
Physical vapour deposition of vanadium dioxide for thermochromic smart window applications
 *ce
and
Yi
Long
*af
*ce
and
Yi
Long
*af
Abstract
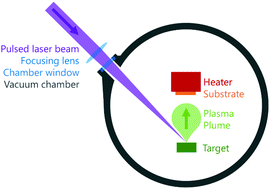
Smart windows are defined by their ability to regulate incoming solar radiation in order to reduce the energy consumption of buildings by modulating the heat intake. Vanadium dioxide (VO2) is one of the most promising potential candidates for smart window materials due to its ability to reversibly transit from monoclinic VO2 (M) to rutile VO2 (R) at near room temperature. As a result of this transition, the infrared radiation (IR) transparent VO2 (M) abruptly becomes IR opaque, effectively regulating the heat intake by solar radiation. Despite their promising potential, VO2-based smart windows have various significant intrinsic limitations: a high transition temperature (τC) of 68 °C; low luminous transmission (Tlum) of around 40% and low solar modulation (ΔTsol) of less than 25%. Currently, various methods have been used to fabricate VO2 thin films in an attempt to improve their intrinsic properties. One of those methods is physical vapour deposition (PVD). In this paper, various PVD techniques, such as pulsed laser deposition (PLD), evaporation decomposition (ED) and sputtering, are examined with respect to their conditions for VO2 fabrication, film quality and the strategies for film improvements. Lastly, some challenges and opportunities for further studies into VO2-based smart windows are discussed.
 Please wait while we load your content...
Please wait while we load your content...

