
A blood cell repelling and tumor cell capturing surface for high-purity enrichment of circulating tumor cells†
 *ab
*ab
Abstract
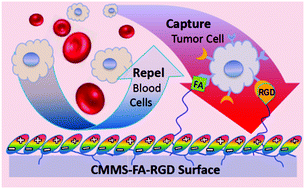
The detection of circulating tumor cells (CTCs), an approach considered to be “liquid biopsy”, is crucial in cancer diagnosis, monitoring and prognosis. However, the extremely large number of blood cells challenges the rare CTC isolation and enrichment. In this report, a red blood cell membrane mimetic surface (CMMS) is fabricated on material-independent substrates to repel blood cell adhesion. Meanwhile, tumor cell targeting ligands, folic acid (FA) and an arginine-glycine-aspartic acid (RGD) peptide, are tethered on the CMMS to give the decorated surface (CMMS–FA–RGD) tumor cell capture ability. The CMMS is composed of a mussel-inspired self-adhesive polydopamine layer and a covalently anchored non-fouling or anti-cell-adhesion layer of a phosphorylcholine zwitterion polymer and poly(ethylene glycol) (PEG). The protruding ends of the PEG chains of the anchored CMMS are further coupled with FA and RGD ligands to endow the tumor cells with specific binding. Furthermore, all the components of the step-by-step constructed surfaces are quantitatively controllable for optimizing the non-specific cell repellence and tumor cell binding performances. Thus, the delicately engineered CTC capture surface enhances the HeLa cell enrichment factor to 19 000-fold by repelling the adhesion of >99.999% blood cells, resulting in high capture efficiency (91%) and capture purity (89%) from the spiked whole blood samples. This substrate independent tumor cell capture and blood cell repellent surface modification strategy may provide a facile, versatile and cost-effective technology solution for more efficient cancer diagnosis and targeted therapy.
 Please wait while we load your content...
Please wait while we load your content...

