore. Fig. 1 depicts the relative energy levels of the possible organic electronic device interlayers. With a HOMO of −5.71 eV and a LUMO of −3.85 eV, Cy3TFSI can be employed as an electron donor in both standard geometry with the hole collecting layer on the substrate anode as well as in inverted geometry with the electron collecting layer on the substrate cathode.
2.2. Factors enabling physical vapour deposition
Physical vapour deposition under vacuum occurs either by sublimation of a solid or by evaporation of a molten substance. Which process takes place is determined by the triple point of the compound. Both mechanisms require a high thermal stability of the compounds. The combined methods of thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC) reveal a melt based evaporation process for the Cy3TFSI dye (Fig. 2). Considering the melting peak onset values, Cy3TFSI has a melting point of 156 °C compared to 165 °C observed for the starting material. Despite the larger molecular weight of Cy3TFSI, the melting point is lowered by 10 °C proving that the bulky anion with its dispersed negative charge reduces intermolecular chromophore interactions. Similar findings regarding the influence of Coulomb interactions on ion pair evaporation were mentioned in the literature on ionic liquids.44 Significant 2% mass losses were obtained at 280 °C for Cy3TFSI while the starting material reaches this onset already at 252 °C. A low melting point and an increased thermal stability favour successful evaporation of an organic salt by widening the thermal window until the decomposition temperature is reached. However, the thermal degradation mechanism is not trivial as it is not sufficient to assign an initial mass loss to its decomposition point, especially for a volatile compound. Additionally, the literature on ionic liquids points towards significant differences in mass loss mechanisms driven either by vaporisation or decomposition or even simultaneous occurrence of both depending on ultra-high vacuum (UHV), high vacuum (HV) or ambient pressure conditions.45 Despite material class differences and a different focus of this work we try to adopt some of the experiments from the literature and reveal first insights into the evaporation mechanism of cyanine salts. While the starting material shows a two-step mass loss in dynamic TGA (Fig. 2) with relative integrals of 46 and 42% up to the range of 450 °C and two endothermal peaks at 265 and 283 °C (peak maxima) in the DSC suggesting a two-step degradation, Cy3TFSI has a more complex TGA curve (Fig. 2). Three well distinguished steps can be identified with relative integrals of 19, 26 and 42% accompanied by a sharp exothermal DSC peak at 285 °C (peak maximum).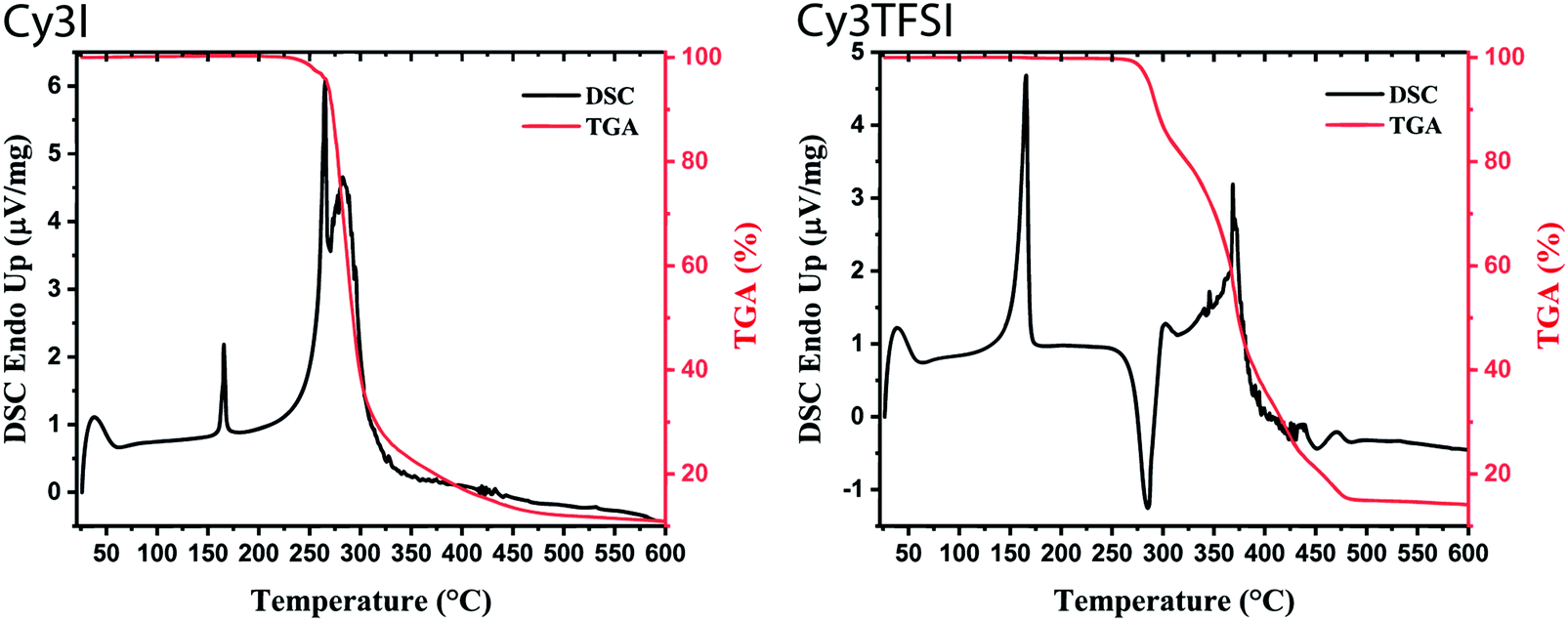 | ||
| Fig. 2 Dynamic TGA (10 °C min−1) with the corresponding DSC signal of starting material (Cy3I) and Cy3TFSI. | ||
Evidence that the evaporated Cy3TFSI that comes to rest on the substrate and the starting material are identical is given by UV-vis spectroscopy. The solution spectrum of a re-dissolved film matches the solution spectra of the starting material. Differently, with Cy3I also the half-dye is observed (Fig. S5, ESI†).
The information content of a dynamic TGA measurement is limited regarding the evaporation mechanism of a compound. To extract the enthalpy of vaporisation  at an average temperature as a physical value describing the evaporation process, a quasi-isothermal TGA measurement was performed (ESI,† Thermal behaviour section).46Fig. 3 and Fig. S6 and S7 (ESI†) display the differences in mass loss of each isothermal step for Cy3I and Cy3TFSI. An evaporation process is expected to have a defined vapour pressure at a given temperature and thus behaves linearly over time while decomposition should occur spontaneously with inconsistent vapour pressure showing a non-linear mass loss over time. The extracted slopes of the isothermal weight loss behave nonlinearly for both materials as a function of temperature. Cy3TFSI possesses a wide linear region from 220–250 °C which can be attributed to the evaporation process. The exponential increase in mass loss of the starting material can be attributed to the simultaneous occurrence of partial evaporation and decomposition mainly visible from the UV-vis spectra of, respectively, heated samples at a particular temperature (Fig. S8, ESI†). Approaching a temperature range above 250 °C both materials show a DSC activity. With the exothermal DSC peak onset at 269 °C for Cy3TFSI the mass loss decreases sharply reaching a local minimum at 270 °C pointing towards partial decomposition forming non-volatile degradation products as visible in the UV-vis spectrum (Fig. S8, ESI†). The unpredictable mass loss decrease over time especially during isothermal steps at 250 and 260 °C with the corresponding broad endothermal DSC peak onset at 260 °C and random distribution of dm/dt vs. T indicate that Cy3I undergoes unpredictable thermal decomposition only (Fig. 3 and Fig. S6–S8, ESI†). The decomposition only pathway is supported by observing a 75% total weight loss for Cy3I compared to only 27% obtained for Cy3TFSI during the measurement. This points towards the assumption that Cy3I forms volatile decomposition species while Cy3TFSI is more stable and decomposes only partially by forming less volatile decomposition species.
at an average temperature as a physical value describing the evaporation process, a quasi-isothermal TGA measurement was performed (ESI,† Thermal behaviour section).46Fig. 3 and Fig. S6 and S7 (ESI†) display the differences in mass loss of each isothermal step for Cy3I and Cy3TFSI. An evaporation process is expected to have a defined vapour pressure at a given temperature and thus behaves linearly over time while decomposition should occur spontaneously with inconsistent vapour pressure showing a non-linear mass loss over time. The extracted slopes of the isothermal weight loss behave nonlinearly for both materials as a function of temperature. Cy3TFSI possesses a wide linear region from 220–250 °C which can be attributed to the evaporation process. The exponential increase in mass loss of the starting material can be attributed to the simultaneous occurrence of partial evaporation and decomposition mainly visible from the UV-vis spectra of, respectively, heated samples at a particular temperature (Fig. S8, ESI†). Approaching a temperature range above 250 °C both materials show a DSC activity. With the exothermal DSC peak onset at 269 °C for Cy3TFSI the mass loss decreases sharply reaching a local minimum at 270 °C pointing towards partial decomposition forming non-volatile degradation products as visible in the UV-vis spectrum (Fig. S8, ESI†). The unpredictable mass loss decrease over time especially during isothermal steps at 250 and 260 °C with the corresponding broad endothermal DSC peak onset at 260 °C and random distribution of dm/dt vs. T indicate that Cy3I undergoes unpredictable thermal decomposition only (Fig. 3 and Fig. S6–S8, ESI†). The decomposition only pathway is supported by observing a 75% total weight loss for Cy3I compared to only 27% obtained for Cy3TFSI during the measurement. This points towards the assumption that Cy3I forms volatile decomposition species while Cy3TFSI is more stable and decomposes only partially by forming less volatile decomposition species.
 | ||
| Fig. 3 The upper row represents extracted weight losses as a function of time occurring during each isothermal step. The lower row represents the extracted slopes of the isothermal weight loss curves as a function of temperature. | ||
Applying the developed model for ionic liquids we obtain a value for 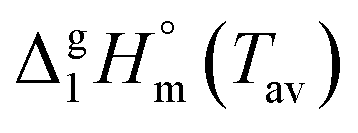 of 152.8 kJ mol−1 for Cy3TFSI within a temperature range of 220–250 °C (Tables S5–S7 and Fig. S9, ESI†).46 With this approach we provide first experimental results quantifying the thermal evaporation process of Cy3TFSI which will be helpful in deeper future thermodynamic investigations.
of 152.8 kJ mol−1 for Cy3TFSI within a temperature range of 220–250 °C (Tables S5–S7 and Fig. S9, ESI†).46 With this approach we provide first experimental results quantifying the thermal evaporation process of Cy3TFSI which will be helpful in deeper future thermodynamic investigations.
In summary it can be concluded that the literature of thermogravimetric experiments of cyanine dyes has to be revised. The thermal behaviour of cyanine dyes containing weakly coordinating anions is more complicated than assigning the first significant mass loss to a decomposition temperature. Furthermore, the thermal behaviour under ambient pressure may strongly deviate from the conditions inside a HV evaporation chamber, making conclusions even more complex.
The thermal properties of an organic salt are closely linked to its molecular packing as well as with the strength of electrostatic interactions in the solid state. Therefore we link the crystal structures of Cy3TFSI and Cy3I to the physical vapour deposition phenomenon and define the lattice energy as a key parameter. Single crystals were obtained by cooling saturated ethanol solutions. The starting material crystallizes in a monoclinic lattice with the space group P21/c while Cy3TFSI belongs to the orthorhombic system with the P212121 space group. The shortest observed anion coordination distances of 7.65 Å for Cy3TFSI and 5.07 Å for Cy3I in Fig. 4 give qualitative insights into reduced electrostatic interactions within Cy3TFSI. Relative lattice energies of 359.09 kJ mol−1 for Cy3TFSI and 376.93 kJ mol−1 for Cy3I and the Coulomb interactions of 0.67 eV for Cy3TFSI and 0.77 eV for Cy3I, respectively, were estimated according to a literature method (Tables S8 and S9, ESI†).47 The estimated lattice and Coulomb energy values are well below the ones obtained for inorganic molecular ions as well as smaller organic ionic liquids and visualise well the strong influence of the bulky bistriflylimide on the thermal stability of the Cy3TFSI chromophore.
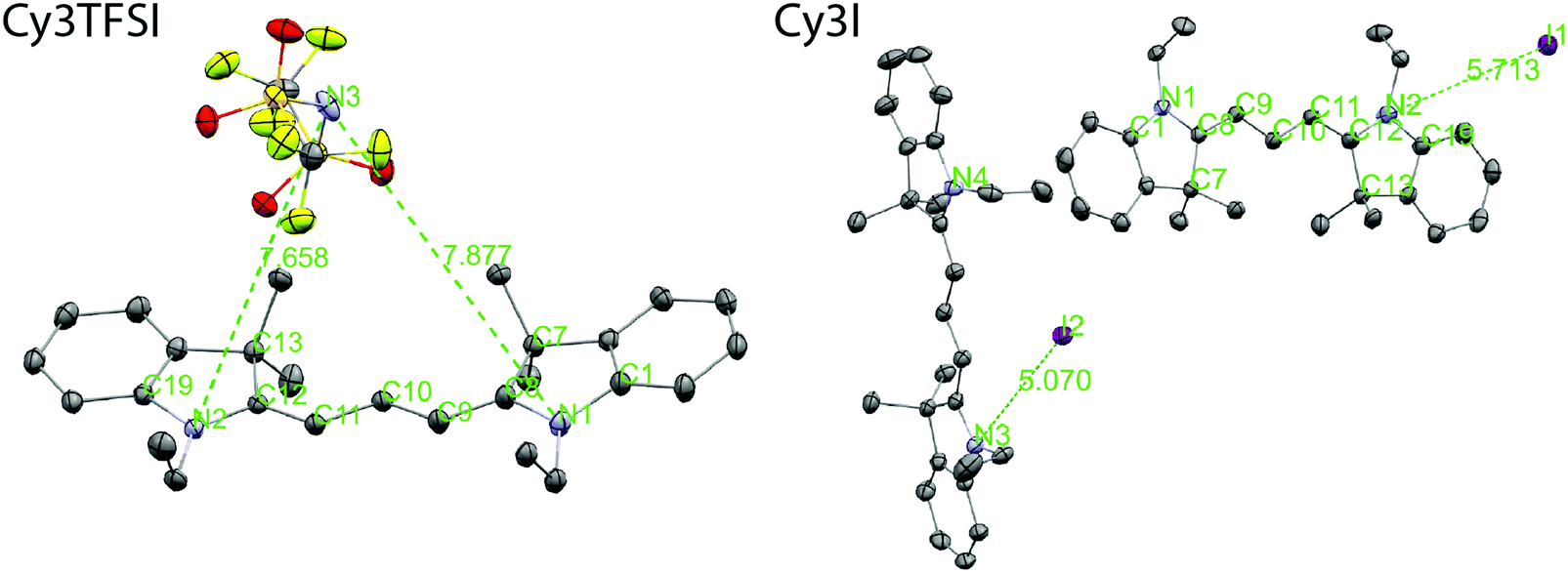 | ||
| Fig. 4 Molecular structure and anion coordination environment observed in the single crystal structures of Cy3I and Cy3TFSI. The shortest anion–chromophore distances are given in Å. The thermal ellipsoids represent 50% probability levels. The disorder in Cy3TFSI as well as the hydrogen atoms are omitted for clarity. Color code: C: grey, N: violet, S: yellow, O: red, F: yellow-green, I: purple. | ||
The bistriflylimide anion prevents short distance intermolecular chromophore interactions since no regular packing motif can be observed (Fig. 5). In contrast the chromophores in Cy3I are packed in two symmetry equivalents; see Fig. 5: the blue layers form a plane that sandwiches the cross bone packed yellow symmetry equivalent layer. The iodide anions are closely distributed along the plane of blue chromophores pointing towards complex shared anion–cation interactions within the two symmetry operation separated layers as observed by the highly asymmetric iodide–chromophore coordination distance (Fig. 4). Despite the well-ordered symmetry close π-stacking or long range heteroatom interactions are prevented, since the closest chromophore distances exceed 5 Å. The packing motifs are in agreement with the 10 °C difference in the melting point between Cy3TFSI and Cy3I.
 | ||
| Fig. 5 Molecular arrangement over 2 × 2 × 2 unit cells with views along the a axis (left) and b axis (right). For clarity the Cy3TFSI packing is depicted in the wire frame model, while Cy3I contains the chromophore in the wire frame and the iodide as a thermal ellipsoid with a 50% probability level. The disorder in Cy3TFSI as well as the hydrogen atoms are omitted for clarity. Color code: Cy3TFSI symmetry equivalent chromophores: red, anion: blue. Cy3I symmetry equivalent chromophores: blue and green, iodide: red or purple. | ||
2.3. Substrate dependent thin film morphology
The concept of “physical vapour deposited cyanine organic electronics” was proven in devices with standard geometry containing hole transport layers such as MoO3 or PEDOT:PSS as well as inverted geometry devices with compact and mesoporous TiO2, using commercially available indium-tin-oxide (ITO) on glass as a substrate. Thin film formation during vacuum deposition is mainly determined by the surface energy of the substrate, substrate temperature and the evaporation rate which allows the formulation of different growth mechanisms.48 Depending on the substrate used, significantly different morphologies of the Cy3TFSI dye were obtained (Fig. 6). The glass substrate leads to the formation of well isolated up to 2 micrometre sized islands of 70 nm height (Fig. S14, ESI†). It provides also the roughest surface of the investigated thin films, such droplet formation results from dewetting phenomena. MoO3 provides a better surface energy match by firstly adsorbing a thin dye layer followed by an irregularly shaped micrometre sized island formation indicating possible continuous film formation at higher thicknesses but with increased surface roughness comparable to a mechanism formulated by Stranski–Krastanov.48 When PEDOT:PSS is employed, the amorphous film with low overall surface roughness forms over areas of more than 10 μm2. However, on a larger scale dewetting holes up to 6 μm in diameter (Fig. S14, ESI†) as well as local dye droplet formation on the amorphous bottom layer is observed, indicating an additional abrupt organic material erosion from the crucible. The high surface energy mismatch can be bypassed when naturally rough mesoporous TiO2 with large surface volume is used. The atomic force microscopy (AFM) scanned area reveals spherical particles smaller than 500 nm in diameter close to native mesoporous TiO2 morphology suggesting a continuous dye coverage of the TiO2 particles. Together with MoO3 the investigated dye film on mesoporous TiO2 provides the smallest surface roughness (Fig. S14, ESI†).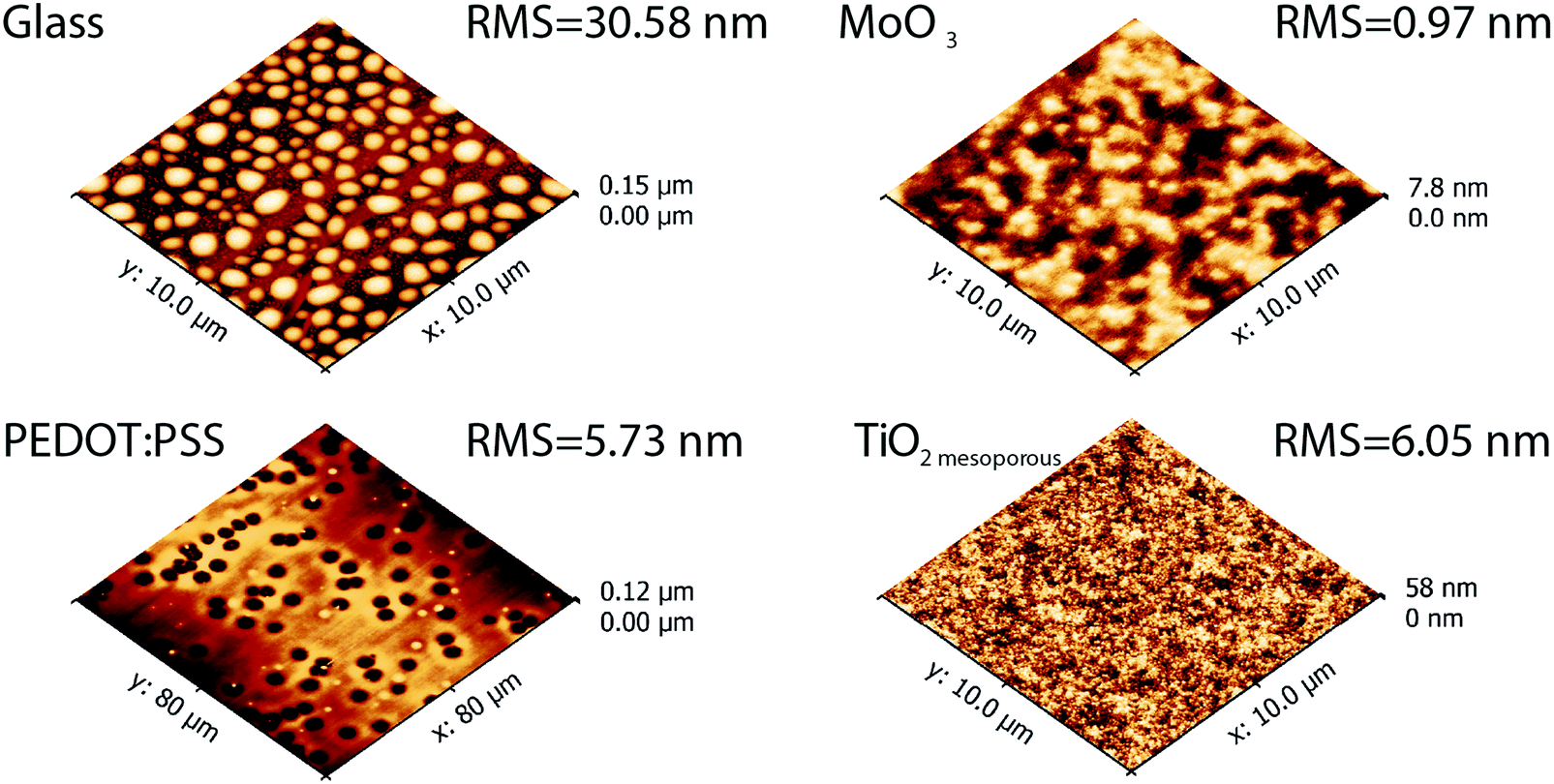 | ||
| Fig. 6 Topography of vacuum deposited Cy3TFSI films on different substrates. | ||
We can assume that the films formed after the melt based evaporation are amorphous in nature. In this sense our evaporation process is different from a sublimation process. Neither AFM nor UV-vis spectroscopy points to the presence of crystalline domains.
We here also want to point to a drawback of films processed from solution. We had observed earlier that these films contain considerable amounts of residual solvents.49 This leads to increased mobility of dye molecules and may account for the ability of the molecules in the film to crystallize over time. We also demonstrated that the solvent molecules have a significant influence on electronic processes at interfaces in solar cells.
Further trials to form a bulk heterojunction by co-evaporation of Cy3TFSI and fullerene C60 blends on MoO3 and compact TiO2 substrates (Fig. 7) have led to a better surface energy match. The surface roughness in the order of 2 nm in both blends is low and should allow applications in either standard or inverted geometry bulk heterojunction devices. The surface profile exhibits mixed random amorphous features suggesting suppressed phase separation below 1 μm in diameter. A spinodal pattern is recognisable on the TiO2 substrate. However, local organic material accumulation ascending several nanometres from the surface can also be observed in both blends, mainly caused by an abrupt Cy3TFSI ejection from the crucible.
 | ||
| Fig. 7 Topography of co-evaporated Cy3TFSI and C60 on TiO2 and MoO3 substrates. | ||
Solution processed blend films of cyanine dyes and fullerenes on the other hand frequently phase separate into large scale structures by liquid–liquid dewetting.50 Here co-evaporating Cy3TFSI and a fullerene leads to a more favourable morphology for solar cell application than the spincoated samples (see Section 4.2. Device fabrication).
In summary, wetting phenomena observed during spincasting from solution also apply to evaporated cyanine films making extended film tuning on different substrates an important issue. The substrate coverage can be improved by blending Cy3TFSI or incorporating it as a dopant into a carrier matrix for OLED application overcoming the surface energy mismatch.
2.4. Proof of concept in organic electronic devices
The Cy3TFSI salt enables a wide variety of electronic device fabrication methods not seen in cyanine dye semiconductors so far. First proof-of-concept devices are presented with pros and cons regarding their further perspectives. With an open circuit voltage of 1 V, a solution based bilayer device of Cy3TFSI and C60 shows one of the highest potentials among all organic semiconductors (Table S13, ESI†). One of the major existing limitations in cyanine dye semiconductors for organic photovoltaic applications was the so far only poorly performing bulk-heterojunction morphology.51 By blending cyanine dyes with fullerene based electron acceptors in solution, typically micrometre sized phase separated domains of pure cyanine/fullerene rich phases form as demonstrated in Fig. S15 (ESI†). Such a morphology leads to short circuited organic photovoltaic devices (Table S14, ESI†). However our new approach of simultaneous dye/fullerene evaporation has led to the first vacuum processed cyanine dye based bulk heterojunction device.Cy3TFSI was employed as an electron donor material in a fully vacuum processed inverted geometry bulk heterojunction OPV device. In total 32 cells with 8 cells per substrate were prepared. 13 cells showed short circuit behaviour, therefore 19 cells could be measured. Fig. 8 represents the best performing device of the batch. The OPV device exhibits a dark current of 0.34 mA cm−2 at −0.8 V. One origin of the dark current could be alternative pathways through the thin active layer formed by a continuous electron acceptor channel ranging from the hole collecting layer to TiO2. This could partially explain the relative low fill factor (FF) of 41.5% and the open circuit voltage (Voc) of 0.67 V in the device. The relatively low Voc points also towards the not optimal morphology of the active layer missing the well-defined phase separated networks between the dye and electron acceptor in the order of 10–20 nm. The AFM analysis in Fig. 7 shows the absence of micrometre sized phase separated domains in freshly prepared blends, which typically would occur when cyanine dye fullerene blends are coated from solution. However, a repeated measurement after 3 weeks of storage in the glovebox shows several micrometre sized dendritic crystals (Fig. S16, ESI†) pointing towards ongoing kinetic processes, driven by dye crystallization in the blend.
 | ||
| Fig. 8 JV and EQE curves of the best performing OPV cell. The EQE represents a sharp C60 peak at shorter wavelengths followed by an overlap of the second C60 absorption peak with Cy3TFSI. | ||
Cy3TFSI proves itself as a strong absorber capable of generating a sufficient short circuit current density (Jsc) of 4.02 mA cm−2 since an active layer thickness of only 10 nm is used. The values lead to a champion performance (η) of 0.96%. However, the experimentally obtained Jsc for the best device does not match the calculated Jsc of 0.90 mA cm−2 from the external quantum efficiency (EQE) spectrum. This deviation originates mainly from differences in the two measurements and can only be compared when no light intensity related effects play a role. In fact, while the average Voc of 0.63 ± 0.06 V and FF of 38.98 ± 2.41% are stable for all 19 cells, the Jsc of 1.46 ± 0.98 mA cm−2 shows a large variation between minima and maxima. The Jsc calculated from the EQE spectrum matches the measured average value much better, pointing towards quick degradation mechanisms within the best cell. This effect also leads to a broad distribution in an average device performance of 0.36 ± 0.25%. The EQE data prove that both components of the blend, Cy3TFSI and C60, contribute to the photovoltaic effect with an external quantum efficiency of 8%. The contribution of the dye to the external quantum efficiency, however, is less than that of C60 suggesting either an extraction barrier for holes formed at the heterojunction/MoO3 surface or a partial chromophore degradation or is simply the result of the very thin active layer used. Furthermore, the co-evaporation of Cy3TFSI and C60 prevents aggregate formation of the cyanine dye since a peak/shoulder ratio of 1:0.55 is comparable to the one obtained from acetonitrile solution although the absorbance of C60 may overlap with the shoulder at shorter wavelengths of Cy3TFSI.
Further vacuum processed devices should demonstrate the introduced flexibility by Cy3TFSI in fabricating varying device stacks. The bilayer geometry cells realized by physical vapour deposition suffer from bad morphology of the neat evaporated films as discussed in Fig. 6. A further promising device stack is the inverted bilayer cell (Fig. S17–S19 and Table S16, ESI†). With a high Voc in the range of 0.8 V, the inverted bilayer device approaches Voc values also found in a solution spin-cast bilayer reference device. The regular geometry bulk heterojunction device cannot compete with the implemented inverse structure. Furthermore the solid state dye sensitized solar cell approach seems to be less promising from all trials.
3. Conclusions
By employing the bistriflylimide anion with a well dispersed negative charge over seven atoms into the 1-ethyl-2-[3-(1-ethyl-1,3-dihydro-3,3-dimethyl-2H-indol-2-ylidene)-1-propen-1-yl]-3,3-dimethyl-3H-indolium chromophore we introduce volatility and thermal stability to an organic salt of high molecular weight. The properties of the solid state structure from single crystals and thermo-analytical methods were linked to reveal the evaporation mechanism of the dye. The lattice energy and enthalpy of vaporization were identified to be the key parameters of the first volatile organic cyanine salt. We give evidence that a weakly coordinating anion tailored for a specific chromophore regarding the number of atoms for dispersed negative charge can introduce quasi gas phase conditions to the chromophore. The semiconducting properties of the cyanine salt allowed its application in organic electronic devices as a light absorbing or emitting layer. The proof of concept was demonstrated by fabricating fully vacuum deposited OPV devices. This work aims to pave the way to development strategies of cyanine based volatile organic salts covering the full visible and infrared spectral absorption range.4. Experimental section
4.1. Materials and methods
All chemicals and solvents were purchased from commercial sources (Aldrich, VWR, FEW Chemicals, Kurt J. Lesker) and used as received, unless otherwise stated. Reactions were carried out under an air atmosphere using common lab glass ware. NMR multicore spectra were recorded on a Bruker AV-400 spectrometer (1H-NMR: 400 MHz, 13C{1H}-NMR: 100 MHz, 19F{1H}-NMR: 377 MHz). Chemical shifts (δ) are reported in ppm (parts per million) with the solvent residual signal (1H/13C{1H}: 7.20/79 for CDCl3) as a reference. J coupling constants are given in Hz. Multiplicities are reported as singlet (s), doublet (d), triplet (t), quartet (q), and multiplet (m). Elemental analysis data were obtained from the Micro Laboratory of ETH Zürich with the instrument Leco TruSpec Micro for C, H, N, S, F and O, while I was determined by ion chromatography. Thermal gravimetric analysis and differential scanning calorimetry were performed on a Netzsch TG 209 F1 and Netzsch STA449 F3 Jupiter, respectively.4.2. Device fabrication and characterisation
Glass/ITO substrates were cleaned in acetone (VWR, 99.5% GPR RECTAPUR), isopropanol (VWR, EMPLURA), ethanol (VWR, 99.5% AnalaR NORMAPUR®), detergent (Hellmanex® III, 2 wt% water solution) and finally washed four times with deionised water. Subsequently a precursor sol yielding a 50 nm thick TiO2 layer was spincast according to a literature protocol.52 The vacuum deposited bulk heterojunction was fabricated by adjusting a constant rate of 0.12 Å s−1 from the C60 source followed by evaporation of Cy3TFSI at 128 °C with a combined rate of 0.24 Å s−1. Finally after stabilization of the deposition rate the substrate shutter was opened to deposit the bulk heterojunction layer by co-evaporation. Evaporation of MoO3 (99.97%) as the hole transport layer and silver (99.99%) as the top electrode followed immediately after with a deposition rate of 0.1 Å s−1. The pressure in the evaporation chamber did not exceed 6 × 10−6 mbar during all evaporation steps. The OPV cell areas were defined as 3.1 and 7.1 mm2 by using a mask for cathode deposition. Further details about device fabrication can be found in the ESI.† The solar cells were characterised under an inert gas atmosphere on a calibrated solar simulator (Spectra Nova) using a Xe lamp with 100 mW cm−2 simulated AM1.5G solar irradiation. The light intensity was adjusted using a calibrated silicon reference cell from Rera Solutions. External quantum efficiency measurements were performed on a SpeQuest RR-2100.4.3. Further characterisation
UV-vis spectra were measured on a Varian Cary 50. Measurement of n and k was performed using a spectroscopic ellipsometer M2000-VI (J. A. Woollam). The AFM analysis was performed on a scanning probe microscope Bruker Icon3 in the tapping mode. Cyclic voltammetry (CV) measurements were performed on a PGStat 30 potentiostat (Autolab) using a three cell electrode system (Au working electrode, Pt counter electrode and an Ag/AgCl reference electrode). Single crystal diffraction patterns were recorded on a Stoe Mark II-Imaging Plate Diffractometer System (Stoe & Cie GmbH, Darmstadt, Germany) equipped with a graphite-monochromator. Data collection was performed at −100 °C using Mo-Kα radiation (λ = 0.71073 Å). The structures were solved by direct methods using the program SHELXS and refined by full matrix least squares on F2 with SHELXL.53 The hydrogen atoms were included in calculated positions and treated as riding atoms using SHELXL-97 default parameters.53 All non-hydrogen atoms were refined anisotropically. All cartoons depicted in this work as well as chromophore–anion distances were prepared and measured using MERCURY CSD 3.9.54 CCDC 1834037 and 1834038 contain the supplementary crystallographic data for Cy3I and Cy3TFSI, respectively.† Detailed information about the background of the described experiments can be found in the ESI.†Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The use of the Scanning Probe Microscopy User Laboratory at Empa is gratefully acknowledged. We thank the Swiss National Science Foundation for financial support under grant numbers 160116 and 152909.References
- N. Karl, Mol. Cryst. Liq. Cryst. Incorporating Nonlinear Opt., 1989, 171, 31–51 CrossRef.
- S. R. Forrest, IEEE J. Sel. Top. Quantum Electron., 2000, 6, 1072–1083 CAS.
- D. Angmo, T. T. Larsen-Olsen, M. Jørgensen, R. R. Søndergaard and F. C. Krebs, Adv. Energy Mater., 2013, 3, 172–175 CrossRef CAS.
- M. Hösel, D. Angmo, R. R. Søndergaard, G. A. dos Reis Benatto, J. E. Carlé, M. Jørgensen and F. C. Krebs, Adv. Sci., 2014, 1, 1400002 CrossRef PubMed.
- R. R. Søndergaard, M. Hösel and F. C. Krebs, J. Polym. Sci., Part B: Polym. Phys., 2013, 51, 16–34 CrossRef.
- F. C. Krebs, Sol. Energy Mater. Sol. Cells, 2009, 93, 394–412 CrossRef CAS.
- H. Youn, H. J. Park and L. J. Guo, Small, 2015, 11, 2228–2246 CrossRef CAS PubMed.
- N. T. Kalyani and S. J. Dhoble, Renewable Sustainable Energy Rev., 2012, 16, 2696–2723 CrossRef.
- K. Norrman, A. Ghanbari-Siahkali and N. B. Larsen, Annu. Rep. Prog. Chem., Sect. C: Phys. Chem., 2005, 101, 174 RSC.
- D. M. Taylor, Semicond. Sci. Technol., 2015, 30, 054002 CrossRef.
- H. Sasabe and J. Kido, Chem. Mater., 2011, 23, 621–630 CrossRef CAS.
- F. So, J. Kido and P. Burrows, MRS Bull., 2008, 33, 663–669 CrossRef CAS.
- C. D. Dimitrakopoulos and P. R. L. Malenfant, Adv. Mater., 2002, 14, 99–117 CrossRef CAS.
- M. A. Baldo, D. F. O’Brien, Y. You, A. Shoustikov, S. Sibley, M. E. Thompson and S. R. Forrest, Nature, 1998, 395, 151–154 CrossRef CAS.
- M. A. Baldo, S. Lamansky, P. E. Burrows, M. E. Thompson and S. R. Forrest, Appl. Phys. Lett., 1999, 75, 4 CrossRef CAS.
- J. Roncali, P. Leriche and P. Blanchard, Adv. Mater., 2014, 26, 3821–3838 CrossRef CAS PubMed.
- D. Saccone, S. Galliano, N. Barbero, P. Quagliotto, G. Viscardi and C. Barolo, Eur. J. Org. Chem., 2016, 2244–2259 CrossRef CAS.
- J. L. Bricks, A. D. Kachkovskii, Y. L. Slominskii, A. O. Gerasov and S. V. Popov, Dyes Pigm., 2015, 121, 238–255 CrossRef CAS.
- V. Z. Shirinian and A. A. Shimkin, in Heterocyclic Polymethine Dyes, ed. L. Strekowski, Springer, Berlin, Heidelberg, 2008, pp. 75–105 Search PubMed.
- C. H. G. Williams, Trans. – R. Soc. Edinburgh, 1856, 21, 377 CrossRef.
- K. Y.-J. Liao, W.-Y. Huang, C.-L. Huang, D.-R. Chiang, D.-Y. Hu, A. T. Lee and S.-j. Ye, Preparation of cyanine dye for high density optical recording disk, US Pat. US5958087 A, 1999 Search PubMed.
- H. Tian and F. Meng, in Functional Dyes, ed. S.-H. Kim, Elsevier, Amsterdam, 2006, pp. 47–84 Search PubMed.
- J. Tan, D. Ma, X. Sun, S. Feng and C. Zhang, Dalton Trans., 2013, 42, 4337–4339 RSC.
- H. Mustroph, M. Stollenwerk and V. Bressau, Angew. Chem., Int. Ed., 2006, 45, 2016–2035 CrossRef CAS PubMed.
- C. Shi, J. B. Wu and D. Pan, J. Biomed. Opt., 2016, 21, 050901 CrossRef PubMed.
- W. Sun, S. Guo, C. Hu, J. Fan and X. Peng, Chem. Rev., 2016, 116, 7768–7817 CrossRef CAS PubMed.
- U. Lawrentz, W. Grahn, K. Lukaszuk, C. Klein, R. Wortmann, A. Feldner and D. Scherer, Chem. – Eur. J., 2002, 8, 1573–1590 CrossRef CAS.
- R. V. Markov, P. A. Chubakov, A. I. Plekhanov, Z. M. Ivanova, N. A. Orlova, T. N. Gerasimova, V. V. Shelkovnikov and J. Knoester, Nonlinear Opt., 2000, 25, 365–371 CAS.
- M. Young, J. Suddard-Bangsund, T. J. Patrick, N. Pajares, C. J. Traverse, M. C. Barr, S. Y. Lunt and R. R. Lunt, Adv. Opt. Mater., 2016, 4, 1028–1033 CrossRef CAS.
- J. Zhang, P. J. Pellechia, J. Hayat, C. G. Hardy and C. Tang, Macromolecules, 2013, 46, 1618–1624 CrossRef CAS.
- O. Malinkiewicz, T. Grancha, A. Molina-Ontoria, A. Soriano, H. Brine and H. J. Bolink, Adv. Energy Mater., 2013, 3, 472–477 CrossRef CAS.
- A. C. Véron, H. Zhang, A. Linden, F. Nüesch, J. Heier, R. Hany and T. Geiger, Org. Lett., 2014, 16, 1044–1047 CrossRef PubMed.
- G. Wicht, S. Bücheler, M. Dietrich, T. Jäger, F. Nüesch, T. Offermans, J. N. Tisserant, L. Wang, H. Zhang and R. Hany, Sol. Energy Mater. Sol. Cells, 2013, 117, 585–591 CrossRef CAS.
- J. Roncali, P. Leriche and P. Blanchard, Adv. Mater., 2014, 26, 3821–3838 CrossRef CAS PubMed.
- M. Bates and R. R. Lunt, Sustainable Energy Fuels, 2017, 1, 955–968 RSC.
- J. Suddard-Bangsund, C. J. Traverse, M. Young, T. J. Patrick, Y. Zhao and R. R. Lunt, Adv. Energy Mater., 2016, 6, 1501659 CrossRef.
- C. J. Traverse, M. Young, J. Suddard-Bangsund, T. Patrick, M. Bates, P. Chen, B. Wingate, S. Y. Lunt, A. Anctil and R. R. Lunt, Sci. Rep., 2017, 7, 16399 CrossRef PubMed.
- H. Zhang, B. Niesen, E. Hack, S. Jenatsch, L. Wang, A. C. Véron, M. Makha, R. E. Schneider, Y. Arroyo, R. Hany and F. Nüesch, Org. Electron., 2016, 30, 191–199 CrossRef CAS.
- R. Hany, B. Fan, F. A. de Castro, J. Heier, W. Kylberg and F. Nüesch, Prog. Photovoltaics Res. Appl., 2011, 19, 851–857 CrossRef CAS.
- V. E. Shershov, M. A. Spitsyn, V. E. Kuznetsova, E. N. Timofeev, O. A. Ivashkina, I. S. Abramov, T. V. Nasedkina, A. S. Zasedatelev and A. V. Chudinov, Dyes Pigm., 2013, 97, 353–360 CrossRef CAS.
- E. K. Lindenberg and G. N. Patey, J. Chem. Phys., 2015, 143, 024508 CrossRef CAS PubMed.
- D. Gesevičius, A. Neels, S. Jenatsch, E. Hack, L. Viani, S. Athanasopoulos, F. Nüesch and J. Heier, Adv. Sci., 2018, 5, 1700496 CrossRef PubMed.
- D. M. Sturmer, Chemistry of Heterocyclic Compounds: Special Topics in Heterocyclic Chemistry, John Wiley & Sons, Inc., 2008, vol. 30, pp. 441–587 Search PubMed.
- J. P. Armstrong, C. Hurst, R. G. Jones, P. Licence, K. R. J. Lovelock, C. J. Satterley and I. J. Villar-Garcia, Phys. Chem. Chem. Phys., 2007, 9, 982 RSC.
- F. Heym, B. J. M. Etzold, C. Kern and A. Jess, Phys. Chem. Chem. Phys., 2010, 12, 12089 RSC.
- J. Wei, B. H. Fan, Y. Pan, N. N. Xing, S. Q. Men, J. Tong and W. Guan, J. Chem. Thermodyn., 2016, 101, 278–284 CrossRef CAS.
- H. D. B. Jenkins, H. K. Roobottom, J. Passmore and L. Glasser, Inorg. Chem., 1999, 38, 3609–3620 CrossRef CAS PubMed.
- J. A. Venables, Introduction to Surface and Thin Film Processes, Cambridge University Press, Cambridge, 2000 Search PubMed.
- S. Jenatsch, R. Hany, A. C. Véron, M. Neukom, S. Züfle, A. Borgschulte, B. Ruhstaller and F. Nüesch, J. Phys. Chem. C, 2014, 118, 17036–17045 CrossRef CAS.
- J. Heier, J. Groenewold, S. Huber, F. Nüesch and R. Hany, Langmuir, 2008, 24, 7316–7322 CrossRef CAS PubMed.
- J. Heier, C. Peng, A. C. Véron, R. Hany, T. Geiger, F. A. Nüesch, M. V. G. Vismara and C. F. O. Graeff, International Society for Optics and Photonics, ed. Z. H. Kafafi, P. A. Lane and I. D. W. Samuel, 2014, vol. 9184, p. 918408 Search PubMed.
- P. Vivo, A. Ojanperä, J.-H. Smått, S. Sandén, S. G. Hashmi, K. Kaunisto, P. Ihalainen, M. T. Masood, R. Österbacka, P. D. Lund and H. Lemmetyinen, Org. Electron., 2017, 41, 287–293 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- C. F. Macrae, I. J. Bruno, J. A. Chisholm, P. R. Edgington, P. Mccabe, E. Pidcock, L. Rodriguez-Monge, R. Taylor, J. Van De Streek and P. A. Wood, J. Appl. Crystallogr., 2008, 41, 466–470 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1834037 and 1834038. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8tc05286g |
| This journal is © The Royal Society of Chemistry 2019 |