
Continuous multi-channel sensing of volatile acid and organic amine gases using a fluorescent self-assembly system†
Xinhua
Cao
 *ab,
Qianqian
Ding
a,
Yiran
Li
a,
Aiping
Gao
a and
Xueping
Chang
a
*ab,
Qianqian
Ding
a,
Yiran
Li
a,
Aiping
Gao
a and
Xueping
Chang
a
aCollege of Chemistry and Chemical Engineering & Henan Province Key Laboratory of Utilization of Non-metallic Mineral in the South of Henan, Institute for Conservation and Utilization of Agro-bioresources in Dabie Mountains, Xinyang Normal University, Xinyang 464000, China. E-mail: caoxhchem@163.com
bState Key Laboratory of Chemo/Biosensing and Chemometrics, Hunan University, Changsha 410082, P. R. China
First published on 24th November 2018
Abstract
A new gelator (1) was designed and synthesized through a Knoevenagel condensation reaction between 4-dimethylaminocinnamaldehyde and a gallic acid derivative containing a carbanion. This gelator could form stable organogels in hexane, petroleum ether, DMSO and acetonitrile. The self-assembly processes of 1 in four solvents were carefully investigated via UV-vis absorption, fluorescence, FTIR, XRD, water contact angles and field emission scanning electron microscopy (FESEM). Nanofibre and microsphere structures were observed in their self-assembly systems. The xerogel 1 film surfaces exhibited different hydrophobicities with water contact angles of 113.5–155°. The J-type aggregation mode was employed in the self-assembly process, and hydrogen bonding and π–π stacking were the main driving forces for organogel formation. Fluorescence emission of organogel 1 from acetonitrile was shifted from 558 nm of its corresponding diluted acetonitrile solution to 601 nm with a red-shift of 43 nm. Interestingly, compound 1 could sensitively respond to volatile acid, and further to organic amine gases along with obvious color changes. The detection limit for trifluoroacetic acid (TFA) by solution 1 in acetonitrile was 1.03 × 10−8 M with a corresponding association constant (K) of 6.85 × 104 M−1. The detection limit for triethylamine (TEA) by solution 1 in acetonitrile with 1.8 eq. of TFA was 1.27 × 10−8 M with a corresponding association constant (K) of 4.027 × 104 M−1. The yellow color of solution 1 could be reversibly changed to colorless in the titration process. At the same time, xerogel 1 exhibited sensitive response abilities for volatile acids and organic amines. The fluorescence of xerogel 1 could be quenched when in contact with a volatile acid within 7 s and recovered under further contact with a volatile amine within 15 s. The detection limit of xerogel film 1 towards trifluoroacetic acid (TFA) gases was 3.2 ppb. Organogel 1 exhibited sensitive response abilities towards TFA and TEA, and then rapidly expressed fluorescence, a gel state and a color change. This research would provide a new window into fast and sensitive detection of volatile acids and organic amines.
Introduction
Volatile acids, including inorganic and organic acids and organic amines, are important raw materials for the chemical industry. However, volatile acids and organic amines are important gaseous pollutants due to their serious damage to the ecological environment and threats to human health.1 For example, they can give rise to bronchial spasms, inflammation, oedema, chemical pneumonia, pulmonary oedema and death if inhaled.2,3 It is decisively essential for effective sensing systems for volatile acids and amines due to the human nose failing to detect low concentrations of volatile acids and amines.4 Several approaches and materials have been developed to detect volatile acids and amines in the environment, such as electrochemical devices, ion selective electrodes, chemical sensors, chromatography, enzymatic methods, molecularly imprinted polymers, metal–organic frameworks (MOFs), QDs, self-assembly systems, metal nanoparticles, and so on.5–9 However, it is a major challenge for reversible, instant and visual detection of volatile acids and amines, and few such applications have been explored.In the past few decades, low molecular weight organogels as supramolecular soft materials have recently been widely developed due to their potential application in many fields such as materials science, tissue engineering, separation technologies, catalysis, regenerative medicine, bioimaging, drug delivery, light harvesting systems, super wettability materials, sensors and others.10–17 Organogels can be formed under non-covalent bonding interactions including hydrogen bonding, hydrophobic/hydrophilic interactions, electrostatic interactions, coordination interactions, metal–metal interactions, π–π stacking, van der Waals’ forces, etc.18–27 Organic low molecular weight compounds can be self-assembled into various nanostructures, such as fibers, nanorings, rods, spheres, sheets, vesicles, and nanotubes.28–31 Supporting the above features, supramolecular organogels can be designed as stimuli-responsive smart materials which can change their chemical and physical properties under external stimuli, such as light, heat, chemicals, biomolecules, DNA, mechanical force, and magnetic fields.32–41 Under external stimuli, organogels can exhibit different signal representations through color, emission, or physical phase changes. At the same time, the xerogel film can also rapidly respond to gaseous analytes with low detection limits due to their large surface area and porous microstructure with the performance of fast absorption and accumulation of analytes.42,43 For example, Xue and coworkers have reported an aggregation-induced emission self-assembly system as a dual sensor for aromatic amines and acid vapor.44 We have reported a strong blue emissive supramolecular self-assembly system based on naphthalimide derivatives which can sensitively and selectively detect 2,4,6-trinitrophenol, and further remove it through absorption.45
In this study, we attempt to design a fluorescent organic molecule that can self-assemble into an organogel with the ability of reversibly responding to volatile acids and organic amines. 4-Dimethylaminocinnamaldehyde is confirmed to be an excellent molecule for constructing functional organogelators due to its proton-binding unit and large conjugated skeleton which provide the building blocks to prepare a sensor for acid vapors with intermolecular π–π stacking interactions. The good emission properties can be released through a Knoevenagel condensation reaction between 4-dimethylaminocinnamaldehyde and a gallic acid derivative containing a carbanion which is an efficient functional group for the self-assembly of molecules and solvents into organogels. Therefore, a novel fluorescent gelator (1) containing a gallic acid derivative was designed and synthesized (Scheme 1). This compound could gelate certain solvents and self-assemble into different structures with strong red fluorescence. This compound could respond to volatile acids, and further to organic amines, not only in solution but also in the gel phase through color, fluorescence emission or gel phase changes. The color, fluorescence emission and gel state could be reversibly transformed in response to volatile acids and organic amines. Interestingly, the xerogel film also emitted strong red fluorescence, which can be quenched by volatile acid vapors and recovered by organic amine vapors. The detection limit of xerogel film 1 towards trifluoroacetic acid (TFA) gases reaches 3.2 ppb. Thus, compound 1 may act as a smart material for security protection.
 | ||
| Scheme 1 Molecular structure of compound 1. | ||
Results and discussion
The synthesis procedure and characterization data of compound 1 are given in the ESI.† The gelation abilities of 1 were investigated in fourteen frequently-used organic solvents. As shown in Table 1, compound 1 could form stable organogels in hexane, petroleum ether, DMSO and acetonitrile with critical gel concentrations (CGCs) of 5.0, 5.0, 25.0 and 12.5 mg mL−1. The gels could be stable for several months as shown in Fig. 1. The organogels from hexane and petroleum ether were transparent (Fig. 1a, a′, b and b′), and the other two organogels formed in DMSO and acetonitrile were opaque (Fig. 1c, c′, d and d′). It was different that the organogels formed in DMSO and acetonitrile could emit stronger red light than those of the organogels from hexane and petroleum under the excitation of 365 nm light. For toluene, compound 1 did not completely dissolve at the concentration of 5 mg mL−1 even if the temperature was increased up to their corresponding boiling point. On the other hand, compound 1 could form solutions in the other nine solvents under the same experimental conditions (the details are given in Table 1).| Solvent | 1 | Solvent | 1 |
|---|---|---|---|
| I = insoluble; S = solution; G = gel; the values in the brackets denote the CGC. | |||
| DMF | S | 1,4-Dioxane | S |
| Hexane | G (5.0 mg mL−1) | Acetonitrile | G (12.5 mg mL−1) |
| Methanol | S | Ethanol | S |
| Acetone | S | Acetate ether | S |
| Petroleum ether | G (5.0 mg mL−1) | Toluene | I |
| DMSO | G (25.0 mg mL−1) | THF | S |
| CH2Cl2 | S | CHCl3 | S |
 | ||
| Fig. 1 Images of the gels formed in different solvents: (a and a′) for hexane, (b and b′) for petroleum ether, (c and c′) for DMSO, (d and d′) for acetonitrile. (a–d) were under daylight. (a′–d′) were under 365 nm light. | ||
It is well known that various morphologies can be formed in the self-assembly process of low mass organic molecules. The as-synthesized organogels 1 were diluted with the corresponding solvent, then dispersed onto mica plates, and dried using freeze-drying technology. The organogels from hexane and petroleum showed porous networks constructed by irregular nanofibers with widths of about 200 nm and lengths of several microns (Fig. 2a and b). For organogel 1 in DMSO, a nanofiber structure with widths of 100–200 nm and lengths of a dozen microns was observed in Fig. 2c. It was interesting that molecule 1 self-assembled into nanobelts with widths of 1–2 μm and lengths of a dozen microns which further curled into microspheres with diameters of about 4 μm like wool balls (Fig. 2d). From the above results, solvents took part in the self-assembly process through interactions between solvent molecules and molecule 1.
 | ||
| Fig. 2 SEM images of the xerogels in different solvents. (a) For hexane; (b) for petroleum ether; (c) for DMSO; (d) for acetonitrile. The scale bars for (a)–(d) are 2, 2, 8 and 10 μm. | ||
Rheological measurements for the gels in hexane, petroleum ether, DMSO and acetonitrile indicated typical viscoelastic behavior of the gels (Fig. S1, ESI†). Initially, the linear viscoelastic regions (LVRs) of the gels in hexane, petroleum ether, DMSO and acetonitrile at their corresponding CGCs were investigated with an amplitude sweep at a constant angular frequency of 1 Hz. Below the strain of 0.01% as the upper limit of the linear viscoelastic regime, the modulus remained approximately constant, and the storage modulus (G′) was larger than the loss modulus (G′′) which demonstrated an elastic response typical of gels (Fig. S1a, c, e and g, ESI†).48 With the strain increasing gradually, G′ fell sharply and G′′ gradually dominated over G′, which indicated that the organogel began to break up, and finally completely collapsed at the corresponding strain values of 63.7%, 61.6%, 9.0% and 3.3%. Frequency sweeps showed that G′ was always greater than the corresponding G′′ within the range of 0.1–100 Hz, and both moduli exhibited a weak dependence on frequency change which indicated that an energy-storage process occurred without energy dissipation and good elastic character.49,50
UV-vis absorption spectra can provide important information about the self-assembly process, especially for the molecular stacking mode.51 Usually, the blue shift of the π–π* transition peak shows H-type aggregate formation and face-to-face stacking. The red shift of the π–π* transition peak shows J-type aggregate formation and side-by-side stacking.52 The UV-vis absorption spectra of 1 in solution and the gel state in different solvents were investigated one by one and are shown in Fig. 3. The solution of 1 in hexane showed a maximum absorption band at 434 nm belonging to the π–π* electron transition in its UV-vis absorption spectrum (Fig. 3a). After the gel formation in hexane, the absorption band was red-shifted to 448 nm which indicated J-type aggregate formation in the organogel system. Similar changing trends in the absorption spectra also appeared for the other three gel systems. The UV-vis absorption spectrum of solution 1 in petroleum ether showed that the maximum absorption band was at 441 nm, and was red-shifted to 447 nm in the organogel from petroleum ether (Fig. 3b). It was different for the UV-vis absorption spectra of solution 1 in DMSO and acetonitrile which were broadened. The maximum absorption band appeared at 438 nm in the UV-vis absorption spectrum of solution 1 in DMSO which was red-shifted to 451 nm in the organogel state (Fig. 3c). Compared with the solution state, the UV-vis absorption band of the gel from acetonitrile had a red-shift of 27 nm and shifted from 420 nm to 447 nm (Fig. 3d). The above results indicated that the J-type aggregation mode existed in four organogels, and the solvents affected the UV-vis absorption behavior to some extent.
 | ||
| Fig. 3 UV-vis absorption spectra of 1 in solution and gel states in different solvents: (a) for hexane; (b) for petroleum ether; (c) for DMSO and (d) for acetonitrile. The solution concentration was 10−5 M. The gel concentrations were at their corresponding CGC. | ||
Fluorescence emission properties are one of the important optical properties for functional materials which also reflect the self-assembly behavior in organogel systems to some extent.53 The fluorescence emission spectra of 1 in solution and gel states from four solvents were obtained and are shown in Fig. 4. As shown in Fig. 4a, the 1 hexane solution could emit blue light with two emission shoulder peaks at 493 nm and 512 nm under the excitation of 446 nm light. The two emission peaks were changed into one peak and red-shifted to 565 nm for its corresponding gel state which indicated strong intermolecular π–π stacking existing in the gel state (Fig. 4a). For the 1 petroleum ether solution, there were two emission shoulder peaks at 489 nm and 510 nm in its emission spectrum (Fig. 4b). The two emission peaks were also changed into one emission peak at 599 nm with a red-shift of 89 nm in the gel state from petroleum ether (Fig. 4b). For DMSO and acetonitrile, their corresponding solutions exhibited different fluorescence emission behavior (Fig. 4c and d) which showed that the fluorescence emission properties of molecule 1 were affected by the solvent polarity. For example, the 1 DMSO solution only had one emission peak at 564 nm under the same conditions (Fig. 4c). Compared with the fluorescence emission of hexane or petroleum ether, the fluorescence emission of 1 in DMSO had a red-shift of about 50 nm. After the gel formation in DMSO, the emission peak was red-shifted to 598 nm in Fig. 4c. The fluorescence emission peak of the gel in acetonitrile was changed to 601 nm from the emission peak at 558 nm of its corresponding diluted acetonitrile solution with a red-shift of 43 nm (Fig. 4d).
 | ||
| Fig. 4 Fluorescence emission spectra of 1 in solution and gel states in different solvents (λex = 446 nm): (a) for hexane; (b) for petroleum ether; (c) for DMSO and (d) for acetonitrile. The solution concentration was 10−5 M. The gel concentrations were at their corresponding CGC. | ||
In order to further investigate the fluorescence emission changing with the solvent polarity, the fluorescence emission and UV-vis absorption spectra of 1 in different solvents were obtained.45 The maximum absorption bands of 1 in CH2Cl2, DMF, THF, ethyl acetate, hexane, methanol, petroleum ether, DMSO and acetonitrile solution were 462, 448, 442, 443, 441, 453, 440, 448 and 420 nm, respectively (Fig. 5a). The above solution 1 in different solvents could emit light with wavelengths of 511–563 nm under the excitation of 446 nm (Fig. 5b). The fluorescence emission wavelength of solution 1 was gradually red-shifted as the solvent polarity increased. Solutions of 1 in CH2Cl2, DMF, THF, ethyl acetate, hexane, methanol, petroleum ether, DMSO and acetonitrile solution emitted 553, 559, 544, 548, 513, 562, 511, 563 and 558 nm, respectively. The visual changes of solution 1 in different solvents were also investigated (Fig. S2, ESI†). To the naked eye, the color of 1 in different solvents did not have obvious differences. When solutions of 1 in different solvents were placed under 365 nm light, they could emit different lights from blue to yellow light.
 | ||
| Fig. 5 UV-vis absorption (a) and fluorescence emission (b) (λex = 446 nm) changes of 1 in different solvents. The solution concentration was 10−5 M. | ||
Supramolecular self-assembly gels are formed under the driving force of non-covalent bonding interactions including hydrogen bonds, π–π stacking interactions, hydrophobic interactions, van der Waals' forces, electrostatic interactions, and so forth.54 FTIR spectra can provide important information about hydrogen bonds for self-assembly systems. The FTIR spectrum of the xerogel from DMSO demonstrated that the N–H and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching peaks were at 3395 cm−1 and 1668 cm−1, and indicated hydrogen bond interactions existing in the gel system (Fig. 6).55 The xerogel from acetonitrile had the same N–H and CO stretching peaks in its FTIR spectrum. The strong intermolecular C
O stretching peaks were at 3395 cm−1 and 1668 cm−1, and indicated hydrogen bond interactions existing in the gel system (Fig. 6).55 The xerogel from acetonitrile had the same N–H and CO stretching peaks in its FTIR spectrum. The strong intermolecular C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N interaction bands appeared at 2204 cm−1 and 2207 cm−1 in xerogels from DMSO and acetonitrile. The N–H and CO stretching peaks of xerogels from petroleum ether and hexane were at 3361 cm−1 and 1658 cm−1 which demonstrated stronger hydrogen bond interactions than those of xerogels from DMSO and acetonitrile.56 These results well-explained why compound 1 had better gelation abilities in petroleum ether and hexane than in DMSO and acetonitrile. In order to further understand the intermolecular hydrogen bonding in the self-assembly system, a temperature-dependent 1H NMR experiment of the gel 1 in d6-DMSO was carried out (Fig. 6b). The signals of the amide protons NH7 and NH6 of the gel in DMSO shifted upfield from 8.44 to 8.21 ppm and 8.23 to 8.03 ppm, respectively, between 20 and 70 °C which indicated that intermolecular hydrogen bonding played an important role in the formation of the gel.56
N interaction bands appeared at 2204 cm−1 and 2207 cm−1 in xerogels from DMSO and acetonitrile. The N–H and CO stretching peaks of xerogels from petroleum ether and hexane were at 3361 cm−1 and 1658 cm−1 which demonstrated stronger hydrogen bond interactions than those of xerogels from DMSO and acetonitrile.56 These results well-explained why compound 1 had better gelation abilities in petroleum ether and hexane than in DMSO and acetonitrile. In order to further understand the intermolecular hydrogen bonding in the self-assembly system, a temperature-dependent 1H NMR experiment of the gel 1 in d6-DMSO was carried out (Fig. 6b). The signals of the amide protons NH7 and NH6 of the gel in DMSO shifted upfield from 8.44 to 8.21 ppm and 8.23 to 8.03 ppm, respectively, between 20 and 70 °C which indicated that intermolecular hydrogen bonding played an important role in the formation of the gel.56
 | ||
| Fig. 6 (a) FTIR spectra of the xerogels from different solvents and (b) temperature-dependent 1H NMR spectra of the gel in d6-DMSO. | ||
XRD patterns of xerogels can provide information about molecular self-assembly modes in organogel systems. The as-synthesized organogels were freeze-dried and subjected to XRD. The XRD pattern of the xerogel from hexane showed four diffraction peaks at 2θ = 3.37°, 8.70°, 10.25° and 20.98° with the corresponding d-spacing values of 2.62, 1.11, 0.86 and 0.42 nm (Fig. 7a). The d-spacing value of 2.62 nm should correspond to one side of the molecule 1. For the xerogel from petroleum ether, there was a series of diffraction peaks with d-spacing values of 2.67, 1.17, 0.58, 0.52, 0.44, 0.42, 0.41, 0.40, 0.38 and 0.31 nm in its XRD pattern (Fig. 7b). The d-spacing at 0.41 nm was attributed to the contribution of intermolecular π–π stacking interactions.57 There was a series of diffraction peaks in the XRD pattern of the xerogel from DMSO (Fig. 7c). More importantly, the d-spacing values at 2.84, 1.43, and 0.69 nm with a ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :½:¼ were suggestive of a layer organization within the nanofibers of the gel with an interlayer distance of 2.84 nm.58 Similar diffraction peaks also appeared in the XRD pattern of the xerogel from acetonitrile. In the many peaks, there were three peaks with the corresponding d-spacing values of 1.99, 1.15 and 0.57 nm with a ratio of 1:
:½:¼ were suggestive of a layer organization within the nanofibers of the gel with an interlayer distance of 2.84 nm.58 Similar diffraction peaks also appeared in the XRD pattern of the xerogel from acetonitrile. In the many peaks, there were three peaks with the corresponding d-spacing values of 1.99, 1.15 and 0.57 nm with a ratio of 1: :
: which were suggestive of a hexagonal packing array of 1 in the organogel state.
which were suggestive of a hexagonal packing array of 1 in the organogel state.
 | ||
| Fig. 7 XRD patterns of the xerogels formed in different solvents: (a) for hexane; (b) for petroleum ether; (c) for DMSO and (d) for acetonitrile. The gel concentrations were at their corresponding CGC. | ||
Materials with super wettability surfaces have potential applications in many fields such as oil/water emulsion separation, anti-icing surfaces, self-cleaning, anti-fogging, biomedical devices, and so on.59–61 At the same time, self-assembly aggregation surface wettability can afford some information about the self-assembly behavior.62 The diluted organogels were spun onto glass, and the coating should have a certain thickness to avoid the influence of glass. Even if the organogels from hexane and petroleum ether had similar self-assembly morphologies, their corresponding xerogel films still had different surface wettabilities. The xerogel film formed in hexane had a larger hydrophobicity than that of the xerogel film from petroleum ether. The xerogel films formed in hexane and petroleum ether had water contact angles of 142.5° and 113.5°, respectively (Fig. 8). This was possibly due to their different roughnesses. The larger hydrophobicity appeared in the xerogel films from DMSO and acetonitrile. The water contact angles of the xerogel films from DMSO and acetonitrile were 148° and 155°, respectively. From the SEM experimental results, the surface roughnesses of the xerogel films from DMSO and acetonitrile were obviously larger than those of xerogel films from hexane and petroleum ether. This is the reason why the xerogel films from DMSO and acetonitrile had large surface hydrophobicities.
 | ||
| Fig. 8 Water contact angle experimental results of the xerogel films formed in different solvents: (a) for hexane; (b) for petroleum ether; (c) for DMSO and (d) for acetonitrile. The gel concentrations were at their corresponding CGC. | ||
It is well known that irritating, volatile and colorless acids and organic amines have been widely applied in industry. But these volatile acids and amines can cause many environmental problems including acid rain, corrosion effects and health problems such as laryngeal mucosal irritation, nasal erosion ulcers and gastrointestinal diseases. So, the exploration of fast and sensitive detection methods for volatile acid and amine pollutants is quite important.63 Inspired by the excellent fluorescence emission properties of compound 1, we tried to explore its response abilities to acids (Fig. 9). Formic acid, acetic acid, propionic acid and TFA as organic acids and H2SO4 and HCl as inorganic acids were used to verify its response abilities. As shown in Fig. 9a, the absorbance of solution 1 had different degrees of decline except for formic acid. The absorbance of 1 in the visible region absolutely disappeared, especially for TFA, H2SO4 and HCl. The fluorescence emission behavior also changed with the addition of different acids. The fluorescence emission intensity of the 1 solution had some enhancement after the addition of 1.0 eq. of formic acid, acetic acid and propionic acid, possibly due to their too-weak acidity. On the other hand, the fluorescence emission intensity of the 1 solution was almost completely quenched after the addition of 1.0 eq. of TFA, H2SO4 and HCl, indicating 1 had a good response to volatile acids. The whole experimental process was validated through the color and emission changing of the 1 solution (Fig. S3, ESI†). The yellow color of the 1 solution changed to colorless after the addition of TFA, H2SO4 and HCl, and did not change after the addition of formic acid, acetic acid and propionic acid. The 1 solution with the addition of 1.0 eq. of TFA, H2SO4 and HCl almost did not emit light under the portable lamp of 365 nm light, but the 1 solution could still emit yellow light after the addition of formic acid, acetic acid, and propionic acid. More importantly, compound 1 could not only detect TFA from fluorescence changing, but it could also be visually recognized via color changing.
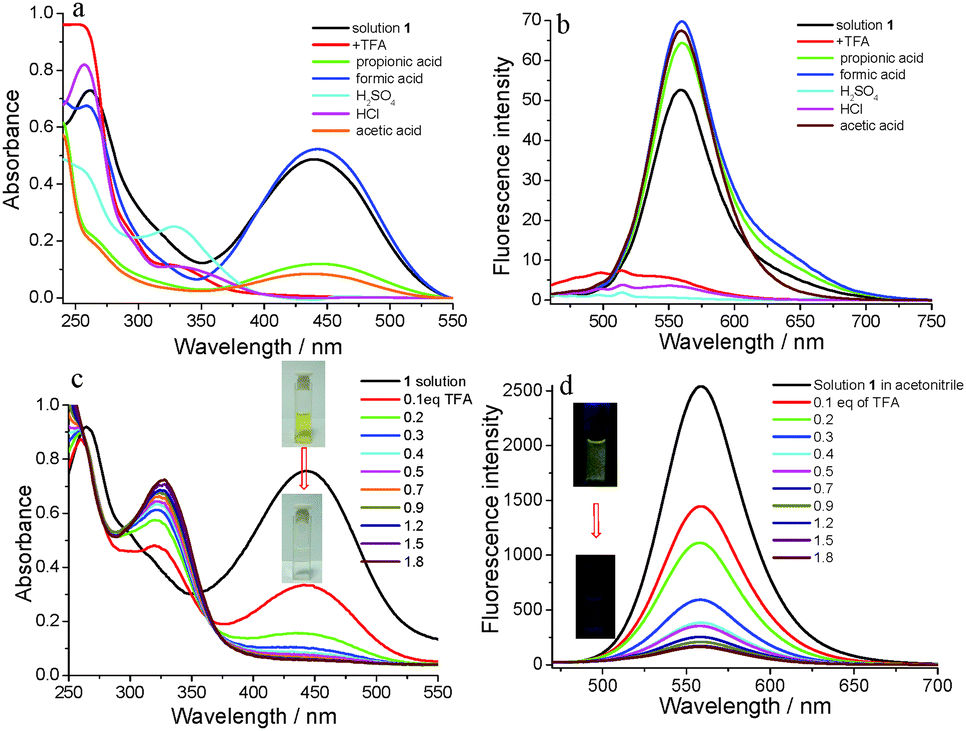 | ||
| Fig. 9 (a) UV-vis absorption spectra change of 1 in acetonitrile with the addition of different acids; (b) fluorescence spectra change of 1 in acetonitrile (λex. = 446 nm) with addition of acids; (c) UV-vis absorption spectra change of 1 in acetonitrile with the gradual addition of TFA; (d) fluorescence titration of 1 in acetonitrile (λex. = 446 nm) with the gradual addition of TFA. The concentration of solution 1 in acetonitrile was 10−4 M. | ||
In order to investigate the detection process and mechanism, TFA was selected as a sample to facilitate quantitative analysis. With the addition of TFA to solution 1, the absorbance gradually decreased (Fig. 9c). At the same time, the yellow color of solution 1 also gradually faded to colorless (inset in Fig. 9c). After the addition amount of TFA reached 1.8 eq., the absorbance of solution 1 at 446 nm reached a minimum value. At the same time, the absorbance in the range of visible light also changed to its minimum value which well-explained why the color changed from yellow to colorless. The fluorescence emission spectra of 1 were obtained and a titration experiment for TFA was carried out. As shown in Fig. 9d, the emission intensity at 558 nm gradually decreased with the increasing concentrations of TFA until the TFA amount was 1.8 eq., and the fluorescence intensity was quenched by 94%. The significant fluorescence changes were observed from yellow light to no light under a 365 nm UV lamp (inset in Fig. 9d). In addition, the plot of the fluorescence intensity ratio of 1/(I0 − I) (I0 = the original fluorescence intensity; I = the real-time fluorescence intensity) at 558 nm with the addition of TFA showed a linear correlation with R2 = 0.99056 (Fig. S4, ESI†). The binding constant was determined using the Benesi–Hildebrand equation for 1:1 stoichiometry (Fig. S4, ESI†).64 The association constant (K) was estimated as 6.85 × 104 M−1. The detection limit was 1.03 × 10−8 M determined using the following equation: LOD = 3σ/b: where σ was the standard deviation of 11 blank sample (10 μmol L−1 of 1) measurements (Table S1, ESI†) and b was the slope between the ratio of emission intensity of 558 nm versus TFA concentration (Fig. S5, ESI†).
In order to further verify the absorption and fluorescence emission spectra changes in the titration process of TFA, theoretical investigations into the difference in the absorption between compound 1 and protonated 1 were performed. The geometry optimizations and harmonic vibrational analyses of simplified compound 1 and protonated 1 were carried out using the hybrid B3LYP-D3/6-31G* and TD-B3LYP-D3/6-311++G** density functional theory method (DFT). The HOMOs and LUMOs of the compound 1 and protonated 1 are presented in Table S2 (ESI†). For compound 1, the HOMO and LUMO primarily reside on the 4-dimethylaminocinnamaldehyde part with slight differences. The HOMO and LUMO of compound 1 were −5.59 eV and 2.5 eV, respectively. After compound 1 was protonated, the HOMO primarily resided on the benzene ring part of the gallic acid as shown in Table S2 (ESI†). On the other hand, the LUMO primarily resided on the 4-dimethylaminocinnamaldehyde part again. The HOMO and LUMO of compound 1 with protonation were −7.29 eV and −5.61 eV, respectively. As shown in Fig. S6a (ESI†), compound 1 has a strong absorption peak in the range of 400–550 nm. However, protonated 1 has a strong absorption peak in the ultraviolet region (Fig. S6b, ESI†). The theoretical calculation results are completely in agreement with the experimental results.
In theory, the fluorescence emission and color of solution 1 with addition of 1.8 eq. of TFA could be recovered through the addition of base through the neutralization reaction between acid and base. The selectivity of solution 1 with the addition of 1.8 eq. of TFA to amine was firstly investigated through UV-vis absorption spectra and fluorescence emission spectra (Fig. 10a and b). As shown in Fig. 10a, the absorption band in the range of 375–525 nm reappeared after 6.0 eq. of amine including ammonium hydroxide, ethylenediamine, propylamine, triethylamine, diethylamine and aniline. Simultaneously, the fluorescence emission peak at 558 nm was also recovered after the addition of aliphatic amine or arylamine to solution 1 with the addition of 1.8 eq. of TFA and had some universality for responses to amines. Triethylamine (TEA) was selected as a representative amine, and titration experiments were carried out (Fig. 10c and d). With the addition of TEA, the maximum absorption band at the absorbance at 450 nm gradually increased until the addition of TEA was 6.0 eq. and the absorbance did not change. The color of the solution changed from colorless to its original yellow along with the absorbance changing (inset in Fig. 10c). The fluorescence emission was also recovered under the addition of TEA. As shown in Fig. 10d, the fluorescence emission intensity gradually increased with the addition of TEA. The significant fluorescence change was observed from no light to yellow light under a 365 nm UV lamp (inset in Fig. 10d). Working on the plot of the fluorescence intensity ratio of 1/(I − I0) (I0 = the original fluorescence intensity; I = the real-time fluorescence intensity) at 560 nm with the addition of TEA, it showed a linear correlation with R2 = 0.99551 (Fig. S7, ESI†). The binding constant was determined using the Benesi–Hildebrand equation for 1:1 stoichiometry.65 The association constant (K) was estimated as 4.027 × 104 M−1. The detection limit of complex 1-TFA towards TEA was 1.27 × 10−8 M (Table S3 and Fig. S8, ESI†).
 | ||
| Fig. 10 (a) UV-vis absorption spectra change of 1 in acetonitrile with the addition of different organic amines; (b) fluorescence spectra change of 1 in acetonitrile (λex. = 446 nm) with the addition of different organic amines; (c) UV-vis absorption spectra change of 1 in acetonitrile with the gradual addition of TEA; (d) fluorescence titration of 1 in acetonitrile (λex. = 446 nm) with the gradual addition of TEA. The concentration of solution 1 in acetonitrile was 10−4 M. | ||
Supramolecular self-assembly gels provide the chance for analyte infiltration into its three-dimensional network due to their driving force of non-covalent bonding interactions. So, gels have another pattern for the manifestation of gel state changes with contact with analytes. Tu and coworkers had reported the platinum–steroid low molecular mass gel system with visual chiral recognition with a chiral binaphthyl derivative through enantioselective metallogel collapsing.65 Firstly, xerogel 1 from hexane was selected and tested to respond to volatile acids due to the gel formed in hexane with a low CGC. The selectivity of the xerogel from hexane towards acid and further amine was firstly investigated and shown in Fig. S9 and S10 (ESI†). As shown in Fig. S9 (ESI†), the fluorescence emission could be quenched after contact with TFA and HCl gases. However, the fluorescence emission did not have obvious changes with the existence of H2SO4, formic acid, acetic acid and propionic acid under the same experimental conditions possibly due to the non-volatility of H2SO4 or the weak acidity of formic acid, acetic acid and propionic acid. The selectivity of the xerogel towards amines was also investigated through contact with different amines. As shown in Fig. S10 (ESI†), the fluorescence emission of the xerogel could be recovered with the gases of ammonium hydroxide, TEA, diethyl amine and ethylenediamine, and could not recover under tripropylamine and aniline possibly due to the weak volatility and alkalinity of tripropylamine and aniline. As shown in Fig. 11, the xerogel had a maximum emission peak at 619 nm with a red-shift of 54 nm compared with that of its gel state. After the xerogel was exposed to TFA atmospheres with different concentrations, the red light emitted from the xerogel could be almost quenched under 30 ppm of TFA atmosphere in 1 s. The limit of detection (LOD) of the xerogel towards TFA was calculated to be 3.2 ppb (Table S4, ESI†), when TFA was quickly removed and the xerogel was again put in the atmosphere of TEA, the fluorescence emission of the xerogel could have recovery to some extent in 15 s. The emission change of the xerogel was well-verified by the images of the xerogel in TFA or TEA atmospheres under a 365 nm light lamp (inset in Fig. 11). The whole change process of the xerogel under contact with TFA and TEA was made into a video as a supporting file (Video S1, ESI†). The cycle times of sensing acids and amine using the xerogel were investigated and further verified the stability of the xerogel in hexane. As shown in Fig. S12 (ESI†), the xerogel from hexane firstly sensing towards TFA and its fluorescence was quenched. Then the xerogel further made contact with TEA gas and its fluorescence was recovered. The xerogel could sense TFA and TEA in turn for at least five times (Fig. S11, ESI†). When the number of cycles was up to five, the fluorescence of the xerogel was not sensitive to TFA and TEA possibly due to the existence of an organic salt between TFA and TEA. Compared with the previous references, the xerogel exhibited a good detection ability and sensing time. The previous references reported that the limit of detection was in the range of 25 ppb–3.3 ppm and the sensing time was in the range of 0.3–10 s.44,66–69 More importantly, the fluorescence of the xerogel could be recovered when TFA was quickly removed and the xerogel was again put in the atmosphere of TEA.
 | ||
| Fig. 11 (a) Fluorescence emission changes of the xerogel in TFA or TEA atmospheres. The inset pictures are images of the xerogel under a 365 nm light lamp; (b) the curves of the fluorescence emission intensity at 619 nm of xerogel versus the concentration of TFA. | ||
Secondly, the organogels from acetonitrile and hexane were used to respond to TFA and TEA (Fig. S12, ESI†). The organogel from acetonitrile did not completely transform into a sol after 3 μL of TFA due to its compact stacking structure and high mechanical strength. On the other hand, the organogel formed in hexane exhibited sensitive responses towards TFA and TEA, and expressed versatility. At the same time, the organogel from hexane exhibited a much higher quantum yield of 12.16% than the diluted 1 hexane solution with a quantum yield of 0.86% which brought convenience for its application as a sensor. With the addition of 2 μL of TFA, the organogel was fast-changed into a sol, and the red self-assembly structure could become molten into the solution in 30 s. The red color of the organogel was changed to pale yellow accompanied by fluorescence quenching. Interestingly, the above sol could be recovered into a gel after the addition of 9 μL of TEA. With the addition of TEA, the original color and fluorescence of the organogel were immediately recovered. It was notable that this sol–gel transition process needed 3 minutes which was a longer time than that of the gel–sol transition, possibly due to energy produced in the neutral reaction between TFA and TEA. From these experimental results, this supramolecular self-assembly system exhibited a multi-approach for the detection of volatile acids and amines.
Conclusions
In conclusion, by an efficient and simple Knoevenagel condensation reaction, we developed a novel supramolecular self-assembly system based on novel fluorescent gelator 1 with interesting and applicable fluorescence properties. Under the main driving forces of hydrogen bonding and π–π stacking, compound 1 could form stable organogels in some solvents and self-assemble into nanofibre and microsphere structures. The xerogel films from the above solvents all exhibited hydrophobic properties. The J-type aggregation mode existed in the self-assembly system. There was a relatively large red-shift in their fluorescence emission between gel and sol states, especially for the gel and sol in hexane with a large red-shift of 89 nm. The UV-vis absorption spectra and fluorescence emission properties of compound 1 were perturbed with the solvent change. More interestingly, compound 1 had a fast and sensitive response ability towards volatile acids and amines. The fluorescence emission, color or gel state of 1 in solution, gel and xerogel states could have obvious changes after contact with volatile acid. At the same time, this transition process could be reversible under further contact with volatile amine. The detection limit for trifluoroacetic acid (TFA) by solution 1 in acetonitrile was 1.03 × 10−8 M. The detection limit for triethylamine (TEA) by solution 1 in acetonitrile with 1.8 eq. of TFA was 1.27 × 10−8 M. In particular, the xerogel could respond towards TFA gases in 7 s and further respond towards TEA in 15 s. The limit of detection (LOD) of the xerogel towards TFA was calculated to be 3.2 ppb. This work would provide a visible and fast detection strategy towards volatile acids and amines.Experimental section
Reagents and materials
Methyl gallate, 4-dimethylaminocinnamaldehyde and cyanoacetic acid were all purchased from Shanghai Titan technology Co., Ltd. 1-Bromododecane and ethanediamine were purchased from Shanghai Darui Fine Chemicals Co., Ltd. All inorganic metallic salts were provided by Zhengzhou Alfachem Co., Ltd. All other reagents were analytically pure. Water used throughout was deionized and then triply distilled.Gelation test
The gelator and solvent were put in a septum-capped test tube and heated (>80 °C) until the solid dissolved. The sample vial was then cooled to 25 °C (room temperature). Qualitatively, gelation was considered a success if no sample flow was observed upon inversion of the container at room temperature (the inverse flow method).46 Xerogel films were obtained by evaporation of the solvent from the gel via freeze-drying.Instrumentation conditions
1H NMR and 13C NMR spectra were recorded on a Bruker-Avance (Bruker, Ltd, Switzerland), at 600 and 150 MHz, respectively. Proton chemical shifts were reported in parts per million downfield from tetramethylsilane. HRMS were recorded on a LTQ-Orbitrap mass spectrometer (ThermoFisher, San Jose, CA, USA). The temperature dependent 1H NMR of 1 was performed in d6-DMSO from 20 to 70 °C. Field emission scanning electron microscopy (FESEM) images were obtained using a FE-SEM S-4800 instrument (Hitachi, Ltd, Tokyo, Japan). Samples were prepared by spinning the samples on glass slides and coating with Pt. Powder X-ray diffractions were generated using a Philips PW3830 (Philips, Ltd, Eindhoven, Holland) with a power of 40 kV at 40 mA (Cu target, λ = 0.1542 nm). UV-vis absorption spectra were recorded on a UV-vis 2550 spectroscope (Shimadzu, Ltd, Tokyo, Japan). Fluorescence spectra were recorded on an Edinburgh Instruments FLS 900 (Edinburgh Instruments, Ltd, Livingston, UK). The structure of the HOMO and LUMO states of 1 with simplification were determined with the help of theoretical calculations, in the framework of density functional theory (DFT) calculations at the level of B3LYP/6-31G* in a suite of the Gaussian 09 programs.47Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the financial support from the National Natural Science Foundation of China (U1704164 and 21401159), the Funding Scheme for the Young Backbone Teachers of Higher Education Institutions in Henan Province (2015GGJS-141), the Science & Technology Innovation Talents in Universities of Henan Province (No. 17HASTIT005) and the Nanhu Scholars Program for Young Scholars of XYNU.Notes and references
- H. W. Lin, M. Jang and K. S. Suslick, J. Am. Chem. Soc., 2011, 133, 16786–16789 CrossRef CAS PubMed.
- Z. L. Tang, J. H. Yang, J. Y. Yu and B. Cui, Sensors, 2010, 10, 6463–6476 CrossRef CAS PubMed.
- X. L. Pang, X. D. Yu, H. C. Lan, X. T. Ge, Y. J. Li, X. L. Zhen and T. Yi, ACS Appl. Mater. Interfaces, 2015, 7, 13569–13577 CrossRef CAS PubMed.
- J. W. Erisman, R. Otjes, A. Hensen, P. Jongejan, P. Van Den Bulk, A. Khlystov, H. Möls and S. Slanina, Atmos. Environ., 2001, 35, 1913–1922 CrossRef CAS.
- W. P. Lustig, S. Mukherjee, N. D. Rudd, A. V. Desai, J. Li and S. K. Ghosh, Chem. Soc. Rev., 2017, 46, 3242–3285 RSC.
- S. S. Babu, V. K. Praveen and A. Ajayaghosh, Chem. Rev., 2014, 114, 1973–2129 CrossRef CAS PubMed.
- R. A. Potyrailo, Chem. Soc. Rev., 2017, 46, 5311–5346 RSC.
- Y. H. Lee, O. Y. Kweon, H. Kim, J. H. Yoo, S. G. Han and J. H. Oh, J. Mater. Chem. C, 2018, 6, 8569–8612 RSC.
- A. L. Wu, P. P. Sun, N. Sun, Y. Yu and L. Q. Zheng, Chem. – Eur. J., 2018 DOI:10.1002/chem.201803800.
- J. Mayr, C. Saldías and D. D. Díaz, Chem. Soc. Rev., 2018, 47, 1484–1515 RSC.
- S. Theis, A. Iturmendi, C. Gorsche, M. Orthofer, M. Lunzer, S. Baudis, A. Ovsianikov, R. Liska, U. Monkowius and I. Teasdale, Angew. Chem., Int. Ed., 2017, 56, 15857–15860 CrossRef CAS PubMed.
- C. J. Lu, M. M. Zhang, D. T. Tang, X. Z. Yan, Z. Y. Zhang, Z. X. Zhou, B. Song, H. Wang, X. P. Li, S. C. Yin, H. Sepehrpour and P. J. Stang, J. Am. Chem. Soc., 2018, 140, 7674–7680 CrossRef CAS PubMed.
- L. Zhang, S. L. Li, M. A. Squillaci, X. L. Zhong, Y. F. Yao, E. Orgiu and P. Samorì, J. Am. Chem. Soc., 2017, 139, 14406–14411 CrossRef CAS PubMed.
- B. O. Okesola and D. K. Smith, Chem. Soc. Rev., 2016, 45, 4226–4251 RSC.
- L. J. Chen, G. Z. Zhao, B. Jiang, B. Sun, M. Wang, L. Xu, J. M. He, Z. Abliz, H. W. Tan, X. P. Li and H. B. Yang, J. Am. Chem. Soc., 2014, 136, 5993–6001 CrossRef CAS PubMed.
- N. Busschaert, C. Caltagirone, W. V. Rossom and P. A. Gale, Chem. Rev., 2015, 115, 8038–8155 CrossRef CAS PubMed.
- Q. Lin, T. T. Lu, X. Zhu, T. B. Wei, H. Li and Y. M. Zhang, Chem. Sci., 2016, 7, 5341–5346 RSC.
- P. C. Xue, Z. C. Yang and P. Chen, J. Mater. Chem. C, 2018, 6, 4994–5000 RSC.
- Z. C. Liu, G. L. Liu, Y. L. Wu, D. Cao, J. L. Sun, S. T. Schneebeli, M. S. Nassar, C. A. Mirkin and J. F. Stoddart, J. Am. Chem. Soc., 2014, 136, 16651–16660 CrossRef CAS PubMed.
- K. Tao, A. Levin, L. Adler-Abramovich and E. Gazit, Chem. Soc. Rev., 2016, 45, 3935–3953 RSC.
- X. Q. Wang, W. Wang, W. J. Li, L. J. Chen, R. Yao, G. Q. Yin, Y. X. Wang, Y. Zhang, J. L. Huang, H. W. Tan, Y. H. Yu, X. P. Li, L. Xu and H. B. Yang, Nat. Commun., 2018, 9, 3190 CrossRef PubMed.
- Y. Lan, M. G. Corradini, R. G. Weiss, S. R. Raghavan and M. A. Rogers, Chem. Soc. Rev., 2015, 44, 6035–6058 RSC.
- G. Q. Yin, H. Wang, X. Q. Wang, B. Song, L. J. Chen, L. Wang, X. Q. Hao, H. B. Yang and X. P. Li, Nat. Commun., 2018, 9, 567 CrossRef PubMed.
- P. F. Wei, X. Z. Yan and F. H. Huang, Chem. Soc. Rev., 2015, 44, 815–832 RSC.
- L. J. Chen, S. J. Chen, Y. Qin, L. Xu, G. Q. Yin, J. L. Zhu, F. F. Zhu, W. Zheng, X. P. Li and H. B. Yang, J. Am. Chem. Soc., 2018, 140, 5049–5052 CrossRef CAS PubMed.
- T. Gorai and U. Maitra, Angew. Chem., Int. Ed., 2017, 56, 10730–10734 CrossRef CAS PubMed.
- X. D. Yu, L. M. Chen, M. M. Zhang and T. Yi, Chem. Soc. Rev., 2014, 43, 5346–5371 RSC.
- X. H. Cao, N. Zhao, R. H. Li, H. T. Lv, Z. W. Zhang, A. P. Gao and T. Yi, Chem. – Asian J., 2016, 11, 3196–3204 CrossRef CAS.
- T. Y. Yuan, Y. Xu, C. Z. Zhu, Z. Y. Jiang, H. J. Sue, L. Fang and M. A. Olson, Chem. Mater., 2017, 29, 9937–9945 CrossRef CAS.
- X. H. Cao, N. Zhao, A. P. Gao, Q. Q. Ding, Y. R. Li and X. P. Chang, Langmuir, 2018, 34, 7404–7415 CrossRef CAS PubMed.
- V. K. Praveen, C. Ranjith and N. Armaroli, Angew. Chem., Int. Ed., 2014, 53, 365–368 CrossRef CAS PubMed.
- Y. Wu, Y. Hirai, Y. Tsunobuchi, H. Tokoro, H. Eimura, M. Yoshio, S. I. Ohkoshi and T. Kato, Chem. Sci., 2012, 3, 3007–3010 RSC.
- S. Takeshita, Y. Takebayashi, H. Nakamura and S. Yoda, Chem. Mater., 2016, 28, 8466–8469 CrossRef CAS.
- J. W. Seo, J. W. Chung, J. E. Kwon and S. Y. Park, Chem. Sci., 2014, 5, 4845–4850 RSC.
- K. Akkarachaneeyakorn, M. Li, J. Harris, S. A. Davis and S. Mann, Chem. Mater., 2014, 26, 5965–5972 CrossRef CAS.
- J. L. L. Tsai, T. T. Zou, J. Liu, T. F. Chen, A. O.-Y. Chan, C. Yang, C. N. Lok and C. M. Che, Chem. Sci., 2015, 6, 3823–3830 RSC.
- S. Ghosh, S. Cherumukkil, C. H. Suresh and A. Ajayaghosh, Adv. Mater., 2017, 1703783 CrossRef PubMed.
- Y. Ma, M. Cametti, Z. Džolić and S. M. Jiang, J. Mater. Chem. C, 2018, 6, 9232–9237 RSC.
- M. Externbrink, S. Riebe, C. Schmuck and J. Voskuhl, Soft Matter, 2018, 14, 6166–6170 RSC.
- P. Y. Xing, C. L. Yang, Y. Wang, S. Z. F. Phua and Y. L. Zhao, Adv. Funct. Mater., 2018, 1802859 CrossRef.
- C. Jiao, Y. Y. Chen, T. Q. Liu, X. Peng, Y. X. Zhao, J. A. Zhang, Y. Q. Wu and H. L. Wang, ACS Appl. Mater. Interfaces, 2018 DOI:10.1021/acsami.8b11391.
- X. H. Cao, Q. Q. Ding, N. Zhao, A. P. Gao and Q. S. Jing, Sens. Actuators, B, 2018, 256, 711–720 CrossRef CAS.
- R. K. Mishra, S. Das, B. Vedhanarayanan, G. Das, V. K. Praveen and A. Ajayaghosh, Stimuli-responsive Supramolecular Gels Molecular Gels: Structure and Dynamics, 2018, pp. 190–226 Search PubMed.
- P. C. Xue, J. P. Ding, Y. B. Shen, H. Q. Gao, J. Y. Zhao, J. B. Sun and R. Lu, J. Mater. Chem. C, 2017, 5, 11532–11541 RSC.
- X. H. Cao, N. Zhao, H. T. Lv, Q. Q. Ding, A. P. Gao, Q. S. Jing and T. Yi, Langmuir, 2017, 33, 7788–7798 CrossRef CAS PubMed.
- T. Wang, X. D. Yu, Y. Y. Li, J. J. Ren and X. L. Zhen, ACS Appl. Mater. Interfaces, 2017, 9, 13666–13675 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, J. J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian (Revision A.02), Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- J. Feng, L. H. Zeng, K. Y. Chen, H. B. Fang, J. Y. Zhang, Z. G. Chi and C. Y. Su, Langmuir, 2016, 32, 12184–12189 CrossRef CAS PubMed.
- K. Hyun, S. H. Kim, K. H. Ahn and S. J. Lee, J. Non-Newtonian Fluid Mech., 2002, 107, 51–65 CrossRef CAS.
- Q. Wang, J. C. Wu, Z. G. Gong, Y. Zou, T. Yi and C. H. Huang, Soft Matter, 2010, 6, 2679–2684 RSC.
- S. Yagai, T. Seki, T. Karatsu, A. Kitamura and F. Wurthner, Angew. Chem., Int. Ed., 2008, 47, 3367–3371 CrossRef CAS.
- P. Bairi, B. Roy, P. Routh, K. Sen and A. K. Nandi, Soft Matter, 2012, 8, 7436–7445 RSC.
- K. K. Kartha, V. K. Praveen, S. S. Babu, S. Cherumukkil and A. Ajayaghosh, Chem. – Asian J., 2015, 10, 2250–2256 CrossRef CAS PubMed.
- R. H. Li, S. Wang, Q. Li, H. C. Lan, S. Z. Xiao, Y. Q. Li, R. H. Tan and T. Yi, Dyes Pigm., 2017, 137, 111–116 CrossRef CAS.
- F. Aparicio, S. Cherumukkil, A. Ajayaghosh and L. Sánchez, Langmuir, 2016, 32, 284–289 CrossRef CAS PubMed.
- X. D. Yu, Q. Liu, J. C. Wu, M. M. Zhang, X. H. Cao, S. Zhang, Q. Wang, L. M. Chen and T. Yi, Chem. – Eur. J., 2010, 16, 9099–9106 CrossRef CAS PubMed.
- M. H.-Y. Chan, M. Ng, S. Y.-L. Leung, W. H. Lam and V. W.-W. Yam, J. Am. Chem. Soc., 2017, 139, 8639–8645 CrossRef CAS PubMed.
- H. Q. Xu, J. Song, T. Tian and R. X. Feng, Soft Matter, 2012, 8, 3478–3486 RSC.
- A. Sutton, T. Shirman, J. V. I. Timonen, G. T. England, P. Kim, M. Kolle, T. Ferrante, L. D. Zarzar, E. Strong and J. Aizenberg, Nat. Commun., 2017, 8, 14700 CrossRef PubMed.
- H. W. Chen, P. F. Zhang, L. W. Zhang, H. L. Liu, Y. Jiang, D. Y. Zhang, Z. W. Han and L. Jiang, Nature, 2016, 532, 85–89 CrossRef CAS PubMed.
- C. L. Gao, L. Wang, Y. C. Lin, J. T. Li, Y. F. Liu, X. Li, S. L. Feng and Y. M. Zheng, Adv. Funct. Mater., 2018, 1803072 CrossRef.
- J. P. Hill, W. S. Jin, A. Kosaka, T. Fukushima, H. Ichihara, T. Shimomura, K. Ito, T. Hashizume, N. Ishii and T. Aida, Science, 2004, 304, 1481–1483 CrossRef CAS PubMed.
- H. L. Huang, D. E. Gross, X. M. Yang, J. S. Moore and L. Zang, ACS Appl. Mater. Interfaces, 2013, 5, 7704–7708 CrossRef CAS PubMed.
- H. A. Benesi and J. H. Hildebrand, J. Am. Chem. Soc., 1949, 71, 2703–2707 CrossRef CAS.
- T. Tu, W. W. Fang, X. L. Bao, X. B. Li and K. H. Dötz, Angew. Chem., Int. Ed., 2011, 50, 6601–6605 CrossRef CAS PubMed.
- P. C. Xue, B. Q. Yao, P. P. Wang, P. Gong, Z. Q. Zhang and R. Lu, Chem. – Eur. J., 2015, 21, 17508–17515 CrossRef CAS PubMed.
- P. C. Xue, J. B. Sun, B. Q. Yao, P. Gong, Z. Q. Zhang, C. Qian, Y. Zhang and R. Lu, Chem. – Eur. J., 2014, 21, 4712–4720 CrossRef PubMed.
- P. C. Xue, B. Q. Yao, Y. B. Shen and H. Q. Gao, J. Mater. Chem. C, 2018, 5, 11496–11503 RSC.
- N. Xu, R. L. Wang, D. P. Li, Z. Y. Zhou, T. Zhang, Y. Z. Xie and Z. M. Su, New J. Chem., 2018, 42, 13367–13374 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8tc04964e |
| This journal is © The Royal Society of Chemistry 2019 |