
“Reversible” photochromism of polyoxomolybdate–viologen hybrids without the need for proton transfer†
Li
Li
 ,
Jun-Ren
Wang
,
Yang
Hua
,
Yu
Guo
,
Chen
Fu
,
Ya-Nan
Sun
and
Hong
Zhang
*
,
Jun-Ren
Wang
,
Yang
Hua
,
Yu
Guo
,
Chen
Fu
,
Ya-Nan
Sun
and
Hong
Zhang
*
Institute of Polyoxometalate Chemistry, Department of Chemistry, Northeast Normal University, Changchun, Jilin 130024, P. R. China. E-mail: hope20130122@163.com; zhangh@nenu.edu.cn
First published on 25th October 2018
Abstract
A case study of two new covalently bonded polyoxomolybdate–viologen hybrids revealed that “reversible” photochromic properties can be achieved without the need for proton transfer, and the weak electron-withdrawing ability of the coordinated ligand favors photoreduction of Mo(VI) to Mo(V).
Photochromic materials have appealing applications in smart windows,1 photomasks,2 data storage,3 photocatalysis,4 energy conversion,5etc. Polyoxometalates (POMs) are a class of very important photochromic materials. Polyoxomolybdates6 are a large subgroup of polyoxometalates that easily undergo photoinduced transformation into Mo(V)-containing heteropolyblue.6e To date, the “reversible” photochromic properties of polyoxomolybdates are mostly based on organoammonium cations.6a,c Yamase proposed that proton transfer plays a key role in the photochromism of these compounds.6a Some examples have witnessed the effectiveness of this proposition, but more evidence is still needed. On the other hand, Gamelas et al. found that the photochromic reaction was not fully reversible if an aromatic ammonium was present.7 It is required to further demonstrate whether this phenomenon is popular. The aim of this work was to probe the above two issues and explore the structure–property relationship when different ligands were introduced into polyoxomolybdates.
To fulfil this aim, we synthesized two new crystalline polyoxomolybdate–viologen hybrids, [(Bpyen)2(Mo8O26)]·2H2O (1; Bpyen = 1,2-bis(4,4′-bipyridinium)ethane) and [(Pbpy)2(Mo8O26)]·4H2O (2; Pbpy = 1,1′-[1,4-phenylenebis-(methylene)]bis(4,4′-bipyridinium)), and studied their photochromic behaviour. Viologens have been applied to the syntheses of a lot of photo- or thermochromic materials because of their excellent electron-accepting ability. In this paper, we chose the above two viologen ligands for the following reasons: (1) compounds based on POMs linked by covalent bonds are very rare, and they have two terminal pyridyl N atoms that may coordinate to open Mo sites; (2) they have different electron-withdrawing abilities, which may result in different photochromic behaviors of polyoxomolybdates; (3) we want to understand the competition mechanism between viologen and polyoxomolybdates. The experimental results revealed that both 1 and 2 easily exhibited photoinduced coloration, and their anhydrous products showed a “reversible” photochromic phenomenon. Moreover, we also found that the weak electron-withdrawing ability of the coordinated viologen ligand favors photoreduction of Mo(VI) to Mo(V). A single crystal X-ray diffraction analysis indicates that compound 1 crystallizes in a triclinic crystal system with space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) . It is built of neutral coordination molecules [(Bpyen)2(Mo8O26)] (Fig. 1a) and lattice water molecules. In the neutral coordination molecule, one γ-[Mo8O26]4− anion is ligated by two Bpyen cations. The nearest distance between OMo–O and the positively charged N atom in the Bpyen cations (OMo–O⋯N+) is 2.6453(4) Å (Table 1). The intermolecular p(OMo–O)⋯π stacking interactions (Fig. S1a, ESI†) observed between OMo–O and pyridine rings are larger than 4 Å and thus weak. In each Bpyen ligand, the two terminal 4,4′-bipyridinium cations rotate along the –CH2–CH2– linker to yield a trans structure, and the dihedral angle between two pyridyl groups of the coordinated bipyridinium cation is ca. 50.1°. Thermogravimetric analysis (TGA) under N2 revealed that compound 1 exhibited excellent thermal stability of the neutral coordination molecule. The weight loss of 2.0 wt% from 89 to 141 °C is attributed to the desorption of two free H2O molecules (calc. 1.9 wt%). The coordination molecule is stable up to 300 °C (Fig. S4a, ESI†).
. It is built of neutral coordination molecules [(Bpyen)2(Mo8O26)] (Fig. 1a) and lattice water molecules. In the neutral coordination molecule, one γ-[Mo8O26]4− anion is ligated by two Bpyen cations. The nearest distance between OMo–O and the positively charged N atom in the Bpyen cations (OMo–O⋯N+) is 2.6453(4) Å (Table 1). The intermolecular p(OMo–O)⋯π stacking interactions (Fig. S1a, ESI†) observed between OMo–O and pyridine rings are larger than 4 Å and thus weak. In each Bpyen ligand, the two terminal 4,4′-bipyridinium cations rotate along the –CH2–CH2– linker to yield a trans structure, and the dihedral angle between two pyridyl groups of the coordinated bipyridinium cation is ca. 50.1°. Thermogravimetric analysis (TGA) under N2 revealed that compound 1 exhibited excellent thermal stability of the neutral coordination molecule. The weight loss of 2.0 wt% from 89 to 141 °C is attributed to the desorption of two free H2O molecules (calc. 1.9 wt%). The coordination molecule is stable up to 300 °C (Fig. S4a, ESI†).
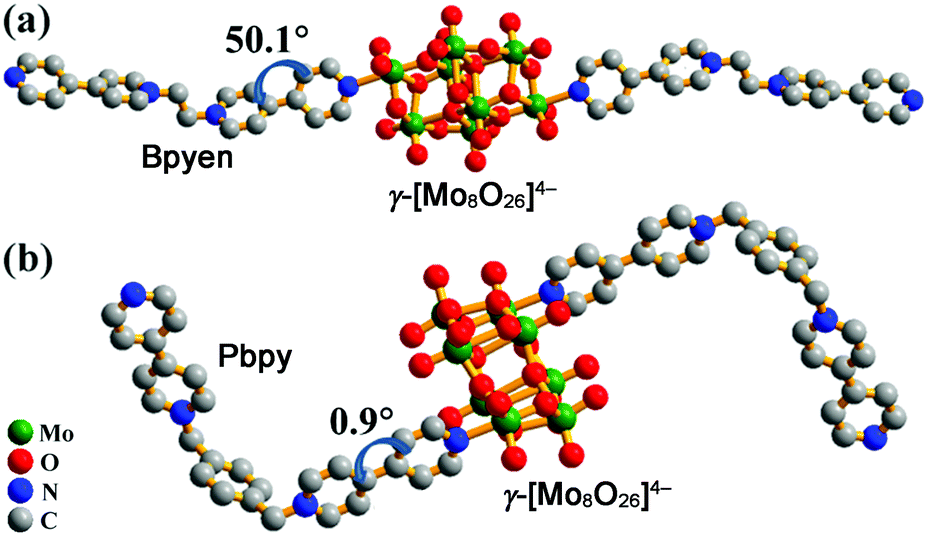 | ||
| Fig. 1 Molecular structures of 1 (a) and 2 (b). H atoms and H2O are omitted for clarity. Dihedrals between two pyridyl rings of the coordinated 4,4′-bipyridinium cations are labelled. | ||
| 1 | 2 | |
|---|---|---|
| a TD-DFT (B3LYP/6-31G+g(d,p)). | ||
| The shortest OMo–O⋯N+ distance (Å) | 2.6453(4) | 2.6540(5) |
| Dihedral angle (Å) between two pyridyl groups of the coordinated 4,4′-bipyridinium | 50.1(1) | 0.9(2) |
| Face-to-face π⋯π distance (Å) between pyridine rings | No | 3.8378(3) |
| Ligand, LUMO energy (eV) | Bpyen, −9.02 | Pbpy, −8.19 |
| Oscillator strength f (radical)a | 0.003 ([Bpyen]˙+) | 0.006 ([Pbpy]˙+) |
Compound 2 crystallizes in a monoclinic crystal system with space group P21/c. It is constituted by neutral coordination molecules [(Pbpy)2(Mo8O26)] (Fig. 1b) and lattice water molecules. The neutral coordination molecule has a similar structure to that in 1. The nearest distance between OMo–O and the positively charged N atom in the Pbpy cations (OMo–O⋯N+) is 2.6540(5) Å, close to that found in 1. In each Pbpy ligand, the two terminal 4,4′-bipyridinium cations rotate along the –CH2–C6H4–CH2– linker to yield a cis structure, and the dihedral angle between two pyridyl groups of the coordinated bipyridinium cation is ca. 0.9°. In 2, the intermolecular p(OMo–O)⋯π and π⋯π stacking interactions (Fig. S1b, ESI†) are smaller than 3.9 Å and thus stronger than those in 1. TGA in a nitrogen flow revealed that 2 exhibits excellent thermal stability. The weight loss of 3.2 wt% from 35 °C to 83 °C is attributed to the desorption of four free H2O molecules (calc. 3.1 wt%). The neutral coordination molecule is stable up to 268 °C (Fig. S4b, ESI†).
Compound 1 is sensitive to light, exhibiting an eye-detectable colour change at ambient temperature in air. As shown in Fig. 2 (left), the as-synthesized pale brown crystals of 1 (named 1a) turned cadmium brown after irradiation with a 300 W Xe lamp (ca. 2.4 W cm−2). It can also change from pale brown to cadmium brown upon irradiation at room temperature in N2 (Fig. S8, left, ESI†). The irradiated sample, named 1b-P, was obviously lighter through annealing at 120 °C for 1 h in air. At this time, the obtained partially decolored sample (labelled p-decolored in Fig. 2) had lost the lattice water molecules, but the coordination molecule was intact. The decolored sample turned the same colour as that of 1b-P after being irradiated again, indicating that the coloration–decoloration of the coordination molecule of 1 is reversible. The UV-visible (UV-vis) diffuse reflectance spectra (Fig. S7a, ESI†) further proved the reversibility.
 | ||
| Fig. 2 Photochromic behaviors of 1 (left) and 2 (right) shown in photographic images. 1a and 2a: before photoirradiation; 1b-P and 2b-P: after exposure to a 300 W Xe lamp; p-decolored (partially decolored) and decolored, after annealing at 120 °C; 1b-P′ and 2b-P′: the decolored samples exposed to the same Xe lamp again. The decolouring process of 1b-P is competitive with thermochromism, so the color of the annealed samples (p-decolored) is slightly different from that of 1a. | ||
In situ electron spin resonance (ESR) experiments were performed to probe the electron-transfer process of 1 (Fig. 3a). Before irradiation, the ESR spectrum showed two very weak signals at g values of 2.0039 and 1.9514, respectively. The former corresponds to the viologen free radicals,8 that is, [Bpyen]˙+ radicals. The latter is consistent with Mo(V)6b,e originating from the partial reduction of the Mo(VI). After irradiation with the Xe lamp, these two signals increased obviously. The presence of weak ESR signals before irradiation indicates that compound 1 is rather sensitive to ambient light. The O atoms from H2O and γ-[Mo8O26]4− anions are potential electron donors, but the decolored sample can change color to cadmium brown after losing water. Because the nearest distance between OMo–O and the N atom in the Bpyen cations (OMo–O⋯N+) is 2.6453(4) Å, we speculated that OMo–O should be the electron donor and Bpyen cations and Mo(VI) atoms are electron acceptors. The electrostatic potential surfaces of the neutral coordination molecule, calculated using the DFT method (6-31g(d,p) for C H N O; Lanl2dz for Mo), further provide some insight into the photo-induced electron transfer behavior. As can be seen from Fig. 4, it is very distinct that the O atoms from γ-[Mo8O26]4− anions have most potential as electron donors.
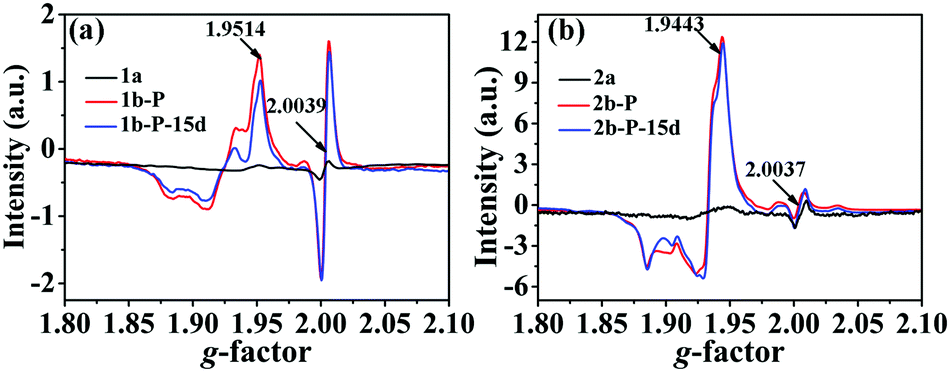 | ||
| Fig. 3 ESR spectra of 1 (a) and 2 (b) with identical mole quantities. Labels: 1a and 1b, before photoirradiation; 1b-P and 2b-P, after photoirradiation; 1b-P-15d and 2b-P-15d, after keeping the irradiated sample in the dark in air for 15 d. | ||
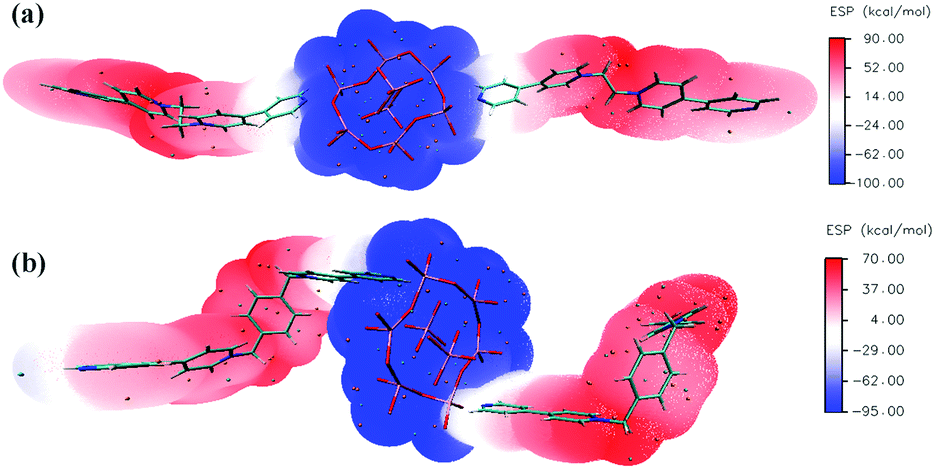 | ||
| Fig. 4 Electrostatic potential (ESP) surfaces of the neutral coordination molecule in 1 and 2 before irradiation. Significant surface local minima and maxima are represented as orange and cyan spheres, respectively. | ||
The ESR spectrum (Fig. S11a, ESI†) further showed that there are no OMo–O radicals. The as-synthesized samples can also change colour after losing H2O, and PXRD (Fig. S2, left, ESI†), IR (Fig. S3, left, ESI†) and H1 NMR (Fig. S9, ESI†) analyses showed that 1 retained the same crystal structure and main components through the coloration process. The nearest distance between H atoms in H2O and OMo–o in compound 1 is 10.169 Å (Fig. S12a, ESI†), which is not enough to provide protons to cause the quenching of the OMo–O radicals. So, H2O is not involved in the reaction. So, it is likely that the OMo–O radicals react with some surrounding unknown substance.9 Compound 1 has no protons to transfer, indicating that the proton-transfer mechanism proposed by Yamase6a is not suitable for 1.
The incomplete decoloration upon annealing at 120 °C in air illustrates that compound 1 may exhibit a thermochromic phenomenon. Indeed, the as-prepared sample 1a changed its color to that of p-decolored after annealing at 90 °C for 5 h in air (Fig. 2, left). Moreover, an ESR study supported that the heating process was also accompanied by the generation of [Bpyen]˙+ radicals and Mo(V), with g values of 2.0039 and 1.9514, respectively (Fig. S5, ESI†). Different from photoinduced coloration, the signal at g = 2.0039 increased clearly, while that at g = 1.9514 showed no obvious change. We assumed that the electrons were mainly transferred to the viologen ligand instead of the Mo(VI) atoms upon heating. That is to say, the heating process is not beneficial to the reduction of Mo(VI).
After thermally induced coloration, the absorption band between 280 and 1652 nm is obviously enhanced. The ESR data (Fig. S5, ESI†) revealed that viologen made the main contribution to the peak at 470.5 nm and the band above 800 nm (Fig. S7b, ESI†).10 After photocoloration, as shown in Fig. 5a, the absorption band between 280 and 1652 nm in the UV-vis diffuse reflectance spectra is also significantly enhanced as the time of exposure. Mo(V) made the main contribution to the peak at 686.8 nm, which refers to Mo(VI)–Mo(V) intervalence charge transfer and d–d transitions.11
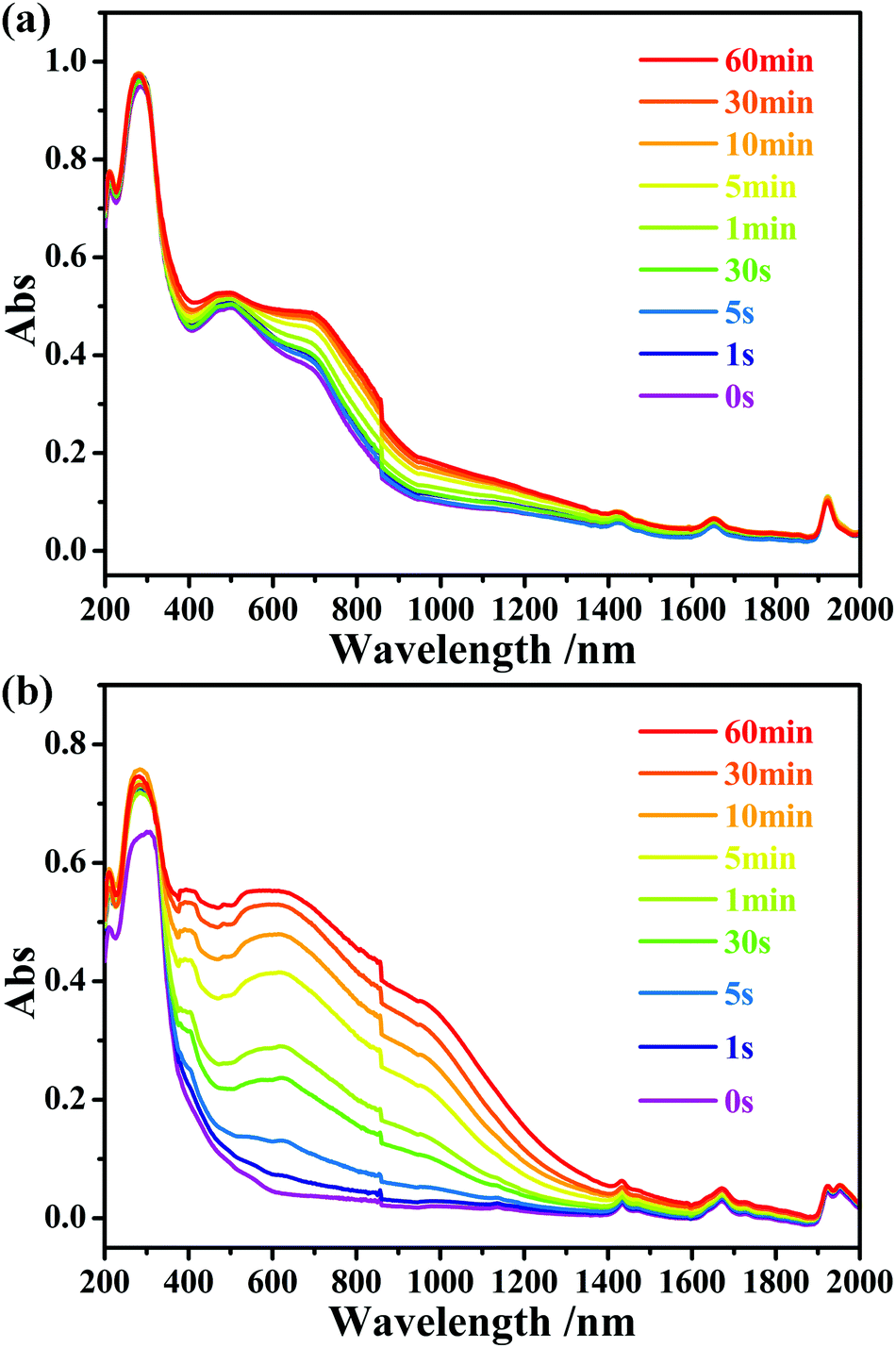 | ||
| Fig. 5 Time-dependent UV-vis diffuse reflectance spectra of compounds 1 (a) and 2 (b) upon irradiation at room temperature in air. | ||
Compound 2 showed a fast photoinduced colour change from antique white to black at ambient temperature (Fig. 2, right). It can also change from antique white to black upon irradiation at room temperature in N2 (Fig. S8, right, ESI†). The irradiated sample, named 2b-P, was completely decolored through annealing at 120 °C for 1.5 h in air, which is different from that of 1b-P. Similar to 1, the obtained decolored sample (labelled decolored in Fig. 2) had also lost the lattice water molecules, but the coordination molecule was still retained. The decolored sample turned the same colour as that of 2b-P under the same light source. This coloration–decoloration process of the coordination molecule of 2 is reversible. The UV-vis diffuse reflectance spectra (Fig. S6, ESI†) also proved the reversibility.
Similarity, the ESR signals at g = 2.0037 and g = 1.9443 also indicate the generation of [Pbpy]˙+ radicals and Mo(V) (Fig. 3b). OMo–O should be the electron donor and Bpyen cations and Mo(VI) atoms are electron acceptors. However, the ESR spectrum (Fig. S11b, ESI†) showed that there are also no OMo–O radicals. The anhydrous products also showed a “reversible” photochromic phenomenon, PXRD (Fig. S2, right, ESI†), IR (Fig. S3, right, ESI†) and H1 NMR (Fig. S10, ESI†) analyses showed that 2 retained the same crystal structure in the whole coloration process. Since compounds 1 and 2 have analogous organic ligands and the same polyoxomolybdate, the photochromic mechanism should also be the same. So, H2O is also not involved in 2. So, it is also likely that OMo–O radicals react with some surrounding unknown substance. The proton-transfer mechanism proposed by Yamase is also not suitable in this case. The irradiated sample, 2b-P, is also very stable. After keeping it in the dark in air for 15 d, the ESR signal showed a minor change (Fig. 3b).
Different from that of 1b-P, the ESR signal of Mo(V) in 2b-P is obviously stronger than that of [Pbpy]˙+ radicals (Fig. 3b). In addition, the UV-vis absorption band around 598.8 nm, assigned to Mo(V),6b is clearly stronger than those at 470.5 nm and above 800 nm, assigned to [Pbpy]˙+ radicals (Fig. 5b). These data revealed that electrons of the OMo–O atoms in 2 are mainly transferred to the Mo(VI) atoms instead of the Pbpy cation upon irradiation. The LUMO level of (Pbpy) is higher than that of (Bpyen) (Table 1), implying that the electron-withdrawing ability of Pbpy is weaker than that of Bpyen. This result accounts for the reason why the electrons of the OMo–O atoms are more inclined to transfer to Mo(VI) in 2. That is to say, the weak electron-withdrawing ability of the coordinated ligand favors photoreduction of Mo(VI) to Mo(V).
It can be seen from Fig. 5 that the UV-vis absorption bands of the viologen radicals in 2b-P are stronger than those found in 1b-P. This phenomenon can be explained from the structural aspect. As described above, the Pbpy ligand in 2 has a better planar character than Bpyen in 1. Thus, it has a larger delocalization degree and is easier to stabilize the electrons received from the OMo–O atoms. Strong intermolecular interactions also benefit the stability of free radicals. As mentioned previously, the intermolecular p⋯π and π⋯π stacking interactions in 2 are stronger than those in 1. Furthermore, the intrinsic absorption ability of the radical contributes to the intensity of the absorption band. A time-dependent density functional density (TD-DFT) calculation at the B3LYP/6-31G+g(d,p) indicated that the oscillator strength f of the [Pbpy]˙+ radical is larger than that of the [Bpyen]˙+ radical (Table 1).
Hitherto, some organic–inorganic hybrids combining POMs and organic photochromic chromophores, such as naphthalene diimides (NDIs),12 viologens,13 and spiropyrane,14 have been studied. To our knowledge, only two photochromic POM–viologen crystalline hybrids, [H2(4,4′-bipyridine)]Mo7O22·H2O and [(AV)(p-AV)(EuW10O36)]n·2nH2O (AV2+ = N,N′-bisaminopropyl-4,4′-bipyridinium bromide hydrobromide; p-AV = poly(1′-ethyl-10-butyl-4,4′-bipyridinium)), were documented.13b,d They are salts where the viologen templates and the POM components are linked by ionic bonds and intermolecular interactions (H-bonds, π–π interactions, etc.). As reported by Marcus,15 the nearer an electron donor and an electron acceptor, the faster the electron transfer. Dispersion of the salts in a solution or film may cause the separation of electron donors and electron acceptors, which does not favor the electron transfer and will weaken the photosensitivity. An applicable method to avoid this possibility is to assemble viologen and POM components in the coordination mode. Photoreactions of 1 and 2 can be detected in 5 s and 1 s, respectively, through UV-vis absorption spectra. Comparably, the shortest time required is 1 min for the two known photochromic POM–viologen hybrids.12b,d This discovery demonstrates that covalent bonding between viologen and POM components does favor electron transfer and the improvement of photosensitivity.
In conclusion, through a case study of two new covalently bonded polyoxomolybdate–viologen hybrids, this work revealed that “reversible” photochromic properties can be achieved without the need for proton transfer. The results also revealed the weak electron-withdrawing ability of the coordinated ligand favors photoreduction of Mo(VI) to Mo(V). Moreover, the heating process is not beneficial to the reduction of Mo(VI) when an electron-accepting ligand exists, such as the viologen ligand described in this work. These discoveries will help in improving performance for the well-known photochromic polyoxomolybdate family.
We gratefully acknowledge financial support from the NSF of China (21571032, 21271038) and the Analysis and Testing Foundation of Northeast Normal University.
Conflicts of interest
There are no conflicts to declare.Notes and references
- C. Bechinǵer, S. Ferrere, A. Zaban, J. Spraǵure and B. A. Gregg, Nature, 1996, 383, 608–610 CrossRef.
- T. L. Andrew, H.-Y. Tsai and R. Menon, Science, 2009, 324, 917–921 CrossRef CAS PubMed.
- S. Kawata and Y. Kawata, Chem. Rev., 2000, 100, 1777–1788 CrossRef CAS PubMed.
- M. Dan-Hardi, C. Serre, T. Frot, L. Rozes, G. Maurin, C. Sanchez and G. Férey, J. Am. Chem. Soc., 2009, 131, 10857–10859 CrossRef CAS PubMed.
- L. A. Vermeulen and M. E. Thompson, Nature, 1992, 358, 656–658 CrossRef CAS.
- (a) T. Yamase, Chem. Rev., 1998, 98, 307–325 CrossRef CAS PubMed; (b) H. Zhang, L. Y. Duan, Y. Lan, E. B. Wang and C. W. Hu, Inorg. Chem., 2003, 42, 8053–8058 CrossRef CAS PubMed; (c) T. He and J. N. Yao, Prog. Mater. Sci., 2006, 51, 810–879 CrossRef CAS; (d) Y. Wang, H. Li, C. Wu, Y. Yang, L. Shi and L. Wu, Angew. Chem., Int. Ed., 2013, 52, 4577–4581 CrossRef CAS PubMed; (e) Y. F. Liang, S. Z. Li, D. H. Yang, P. T. Ma, J. Y. Niu and J. P. Wang, J. Mater. Chem. C, 2015, 3, 4632–4639 RSC; (f) Y. Duan, J. C. Waerenborgh, J. M. Clemente-Juan, C. Giménez-Saiz and E. Coronado, Chem. Sci., 2017, 8, 305–315 RSC.
- J. A. F. Gamelas, A. M. V. Cavaleiro, E. M. Gomes, M. Belsley and E. Herdtweck, Polyhedron, 2002, 21, 2537–2545 CrossRef CAS.
- (a) M. S. Wang, C. Yang, G. E. Wang, G. Xu, X. Y. Lv, Z. N. Xu, R. G. Lin, L. Z. Cai and G. C. Guo, Angew. Chem., Int. Ed., 2012, 51, 3432–3435 CrossRef CAS PubMed; (b) H. Chen, G. Zheng, M. Li, Y. Wang, Y. Song, C. Han, J. Dai and Z. Fu, Chem. Commun., 2014, 50, 13544–13546 RSC; (c) P.-X. Li, M.-S. Wang, M.-J. Zhang, C.-S. Lin, L.-Z. Cai, S. P. Guo and G.-C. Guo, Angew. Chem., Int. Ed., 2014, 53, 11529–11531 CrossRef CAS PubMed; (d) C. Chen, J.-K. Sun, Y.-J. Zhang, X.-D. Yang and J. Zhang, Angew. Chem., Int. Ed., 2017, 56, 14458–14462 CrossRef CAS PubMed.
- W. W. Porter III, T. P. Vaid and A. L. Rheingold, J. Am. Chem. Soc., 2005, 127, 16559–16566 CrossRef PubMed.
- (a) C. Sun, G. Xu, X.-M. Jiang, G.-E. Wang, P.-Y. Guo, M.-S. Wang and G.-C. Guo, J. Am. Chem. Soc., 2018, 140, 2805–2811 CrossRef CAS PubMed; (b) J.-K. Sun, P. Wang, Q.-X. Yao, Y.-J. Chen, Z.-H. Li, Y.-F. Zhang, L.-M. Wu and J. Zhang, J. Mater. Chem., 2012, 22, 12212–12219 RSC; (c) P.-Y. Guo, C. Sun, N.-N. Zhang, L.-Z. Cai, M.-S. Wang and G.-C. Guo, Chem. Commun., 2018, 54, 4525–4528 RSC; (d) X. S. Xing, R. J. Sa, P. X. Li, N. N. Zhang, Z. Y. Zhou, B. W. Liu, J. Liu, M. S. Wang and G. C. Guo, Chem. Sci., 2017, 8, 7751–7757 RSC.
- P. Mialane, G. Zhang, I. M. Mbomekalle, P. Yu, J.-D. Compain, A. Dolbecq, J. Marrot, F. Sécheresse, B. Keita and L. Nadjo, Chem. – Eur. J., 2010, 16, 5572–5576 CrossRef CAS PubMed.
- (a) J.-J. Liu, Y. Wang, Y.-J. Hong, M.-J. Lin, C.-C. Huang and W.-X. Dai, Dalton Trans., 2014, 43, 17908–17911 RSC; (b) J.-Z. Liao, X.-Y. Wu, J.-P. Yong, H.-L. Zhang, W.-B. Yang, R. Yu and C.-Z. Lu, Cryst. Growth Des., 2015, 15, 4952–4958 CrossRef CAS; (c) J.-J. Liu, Y. Wang, M.-J. Lin, C.-C. Huang and W.-X. Dai, Dalton Trans., 2015, 44, 484–487 RSC; (d) J.-Z. Liao, C. Wu, X.-Y. Wu, S.-Q. Deng and C.-Z. Lu, Chem. Commun., 2016, 52, 7394–7397 RSC.
- (a) B. Xu, L. Xu, G. Gao, Y. Yang, W. Guo, S. Liu and Z. Sun, Electrochim. Acta, 2009, 54, 2246–2252 CrossRef CAS; (b) D.-H. Sun, J.-L. Zhang, H.-J. Ren, Z.-F. Cui and D.-X. Sun, Acta Phys. – Chim. Sin., 2010, 26, 1264–1270 CAS; (c) H.-Y. Zhang, A.-J. Miao and M. Jiang, Mater. Chem. Phys., 2013, 141, 482–487 CrossRef CAS; (d) D.-F. Shen, S. Li, H. Liu, W. Jiang, Q. Zhang and G.-G. Gao, J. Mater. Chem. C, 2015, 3, 12090–12097 RSC; (e) H. Liu, Y. Lv, S. Li, F. Yang, S. Liu, C. Wang, J.-Q. Sun, H. Meng and G.-G. Gao, J. Mater. Chem. C, 2017, 5, 9383–9388 RSC.
- (a) J.-D. Compain, P. Deniard, R. Dessapt, A. Dolbecq, O. Oms, F. Sécheresse, J. Marrota and P. Mialane, Chem. Commun., 2010, 46, 7733–7735 RSC; (b) T. Zhang, N. Ma, L. Yan, T. Ma and Z. Su, Dyes Pigm., 2014, 106, 105–110 CrossRef CAS; (c) K. Hakouk, O. Oms, A. Dolbecq, J. Marrot, A. Saad, P. Mialane, H. E. Bekkachi, S. Jobic, P. Deniard and R. Dessapt, J. Mater. Chem. C, 2014, 2, 1628–1641 RSC; (d) A. Parrot, A. Bernard, A. Jacquart, S. A. Serapian, C. Bo, E. Derat, O. Oms, A. Dolbecq, A. Proust, R. Métivier, P. Mialane and G. Izzet, Angew. Chem., Int. Ed., 2017, 56, 4872–4876 CrossRef CAS PubMed.
- R. A. Marcus, J. Chem. Phys., 1956, 24, 966 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1–S10 and Tables S1–S3. CCDC 1554658 and 1545410. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8tc03172j |
| This journal is © The Royal Society of Chemistry 2019 |