
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Nucleic acids presenting polymer nanomaterials as vaccine adjuvants
Alice
Comberlato
,
Kaltrina
Paloja
and
Maartje M. C.
Bastings
 *
*
IMX/IBI, EPFL, EPFL-STI-IMX-PBL MXC 340 Station 12, Lausanne, 1015, Switzerland. E-mail: maartje.bastings@epfl.ch
First published on 8th August 2019
Abstract
Most vaccines developed today include only the antigens that best stimulate the immune system rather than the entire virus or microbe, which makes vaccine production and use safer and easier, though they lack potency to induce acceptable immunity and long-term protection. The incorporation of additional immune stimulating components, named adjuvants, is required to generate a strong protective immune response. Nucleic acids (DNA and RNA) and their synthetic analogs are promising candidates as vaccine adjuvants activating Toll-like receptors (TLRs). Additionally, in the last few years several nanocarriers have emerged as platforms for targeted co-delivery of antigens and adjuvants. In this review, we focus on the recent developments in polymer nanomaterials presenting nucleic acids as vaccine adjuvants. We aim to compare the effectiveness of the various classes of polymers in immune modulating materials (nanoparticles, dendrimers, single-chain particles, nanogels, polymersomes and DNA-based architectures). In particular, we address the critical role of parameters such as size, shape, complexation and release of TLR ligands, cellular uptake, stability, toxicity and potential importance of spatial control in ligand presentation.
 Alice Comberlato | Alice Comberlato is a PhD student at the Programmable Biomaterials Laboratory at École Polytechnique fédérale de Lausanne (EPFL, Switzerland). She works with DNA origami nanoparticles as programmable immune modulators. Previously, she graduated cum laude from the University of Padova (UNIPD, Italy) with a master's degree in Pharmaceutical Biotechnologies. |
 Kaltrina Paloja | Kaltrina Paloja is currently pursuing her PhD degree at École Polytechnique Fédérale de Lausanne (EPFL). Her doctoral research focuses on designing artificial DNA-based T cells to study dendritic cells during infection. As part of her master program in chemical engineering and biotechnology at EPFL, she completed her master's thesis in immune-engineering at the Australian Regenerative Medicine Institute (ARMI), Australia. |
 Maartje M. C. Bastings | Maartje M. C. Bastings, trained as Biomedical Engineer at the Eindhoven University of Technology (TU/e), obtained her PhD in 2012, honored by the University Academic Award. She moved to the Wyss Institute of Harvard University in Boston as a NWO Rubicon and Human Frontier Science Program postdoctoral fellow, where she specialized cellular stability of DNA-origami. Since 2017 she is heading the Programmable Biomaterials Laboratory at EPFL, Switzerland, developing precision-materials as tools to quantify fundamental mechanisms in biology. |
1 Introduction
For decades, the continuous development and use of vaccines helped to save many lives and significantly improved the quality of life. As a method that can control or even eliminate a disease, vaccination can be considered as one of the greatest breakthroughs in modern medicine.1,2 An effective vaccine stimulates both the innate and the adaptive arms of the immune system.3 Innate immunity occurs within hours of pathogen recognition, followed by an adaptive immune response over several days, leading to immune memory. Contrary to the original live or attenuated pathogen vaccines, most vaccines developed today include only the antigens that best stimulate the immune system, such as proteins or peptides, rather than the entire virus or microbe.4 Although this design makes vaccine production and use safer and easier, antigens alone are often not sufficient to induce acceptable immunity and long-term protection. Therefore, the incorporation of additional immune stimulating signals, named adjuvants, are required to generate a strong protective immune response.5Aluminum-containing adjuvants have been used in vaccines since the 1930s. Small amounts of aluminum salts are added to help the body to build stronger immunity against the antigen in the vaccine.6 Newer adjuvants have been developed to target specific components of the body's immune response, yielding a stronger, more specific and long-lasting protection against disease. Among these newer adjuvants, there are nucleotide-polymers DNA, RNA and modifications of both.7 As the cellular receptors for these nucleic acid based-adjuvants reside in the endosomal compartments, protection against extracellular degradation is crucial. Synthetic polymer nanomaterials are rapidly gaining popularity in the field of immune engineering, due to their good biocompatibility, controllable surface properties and biodegradability.8 Through enabling the delivery of functional molecules into cells, as well as protection of DNA and proteins from degradation, nanomaterials can double in function as transport and delivery vehicles as well as vaccine adjuvants themselves.9
In this review, we focus on the recent developments in polymer nanomaterials presenting nucleic acids as vaccine adjuvants. Given the many classes in which polymers can be used as nanomaterial (nanoparticles, dendrimers, single-chain particles, nanogels, polymersomes and DNA-based architectures), we aim to compare the various classes regarding effectiveness for immune modulation. A detailed overview of the respective challenges and advantages is presented, as well as their suitability for translation. While excellent reviews exist on the use of nanoparticles in general in the context of immune engineering,10–15 we specifically focus on polymer-based systems and nucleic acid adjuvants with an emphasis on the materials engineering challenges in terms of properties and stability.
2 TLR-targeting nucleic acid adjuvants in vaccines
2.1 The immune response in vaccines
The immune system is a complex network that has to be manipulated accurately to obtain the desired immunity upon vaccination. Dendritic cells (DCs) are the first players in the vaccination process, as they are the main antigen-presenting cells (APCs), responsible for taking up and presenting antigens within their surface major histocompatibility complexes (MHC) to T cells. Additionally, DCs are the most potent stimulators of the T cell response. They are present in vivo in lymphoid organs and, as a subpopulation, in most tissues to monitor the presence of “danger signals” in the body. DCs express Pattern-Recognition Receptors (PRRs) that detect and bind Pathogen-Associated Molecular Patterns (PAMPs), triggering intracellular signaling pathways associated with DC activation, maturation and migration to the lymph nodes. Mature DCs activate naïve T cells in the lymph nodes through three signals: (A) antigen-specific interaction, in which the antigen-loaded MHCs on DCs bind to T cell receptors (TCR); (B) costimulatory signals, by binding of DC receptors CD80–CD86 to CD28 on T cells; (C) response-specific cytokines (e.g., IL-6, IL-10, IL-12p70, TNF-α).16,17Cytosolic antigens are degraded in the proteasome and loaded on MHC class I that is recognized by CD8+ T cells (cytotoxic T lymphocytes, CTLs), as ubiquitous mechanism of surveillance. Additionally, in APCs extracellular antigens are taken up, degraded in the endolysosomal compartment and loaded on MHC class II that is recognized by CD4+ T cells, activating specific immune responses and antibody production by B cells. Secreting different cytokines in the extracellular environment, DCs can polarize the differentiation of CD4+ T cells into different T helper (TH) cell subtypes, like TH 1 (involved in cell-mediated immunity) or TH 2 (for antibody production), or they can promote the maturation of effector T cells (CTLs). CTLs recognize foreign antigens presented on MHC class I and induce apoptosis of these cells.18
Vaccines can be divided into two categories: “prophylactic vaccines” that produce long-term B cell response against pathogens and “therapeutic vaccines”, mostly for cancer treatment. Cancer therapeutic vaccines require the induction of CD8+ CTL and pro-inflammatory TH 1 response, which is induced by secretion of IFN-γ, IL-12 and TNF-α by activated DCs.19 For cancer therapeutic vaccines, tumor-associated antigens (TAAs), such as oncoviral antigens or neoantigens that are not found in healthy cells, are used to elicit antitumor immunity.20 Cancer cells downregulate antigen-presenting activity so, in new antitumor vaccines, TAAs are delivered to DCs to promote high CTL response against these. By an alternative pathway called “cross-presentation”, DCs uptake extracellular antigens, process and load them on MHC class I to promote CTL response (cross-priming of T cells).21 Whereas the presence of the antigen in the endosomal compartment is useful for MHC class II loading, MHC class I presentation requires it in the cytosol. Following their cellular uptake by endocytosis, antigens should be released in the endosome before lysosome degradation and transferred into the cytoplasm to induce cross-presentation, which is necessary to induce antigen-specific CTLs against tumor antigens but also intracellular pathogens. However, differently from full pathogens, recombinant viral proteins or synthetic peptides that are currently used as antigens in vaccines are not able to trigger sufficiently the immune system. To overcome the weak immunogenicity of these antigens, an effective vaccine should contain both a specific antigen and an “adjuvant” to induce a strong immune response and immunological memory.16
2.2 Old and new adjuvants: why targeting TLRs?
The word “adjuvant” covers a broad range of systems and components, with the common function of enhancing the magnitude of the immune response against co-delivered antigens. One type of adjuvants are antigen delivery systems or depots that protect from degradation increasing antigen stability, provide slow release, enhance uptake (e.g., emulsions, ISCOMs, aluminum salts, liposomes, microspheres and nanoparticles), whereas the second type consists of immunostimulatory agents (e.g., PAMPs).22 Aluminum salts and oil-in-water emulsions such as MF59 (squalene, polysorbate 80, sorbitan trioleate) and AS03 (α-tocopherol, squalene, polysorbate 80), the most common adjuvants present in vaccines approved for human use, induce antibodies production and TH 2 response but they do not induce antigen-specific CTLs and TH 1 response.23 On the other hand, immunostimulatory agents as adjuvants induce TH 1 response, which is required in vaccines against intracellular pathogens and cancer, to specifically activate DCs and fine-tune T cell activation. These molecular adjuvants not only induce maturation and antigen presentation on DCs but can also modulate the quality of the immune response, promoting a specific type of immunity essential for a specific antigen.23 The limited amount of adjuvants available for clinical use but especially the ineffectiveness of alum in inducing cellular responses boosted the research of new immunostimulatory agents. Additionally, it has been shown that aluminum salts compromise long-term antigen stability, possibly due to the different pH on the microenvironment of the aluminum surface.24,25The combination of a specific antigen delivery system with an immunostimulatory molecule can merge the favorable properties of the two adjuvant systems and, in this way, DCs can be programmed towards a tailored response against a pathogen upon the rational choice of a specific “danger signal”. Pathogen-derived molecules bind to PRRs present on DCs, which include receptors of different families, like membrane-associated Toll-like receptors (TLRs), cytosolic nucleotide oligomerization domain (NOD)-like receptors (NLRs) and retinoic acid-inducible gene (RIG)-I-like receptors (RLRs). Their ligand-induced activation and downstream signaling cascade induce upregulation of costimulatory receptors and secretion of pathway-specific cytokines.26 Among PRRs, TLRs on DCs are the best characterized and they have been the target of choice in the last years for synthetic ligands used as vaccine adjuvants. TLRs were preferentially chosen as targets over other PRR classes due to their accessibility by external ligands. While cytosolic delivery requires specific formulations, ligands can easily reach TLRs exposed on the cell membrane or after internalization in the endosome. TLRs have been extensively studied in the past years, as reported in recent outstanding reviews.27,28
2.3 TLRs: structure and function
TLRs are a family of PRRs involved in the first step of the innate immune response, recognizing pathogenic cell components and non-self nucleic acids, such as peptidoglycan, lipoproteins, and bacterial DNA. They are localized on the cell surface (TLR1, TLR2, TLR4, TLR5, TLR6) and on the endosomal membrane (TLR3, TLR7, TLR8 and TLR9) to detect infections and danger signals.29 TLRs are type I transmembrane receptors, with a ligand-binding domain facing the extracellular or endosomal environment. This ectodomain is a horseshoe-shaped solenoid domain, with leucine-rich repeats (LRRs) regions and continuous beta-sheets on the concave inner surface. A transmembrane domain connects this to a cytoplasmic Toll/IL-1 receptor (TIR) domain, involved in the initiation of the intracellular signaling cascade.30 The distinctive cellular location and TLR-type-specific binding site (feature given by the unique LRR side chain arrangements) allow the different classes of TLRs to bind to ligands of different origin and characteristics.31 TLR ligands have been already used in vaccine formulations as adjuvants and, in particular, the first vaccines approved for human use containing a TLR agonist were Cervarix (GlaxoSmithKline) against human papillomavirus (HPV) and FENDrix (GlaxoSmithKline) against hepatitis B virus. They both contain the TLR4 agonist monophosphoryl lipid A (MPL), a lipopolysaccharide (LPS) derivative. AS04 (aluminum hydroxide and 3′-O-deacylated MPL) and AS01 (MPL and saponin QS-21 formulated in liposomes) are adjuvants currently approved for human use in vaccines in Europe and USA.162.4 Nucleic acid-sensing TLRs and signaling
It has been shown that old adjuvants such as alum are triggering the immune response also by the induction of tissue damage, through which the release of endogenous danger signals, namely Damage-Associated Molecular Patterns (DAMPs), is enhanced.32 Since many DAMPs are host nucleic acid-derived molecules, targeting nucleic acids sensing TLRs emerged as a promising strategy for the development of immunostimulatory agents.Nucleic acid-sensing TLRs are TLR3, TLR7/8 and TLR9, activated by dsRNA, ssRNA and unmethylated CpG-containing DNA respectively. They are all localized on the endosomal membrane of immune cells to detect uptaken pathogenic DNA and RNA from the extracellular environment and avoid unwanted recognition of self nucleic acids (Fig. 1). Several nucleic acid sensors are also present in the cytosol (e.g., nucleotide-binding oligomerization domain protein 2 (NOD2), the RIG-I-like receptors (RLRs) and melanoma differentiation-associated protein 5 (MDA5)), which induce the production of proinflammatory cytokines and type I interferons (IFNs).26 Cytosolic nucleic acid receptors are not analyzed in this review, but their activation should be considered in the analysis of possible off-target effects, especially in the case of delivery systems with endosomal escape mechanisms that could bring nucleic acid adjuvants to the cytosol. Indeed, it has been proved that combinatorial activation of cytosolic and endosomal nucleic acid sensors increase the amount of the final immune response, but it can lead to high toxicity and systemic side effects, since many cytosolic nucleic acid receptors are also found in non-immune cells (e.g., fibroblasts, endothelial cells, etc.).33
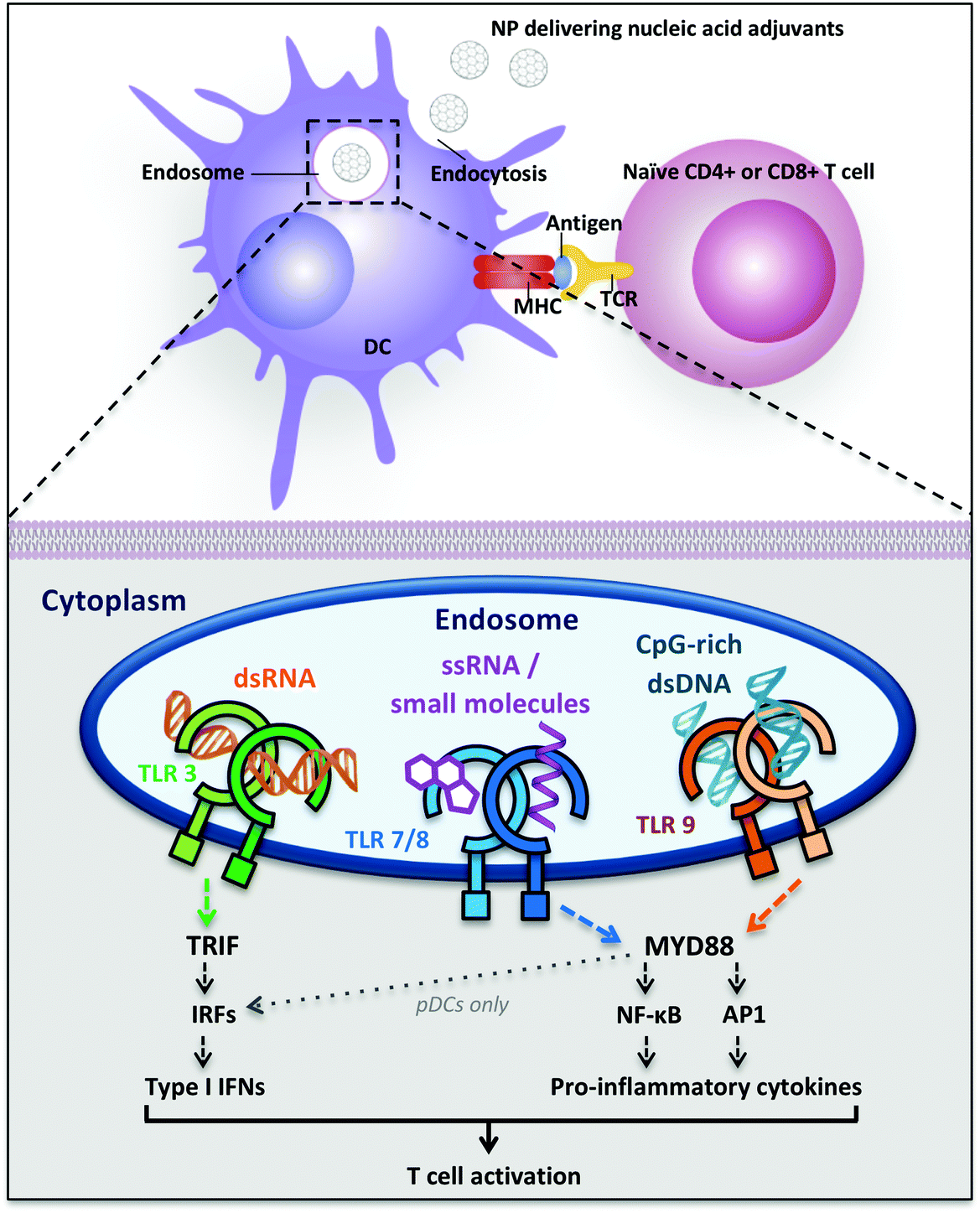 | ||
| Fig. 1 Dendritic cell uptake of nucleic acid-delivering polymer nanomaterials and intracellular Toll-like receptors signaling inducing T cell activation. Abbreviations: AP1 = transcription factor activator protein 1; DC = dendritic cell; IFNs = interferons; IRFs = interferon-regulatory factors; MHC = major histocompatibiliy complex; MYD88 = myeloid differentiation primary-response protein 88; NF-κB = nuclear factor-κB; NPs = nanoparticles; pDCs = plasmacytoid dendritic cells; TCR = T cell receptor; TLR = Toll-like receptor; TRIF = TIR-domain-containing adapter-inducing interferon-β. | ||
Nucleic acid-sensing TLR3 and TLR9 are present as monomers in the inactive form and the ligand binding induces dimerization of these receptors. Ligand-induced dimerization brings in close proximity cytoplasmic Toll/IL-1R domains of two receptors, creating binding sites necessary to recruit adaptor proteins. The main adaptor proteins for the activation of diverse downstream signaling cascade are MyD88 (myeloid differentiation primary-response protein 88) and TRIF (TIR domain-containing adaptor protein inducing IFN-β).31 Differently, TLR7 and TLR8 are already present as dimers in the inactive form and the ligand binding induce conformational changes that are necessary to bring the cytoplasmic TIR domains in proximity for the activation of the downstream signaling. The two major signaling pathways induced following TLR activation are the MyD88-dependent and the MyD88-independent (TRIF-dependent) pathways: they can be distinguished by the different adaptor proteins recruited on the cytosolic domain, which induce separate signaling cascades ending with the activation and migration to the nucleus of transcription factors. The MyD88-dependent pathway activates the mitogen-activated protein kinase (MAPK) cascade leading to the activation of the transcription factor AP1 and induces the activation (and consequent nuclear translocation) of the transcription factor Nuclear Factor-κB (NF-κB) inducing the production of pro-inflammatory cytokines, such as TNF-α, IL-1, IL-6 and IL-12.28,31,34 NF-κB dimers are maintained in the cytoplasm in an inactive form by IκB proteins. TLR signaling cascade activates the IκB kinase (IKK) complex that phosphorylates IκB, inducing its ubiquitylation and subsequent degradation. The proteolysis of IκB releases NF-κB, which is thus activated and free to migrate into the nucleus, where it induces the transcription of several pro-inflammatory genes.34 The MyD88-independent pathway involves the adaptor protein TRIF and leads to the production of type I interferons through the transcription factors Interferon Regulatory Factors (IRFs). In particular, TRIF indirectly induces the activation of the transcription factors IRF3 and IRF7 and also NF-κB.31 TLR7, TLR8 and TLR9 induce MyD88-dependent pathway and pro-inflammatory cytokines production, whereas TLR3 recruits both adaptor proteins MyD88 and TRIF, inducing the activation of the transcription factors NF-κB, AP-1 and IRF, with the subsequent production of pro-inflammatory cytokines, such as IL-12 and IL-6, and also IFNs (IFN-α and IFN-β) (Fig. 1).31
Despite structural similarity and recognition of comparable molecular patterns, TLR7 and TLR8 are expressed on different human DC subsets and their activation induces a dissimilar cytokine array in different locations. TLR7 is expressed on human plasmacytoid dendritic cells (pDCs), which are mainly type I IFN producers, whereas TLR8 is expressed on human myeloid DCs (mDCs, also called conventional DCs (cDCs)) where it stimulates the production of IFNs and pro-inflammatory cytokines with TH 1-polarizing effects (Fig. 1).35 Therefore, the ligand- and location-specific cytokine secretion defines the pattern of polarization of CD4+ T cells, inducing TH 1 or TH 2 immune responses. For example, the pro-inflammatory cytokine IL-12p70 is involved in TH 1 and cell-mediated responses, which are useful against intracellular pathogens and cancer, whereas IL-10 is involved in the TH 2 phenotype and antibodies production against extracellular pathogens.16 Type I interferons are a family of cytokines with antiviral activity, which are able to induce TH 1-type responses.36 Also, their activity has been related to the stimulation of antigen cross-presentation to CD8+ T cells.37,38 Besides the production of cytokines, the maturation of DCs can be detected by the increase in expression of costimulatory cell-surface receptor markers such as CD40, CD80 and CD86, necessary for T cell proliferation.39 Both cytokine production as well as surface-marker analysis can, therefore, be used as an analytical read-out of the effectiveness of a polymeric adjuvant formulation.
2.5 Nucleic acids as adjuvants: three classes
Among TLR ligands, nucleic acids (DNA and RNA) and their synthetic analogs are promising candidates as vaccine adjuvants. They show several favorable drug-like characteristics, such as solubility, easy chemical synthesis and modification and many well-developed analytical methods for characterization. Most of their metabolites are natural components of DNA or RNA, preventing accumulation in the body of toxic compounds, as it is commonly observed as side effect for other small molecule drugs.40 Additionally, the discovery of several receptors for nucleic acids opens the possibility of targeting specific pathways for tailored vaccine design.41 Moreover, nucleic acids are able to induce TH 1 and CTL responses, useful for anticancer and antiviral vaccination.23 In this review, we will focus on three main categories of nucleic acids with adjuvant potential: TLR3-binding analogs of dsRNA, synthetic small molecules agonists of TLR7/8 and CpG-motif-containing DNA sequences binding TLR9 (Fig. 2).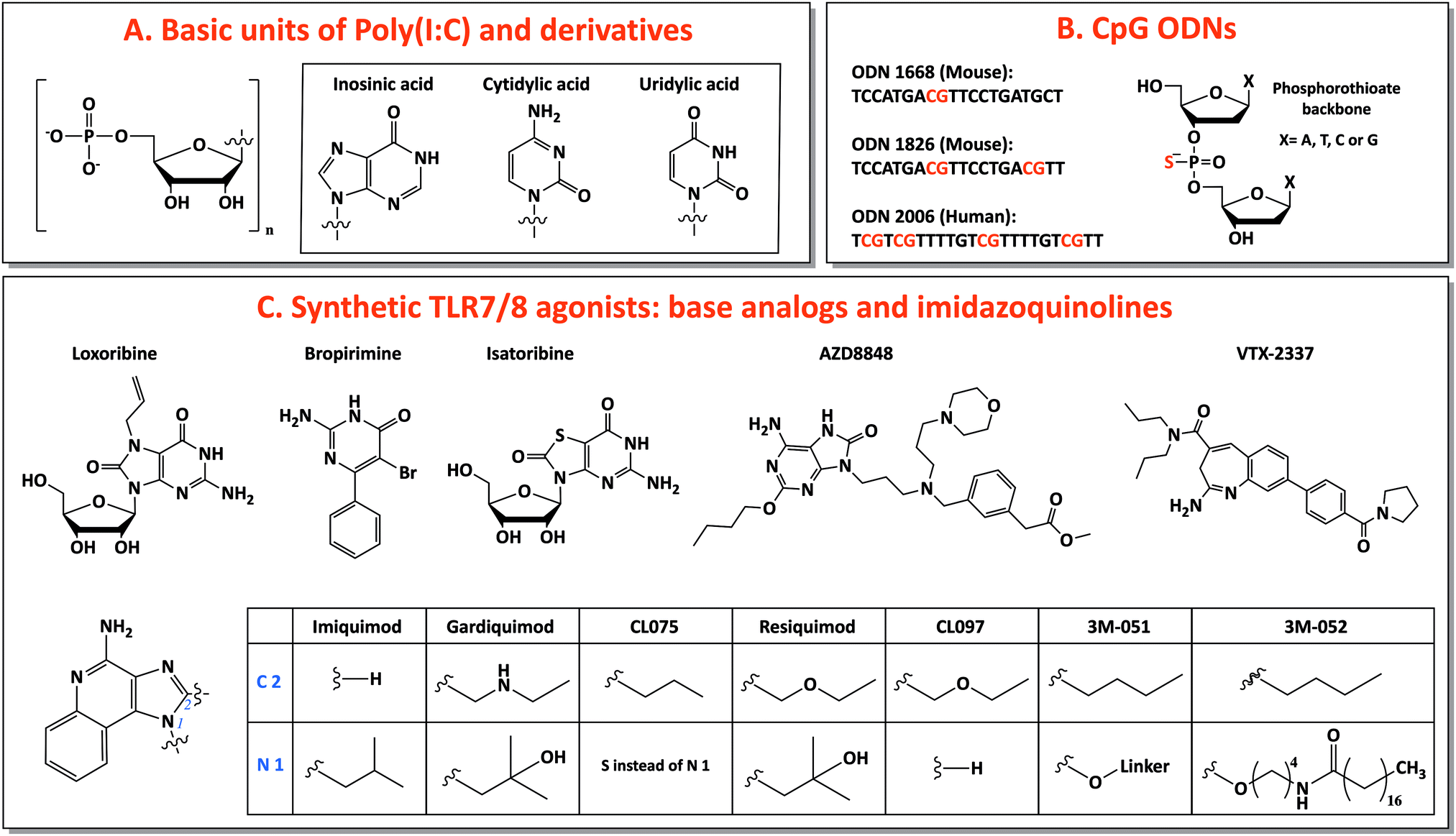 | ||
| Fig. 2 Nucleic acid adjuvants: chemical structures of TLR3, TLR7/8 and TLR9 ligands. | ||
Poly(I:C) was the first synthetic dsRNA analog developed and tested.23 Chemically, it is composed of one strand of riboinosinic acid polymer and of another strand of ribocytidylic acid polymer. Two forms are available, low molecular weight (LMW, length 0.2–1 kb) and high molecular weight (HMW, length 1.5–1 kb) poly(I:C), and both are able to bind TLR3, even if stronger activation was observed for HMW poly(I:C), probably due to the induction of a bigger multivalent complex by the latter.31 Poly(I:C) stimulates one of the strongest TH 1 responses among adjuvants, ideal for antitumor vaccination and against pathogens that require a T cell component, such as HIV.44,45 In particular, it induces high production of interferons and TH 1-specific pro-inflammatory cytokines, together with high expression of costimulatory factors on DC surface, leading to strong activation of DCs and consequently induction of CTLs, memory cells and antibody production.46–48
However, clinical trials revealed the high toxicity of poly(I:C).49 Indeed, poly(I:C) activates not only TLR3 but also cytosolic nucleic acid sensors such as MDA5, which is ubiquitously expressed also in non-immune cells.50,51 By this mechanism, TLR3 ligands increase the expression of MHC class I also in non-immune cells, raising the rate of self-antigen presentation, which can cause tolerance impairment and autoimmunity, especially in the presence of additional inflammatory stimuli due to the induced strong immune response.52 In addition to toxicity, the low in vivo stability of poly(I:C), due to nuclease degradation, slowed down its extensive use as vaccine adjuvant.49 Thus, modified variants of poly(I:C) were developed to reduce its toxicity (poly-IC12U) and increasing its in vivo stability (poly-ICLC), without affecting its potency. Poly-IC12U (trade name Ampligen, generic name rintatolimod) is a modified version of poly(I:C), containing mismatched uracil residues.53 Despite the mismatches reduce its in vivo half-life, this contributes to lower its toxicity compared to poly(I:C), as it has been seen in clinical studies in humans.54,55 Moreover, differently from poly(I:C), poly-IC12U does not bind MDA-5.56 Other dsRNA analogs selective for TLR3 (e.g., IPH 3102 and RGC100) are under development and their clinical efficacy and safety are going to be tested in the following years.23 Poly-ICLC (trade name Hiltonol), the other poly(I:C) derivative, is a complex of poly(I:C) condensed with poly-L-lysine and carboxymethylcellulose.53 This modification increases up to 10-fold the resistance to serum nucleases compared to free poly(I:C).23 As the other components, it promotes a strong immune response with TH 1 profile, high antibody and interferon production.46
Clinical trials including poly(I:C) derivatives used specifically as vaccine adjuvants are limited, since most of the studies exploited them only as antiviral therapeutics. A study comparing the efficacy of poly(I:C) and its derivatives as adjuvants in rhesus macaques using the antigen keyhole limpet hemocyanin as a model, evidenced a 3-fold higher activity of poly-ICLC compared to poly-IC12U, results consistent with the lower stability of poly-IC12U.46 Studies with tumor antigens showed that poly-ICLC and poly(I:C) induce stronger TH 1 immune response compared to other TLR ligands, such as CpG and LPS.45,56,57
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 stoichiometry. However, from crystal structure analysis, it has been discovered that each TLR8 monomer does not bind the full-length ssRNA but rather RNA degradation products, uridine mononucleoside and dinucleotide (GU), in two different sites. TLR7 exhibits a similar activation mechanism, with guanosine, instead of uridine, binding the receptor in the first site.30
:2 stoichiometry. However, from crystal structure analysis, it has been discovered that each TLR8 monomer does not bind the full-length ssRNA but rather RNA degradation products, uridine mononucleoside and dinucleotide (GU), in two different sites. TLR7 exhibits a similar activation mechanism, with guanosine, instead of uridine, binding the receptor in the first site.30
For this reason, base analogs, such as purine and pyrimidine derivatives, and synthetic small molecules (e.g., imidazoquinolines) can activate these receptors and are preferentially used as agonists of TLR7 and TLR8 due to the rapid degradation of natural ssRNA.53 In particular, imidazoquinolines is a class of small molecules represented by imiquimod (R-837), resiquimod (R-848) and derivatives with TLR7- or TLR8-specific or 7/8 bispecific activity. TLR7 can be selectively activated by nucleoside analogs: (1) Loxoribine, a guanosine ribonucleoside analog, specific for human and mouse TLR7; (2) Bropirimine, an aryl pyrimidinone analog; (3) Isatoribine (ANA 245), a guanosine nucleoside analog, initially developed against HCV infection; (4) AZD8848/DSP-3025, initially tested for asthma and hay fever (Fig. 2C).53
Through the synthesis and screening of N1-modified imidazoquinolines, it was discovered that TLR8 selectivity can be obtained with ethyl-, propyl- or butylamino group as N1-substitutions, whereas prolonging the aminoalkyl chain length to pentyl- and p-methylbenzyl-increased affinity for TLR7.58 Additionally, structure–activity relationship (SAR) studies showed that imidazoquinoline potency increases by introducing a C2-alkyl chain: indeed, C2 substituents fit into a hydrophobic pocket present in TLR7/8, as evidenced by X-ray crystal structure analysis (Fig. 2C).59,60
The most used TLR7-selective agonist, which activate pDCs, is Imiquimod (R837: 1-(2-methylpropyl)-1H-imidazo[4,5-c]quinolin-4-amine).53 Gardiquimod (1-(4-amino-2-ethylaminomethylimidozo[4,5-c]quinolon-1-yl)-2-methylpropan-2-ol) is similar to imiquimod and selective for TLR7 when used at a concentration lower than 1 μg ml−1.31 CL075 (3M-002) is a TLR8 selective agonist at a concentration lower than 0.1 μg ml−1 and it is characterized by the absence of the N1-isobutyl group, typical feature of TLR7 agonists.31,61 Among base analogs, VTX-2337 (trade name Motolimod) is a small molecule selective for TLR8, able to stimulate mDCs. Contrarily, resiquimod (R848: 4-amino-2(ethoxymethyl)-α,α-dimethyl-1H-imidazol[4,5-c]quinoline-1-ethanol), a bispecific TLR7/8 agonist, and its derivative CL097 are able to activate both TLR7 and TLR8 (Fig. 2C).53,62,63
Clinical trials using these molecules were initially investigating their antitumor and antiviral activities.53 Later, it has been showed that TLR7/8 agonists are not strong adjuvants when injected with the antigen: the high diffusion capacity of imiquimod, resiquimod and derivatives induce a rapid systemic distribution, bringing the molecules away from the injection site (and away from the antigen), inducing systemic side effects and reduced efficacy. Nevertheless, the adjuvanticity of these compounds has been improved by introducing modifications or a specific formulation able to retain the TLR7/8 agonist at the administration site together with the antigen.64
Thanks to dermal applications, it is possible to obtain a local immune response at the vaccination site, preventing the systemic toxicity induced by rapid diffusion into the blood after injection.64 Imiquimod is already approved for clinical use in topical applications (Aldara, imiquimod 5% cream) as anticancer drug against basal and squamous cell carcinoma and as antiviral agent to treat genital warts.41,65 In recent clinical studies, imiquimod cream Aldara and resiquimod gel have been applied topically at the vaccination site, showing a good immune response with minimal systemic side effects. A second efficient strategy that has been applied to keep the antigen and these small molecules in close proximity is to introduce chemical modifications (e.g., 3M-052, a lipid-modified imidazoquinoline) or to conjugate them directly to the antigen (e.g., 3M-051).65,66
:CpG 2:2).30 It is expressed in pDC, whose activation induces a strong TH 1-polarizing immune response.40 The immunostimulatory features of bacterial CpG DNA were copied to create synthetic CpG-motif-containing oligodeoxynucleotides (ODNs), which are fully or partially modified introducing a phosphorothioate back-bone to confer protection against nuclease degradation, increasing their in vivo half-life.69
Three classes of CpG ODNs, different in structure and function, have been developed. Class A (type D) CpG ODNs have a mixed phosphodiester/phosphorothioate backbone, they are inducers of high levels of IFN-α but they have limited clinical applications because they aggregate in salt solutions forming complex large structures. Class B (type K) CpG ODNs are made of a full phosphorothioate backbone and one or more CpG motifs, they are potent activators of DCs, with high level secretion of IL-12 and polarized TH 1-type cell response: this is the only type involved in clinical use. The last type, class C, exhibits intermediate properties between A and B classes.40,41 Considering species-specificity, the CpG motif sequence for mouse TLR9 is GACGTT (in B-class ODN 1668 and 1826) and for human TLR9 is GTCGTT (in B-class ODN 2006) (Fig. 2B).67 B-class CpG ODN 2006 (also known as Promune, PF-3512676 or CpG 7909) is the most widely utilized CpG ODN. Overall CpG ODNs are so far the nucleic acid adjuvants that have been most extensively characterized in animals and humans, showing a good safety profile.23 In mice, CpG ODNs are able to induce the strongest TH 1-polarized immune responses among different adjuvants and this ability is conserved when CpG is administered together with other adjuvants (e.g., alum) that induce a TH 2 response.70,71
Several clinical trials in human (reported in http://ClinicalTrials.gov) have verified the efficacy of CpG ODNs as adjuvant in vaccines against infectious diseases, such as hepatitis B,72,73 anthrax,74 and malaria.75,76 HEPLISAV is the first approved vaccine containing a CpG ODN, developed to prevent Hepatitis B. It induces a faster, higher and longer seroprotection with fewer immunizations compared to the previous patented vaccine Engerix-B, which contains alum as adjuvant.77 Furthermore, on-going clinical trials are demonstrating the potential of B-class CpG ODNs as adjuvants in cancer therapeutic vaccines using tumor-associated-antigens such as melanoma associated-peptides Melan-A/MART1, MAGE-3 and NY-ESO-1.78–81
2.6 TLR agonists as adjuvants: a comparative analysis
The lack of a comprehensive standardized comparison between adjuvants complicates the evaluation of the best system to enhance the immune response. While the improved efficiency of TLR-specific agents (e.g., CpG ODNs) compared to traditional adjuvants such as alum and complete Freund's adjuvant has been extensively proved, few studies compared the performance of different TLR ligands.82–84Kwissa et al. studied the innate response in rhesus macaques induced by MPL, resiquimod and CpG ODNs, agonists of TLR4, TLR7/8 and TLR9 respectively.85 Interestingly, neither formulations nor synthetic carriers were used in this study, reducing additional mechanisms complicating the analysis. It has been observed that exclusively TLR7/8 and TLR9 ligands mobilized the “inflammatory” CD14+CD16+ and the “patrolling” CD14dimCD16++ monocytes, activated mDCs and pDCs and enhanced the production of type I IFN and chemokines in the blood. Additionally, the sustained effect of CpG ODNs was demonstrated by the observation of secondary expansion of monocytes, which was not detected with other ligands. Moreover, resiquimod and CpG ODNs, but not MPL, increased the expression of costimulatory molecules on mDCs.
Ghosh et al. analyzed the TLR-activation-induced secretion profile of eleven cytokines (IFN-α/β, IFN-γ, IL-12p40/IL-12p70, IL-4, 1L-13, TNF-α, IL-1β, IL-2, and IL-10) and four chemokines (MCP-1, MIP1β, IL-8, and RANTES) using human PDMCs.86 Despite sharing common pathways, TLR agonists induce the production of remarkably diverse cytokine/chemokine profiles. Type I IFN was highly induced by TLR7, TLR7/8, TLR8 and TLR9 in comparable amount, whereas diversity was observed for type II IFN secretion. TH 1 and TH 2 responses are stimulated by IL-12 and IL-4/IL-13 respectively. The TH 1-inducer IL-12 is a heterodimer glycoprotein (IL-12p70) of two subunits, IL-12p40 and IL-12p35. The latter is highly stimulated whereas the former shows low induction by TLR2, TLR4, TLR7/8 and TLR8 agonists, with TLR8 ligands exhibiting the maximal TH 1 polarizing effect. On the other hand, only minimal induction of TH 2 cytokines (IL-4 and IL-13) by TLR agonists was observed. TLR agonists, in particular for TLR2, TLR7/8, TLR8 and TLR9, showed low IL-2 expression, though probably sufficient to reverse the IL-2-dependent CD4+CD25+ regulatory T cell (Treg) suppressive function. TLR ligands induced high levels of proinflammatory cytokines (TNF-α, IL-1β), but also of the anti-inflammatory IL-10, in enough amounts to balance excessive inflammation without preventing its occurrence. About chemokine responses, the endosomal TLRs showed the lowest induction of IL-8 and RANTES. Overall, TLR8 and TLR7/8 agonists induced the highest levels of proinflammatory cytokines (IL-12, TNF-α and IL-1β) and IFN, whereas TLR2, TLR2/6, TLR4 and TLR5 agonists stimulated high production of pro-inflammatory chemokines but variable amount of pro-inflammatory cytokines (maximal for TLR2 and TLR4).
3 Nanomaterials in vaccines
Nucleic acids are negatively charged macromolecules, exhibiting unfavorable biodistribution and pharmacokinetics.87 Indeed, cell membranes bear the same surface charge that makes them impermeable to nucleic acid molecules, as their transport across the membrane often requires the use of transfection agents or external physical forces.88 Moreover, naked nucleic acids traveling in the bloodstream are subject to degradation by endogenous nucleases or excretion through the kidney.89 Therefore, increasing the in vivo half-life, cellular uptake, and stability of nucleic acid adjuvants is the key for their clinical application as therapeutic products.A plethora of nanomaterials, including polymers,90 liposomes,91 silicon-,92 carbon-93 and metal-based94 materials, have been investigated for the delivery of payloads such as drugs, antigens and nucleic acid adjuvants. These nanostructures display very diverse physicochemical properties, sizes, compositions, surface chemistry and payload capacity, which can be tuned according to the application.95 This broad range of characteristics provides flexibility for the rational design of vaccines that can mediate targeted delivery of antigens and adjuvants in ways unachievable with classical vaccination approaches.96 After traditional adjuvants (e.g., alum), nowadays the field of immunotherapy is focusing on cancer nanovaccines (e.g., nanoparticles) as platforms for co-delivering antigens and adjuvants to obtain more specific immune responses mimicking the multivalent presentation of natural pathogens. Over the past years, nanocarrier-based delivery systems have gained attention as a vaccine platform due to their suitability to control the delivery of vaccine components and overcome the limitations in pharmacokinetics and bioavailability.97,98 The incorporation of adjuvants into nanosized particles has the potential to enhance adjuvant stability and facilitate targeted delivery, which in turn increases vaccine potency and minimize undesirable side effects.96 For example, De Titta et al. demonstrated that CpG encapsulation in nanoparticles induces higher DC uptake and maturation and T cell activation compared to injection of soluble CpG ODNs, which is currently the most common way for CpG ODN administration.99 Therefore, engineering and development of smart delivery nanocarriers are critical for successful orchestration of the desired immune response.
3.1 Importance of antigen and adjuvant co-delivery
The co-delivery of antigens and adjuvants is central for specific and efficient antigen-presenting cell activation. These cells have to process and surface-present the antigen on MHC molecules, releasing inflammatory mediators and priming antigen-specific cell population, including effector CD4+ and cytotoxic CD8+ T cells (Fig. 1).26,100,101 Many studies have reported the synergic effects of polymer-based vaccines delivering both nucleic acid adjuvants and antigens targeting APCs in a single delivery system allowing optimal protective immune response against specific pathogen-like components: for example, recently, Weber et al. employed loaded polypept(o)ide-based polymersomes for simultaneous delivery of CpG motif-containing adjuvant and ovalbumin as antigen model.102 Upon particle engulfment, they achieved bone marrow derived dendritic cell (BMDC) stimulation and activation with increased expression of CD80, CD86, and MHC class II surface markers and enhanced inflammatory cytokine levels (TNF-α). The co-encapsulation of CpG and antigen greatly promoted antigen-specific T cell proliferation in contrast to free antigen treatment. In addition, co-delivery of tumor-associated antigens and TLR adjuvants in the context of cancer immunotherapy has been demonstrated. In Liu's group, imiquimod (R837) encapsulating PLGA nanoparticles were coated with mannose-modified cancer cell membranes and were tested for their effectiveness to trigger antitumor immunity.103 Considering that tumor-derived antigens are recognized by APCs via specific binding between mannose and APC receptors, Yang et al. showed that, compared to its antigen-free counterpart, this anticancer vaccine significantly enhances uptake and DC maturation allowing downstream antitumor action. They showed that this nanovaccine can effectively accumulate in the draining lymph nodes and promote tumor-specific effects, characterized by the inhibition and the delay of tumor progression.3.2 Size: a key parameter for lymph node targeting
Robust and long-term vaccine-elicited protection usually requires activation of the adaptive immune system, which involves a highly orchestrated process including antigen capture, processing and internalization by APCs, triggering their activation.104 Once activated, APCs prime T and B cells, mounting downstream immune responses.105,106 The site of vaccination greatly impacts vaccine efficacy. Most traditional vaccines are administered in the periphery, where DC trafficking is slower than for other APCs, reducing the chance to mount an adaptive immune response.107 Thus, very variable immune responses can be achieved in different target tissues.108 The lymph nodes have emerged as an effective target site to precisely control the desirable vaccination outcome, e.g., TH 1- or TH 2-type response. The lymphatic system is designed to filter the lymph fluid, sort foreign materials entering the host and initiate immune reactions to remove these foreign materials.109,110 Targeting lymph nodes with nanoparticles as a vaccine platform can be an efficient strategy, since lymphoid tissues host a large number of DCs, through which adaptive immunity is initiated and regulated.106 Vaccine carriers can be effectively drained to lymph nodes from lymphatic capillaries after interstitial administration via subcutaneous, intramuscular or intradermal injection.106,111 However, from the material perspective, successful lymph node targeting is strongly influenced by critical factors, such as surface charge, composition, shape, and size of the vaccine vehicle.106In particular, the size of the vaccine system deeply influences their access to lymphatic vessels and lymph nodes following injection. Smaller particles (<5 nm) usually results in adsorption into the blood, while particles up to 200 nm can enter the lymphatic system by passively diffusing through the lymph vessels.105 Subsequently, small-sized particles can be taken up by resident immature lymph-node DCs without the need for processing by peripheral DCs in tissues. Therefore, targeting the lymph nodes prevents premature antigen presentation by DCs, which can enhance the vaccine efficacy at low doses and prevent immune cell immunity.14,109 Nanoparticles of size in the range of 20–200 nm can be easily taken up by DCs by clathrin- and caveolae-dependent endocytosis mechanisms.105 The size determines the final success of the uptake by APCs and of the targeting to different endocytosis pathways, influencing antigen presentation and type of immune response. Indeed, nanometer systems preferable induce CD8+ T cell response by cross-priming. It has been verified that 40 nm is the optimal size for nanoparticles to have efficient cross-priming in mice.107
3.3 Classes of polymer nanomaterials suitable for nucleic acid presentation
Over the past decades, a variety of polymeric nanomaterials have been developed as nucleic acid delivery systems in order to achieve cell-specific targeting, including polymer nanoparticles, single-chain particles, dendrimers, polymersomes and recently also DNA-based architectures (Fig. 3A). These materials exhibit superior intrinsic properties such as biodegradability and biocompatibility and have the advantage of being easily manufactured and modified for specific purposes.88 Ideally, polymeric carriers should also present properties that include safekeeping of the biological activity of vaccine components and prevention from chemical and enzymatic degradation, thus avoiding undesirable biological side effects and toxicity. The different classes of polymer architectures with potential for creating nucleic acid adjuvant-presenting nanomaterials are analyzed in the following sections.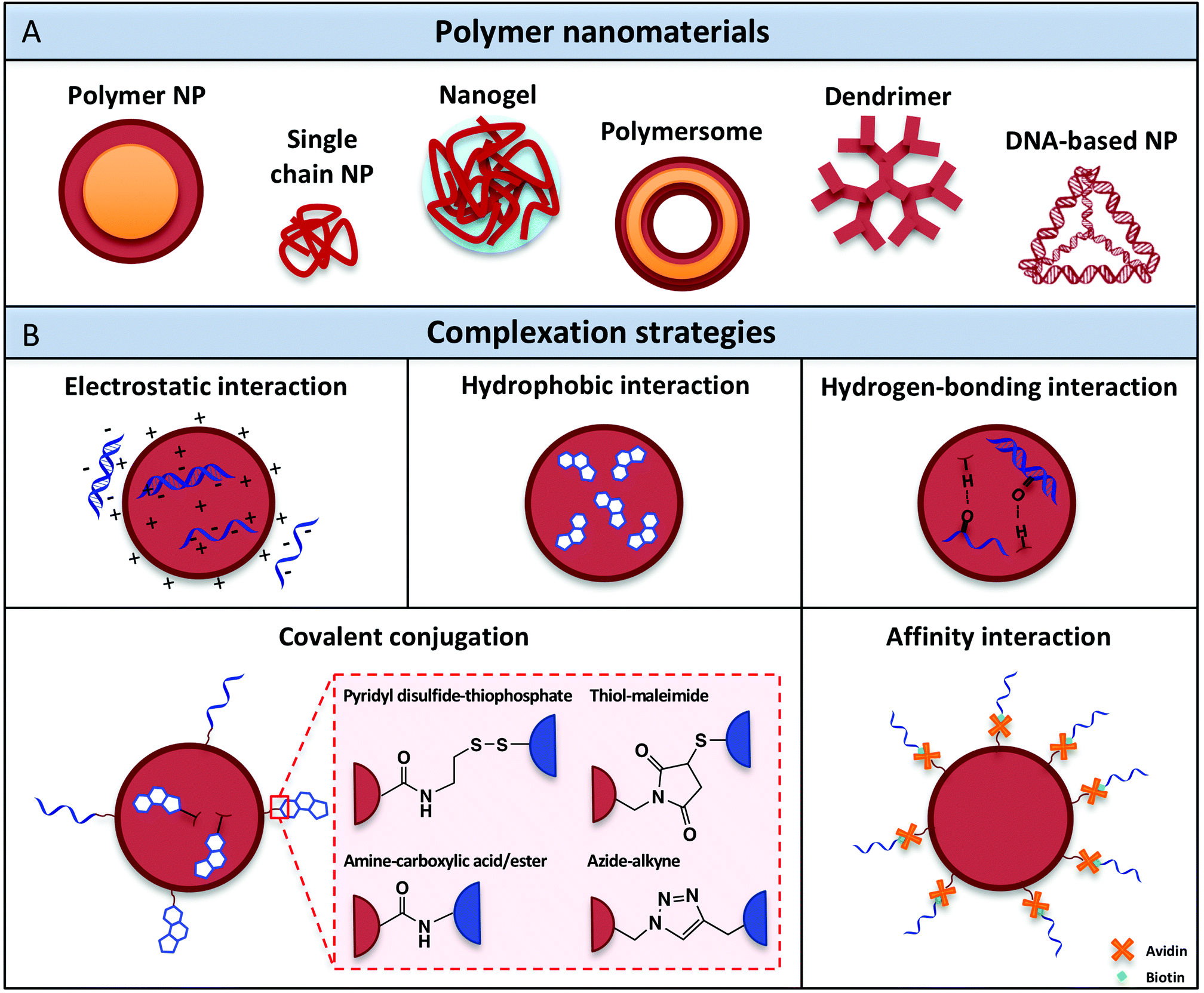 | ||
| Fig. 3 (A) Polymer nanomaterials used for nucleic acid adjuvant delivery. (B) Complexation methods developed to attach nucleic acid adjuvants within or onto the surface of polymeric nanocarriers. | ||
Poly(D,L-lactic-co-glycolic acid) (PLGA) is the most exploited polymer for creating NPs for drug delivery.114 It has been FDA-approved for parental use, thanks to its biodegradability and biocompatibility features. PLGA can be easily metabolized, as it is a copolymer that, upon hydrolysis, releases lactic acid and glycolic acids as metabolites, which are naturally present also in our body.113 This mechanism of hydrolysis in acidic environment can be exploited to fine tune the degradation rate, controlling the monomer ratio in PLGA formulation. NP size and drug release can be regulated by rational design of PLGA properties, such as the ratio of lactide and glycolide components and the addition of groups for functional modification.115 Many groups developed PLGA NPs for nucleic acid adjuvants and antigens delivery (Table 1). For example, Figdor and coworkers studied the kinetics of antigen release in PLGA NPs and demonstrated that coencapsulation of resiquimod and Tetanus Toxoid (TT) peptide antigen in PLGA NPs showed an enhanced T cell response, increasing strength and duration of the immune response compared to soluble components.116 They demonstrated that a slower kinetics of antigen release is more effective for MHC class II and I cross-presentation in human and mouse DCs. Therefore, the prolonged controlled release induced by PLGA NP degradation in the endosome could improve the activity of vaccines. PLGA NPs offer design flexibility in creating new systems with wide loading capacity: Kasturi et al. developed PLGA NPs able to encapsulate both imiquimod and MPL, a TLR4 agonist, showing the advantages of synergistic effects of different adjuvants.117 However, they can have high polydispersity due to the production method and they can be unstable in physiological media over time.
| Nanocarrier | Nucleic acid adjuvant(s) | Target receptor | Antigen(s) | Size (nm) | Mode of adjuvant presentation | Adjuvant complexation method | Ref. |
|---|---|---|---|---|---|---|---|
| Abbreviations: PLGA = poly(D,L-lactic-co-glycolic acid); pLHMGA = poly(D,L-lactic-co-hydroxymethyl glycolic acid); PEG = polyethylene glycol; PEI = polyethylenimine; PCL = poly(ε-caprolactone); PLA = polylactic acid; PPS = poly(propylene sulfide); mTEGMA = methoxy triethylene glycol methacrylate; PFPMA = pentafluorophenyl methacrylate; MEO3MA = tri(ethylene glycol)methyl ether methacrylate; SPG = schizophyllan; CH-CA-DEAE = cholesterol-modified cycloamylose-diethylaminoethane; PGlu(OBn)-b-Psar = poly(benzyl-glutamic acid)-b-polysarcosine; PAMAM = poly(amidoamine); OVA = ovalbumin; H5N1 = hemagglutinin 5 neuraminidase; HPV = human papilloma virus; SLP = synthetic long peptide; gp = glycoprotein; PPE = proline–proline–glutamic acid; SHe = small hydrophobic ectodomain; MUC1 = mucin 1; Ag85B = antigen 85B. | |||||||
| Nanoparticles | |||||||
| PLGA | Poly(I:C) | TLR3 | OVA; HPV-E7 antigen | 195–246 | Encapsulation | — | 203 |
| PLGA | Imiquimod | TLR7 | Mixed tumor-associated antigens of membranes from B16-OVA cancer cells | 120–160 | Encapsulation | Hydrophobic interaction | 103 |
| PLGA | Imiquimod | TLR7 | OVA; Bacillus anthracis protective antigen (PA); haemagglutinin from avian influenza H5N1 virus | 300 | Encapsulation | Hydrophobic interaction | 117 |
| PLGA | Imiquimod | TLR7 | — | 100 | Encapsulation | Hydrophobic interaction | 204 |
| PLGA | Gardiquimod | TLR7 | — | 194 | Encapsulation | Hydrophobic interaction | 205 |
| PLGA | Resiquimod | TLR7/8 | OVA; tetanus toxoid (TT) peptide | 264 | Encapsulation | Hydrophobic interaction | 206 |
| PLGA | Dual-agonist imidazoquinoline | TLR7/8 | OVA | 202 | Encapsulation | Hydrophobic interaction | 194 |
| PLGA | Dual-agonist imidazoquinoline | TLR7/8 | OVA | 156 | Encapsulation | Hydrophobic interaction | 115 |
| PLGA | CpG | TLR9 | West Nile virus envelope protein | 272 | Surface | Biotin–avidin coupling | 202 |
| pLHMGA | Poly(I:C) | TLR3 | HPV-16 E7 SLP peptide | 100–200 | Encapsulation | Electrostatic interaction | 207 |
| PLGA–PEG | Resiquimod | TLR7/8 | — | 270 | Encapsulation | Hydrophobic interaction | 208 |
| PLGA–PEI | Resiquimod; CpG | TLR7/8; TLR9 | OVA | 125–221 | Encapsulation (resiquimod); surface (CpG) | Hydrophobic interaction (resiquimod); electrostatic interaction (CpG) | 198 |
| PLGA–PEG–PCL | Poly(I:C); CpG | TLR3; TLR9 | OVA; melanoma-associated antigens (Melan-A:26, gp100:44, or gp100:209) | 145–190 | Encapsulation | — | 209 |
| mPEG–PLA | Resiquimod | TLR7/8 | — | 205–288 | Encapsulation | Hydrophobic interaction | 200 |
| mPEG–PLA; PLGA/mPEG–PLA | Resiquimod | TLR7/8 | — | 154; 278 | Encapsulation | Hydrophobic interaction | 210 |
| Chitosan | Poly(I:C) | TLR3 | — | 384 | Encapsulation | Electrostatic interaction | 195 |
| Chitosan | Poly(I:C) | TLR3 | OVA | 254 | Encapsulation | Electrostatic interaction | 211 |
| Trimethyl-chitosan | Poly(I:C); CpG | TLR3; TLR9 | Bacillus anthracis protective antigen (PA) | 254 | Encapsulation | Electrostatic interaction | 212 |
| Trimethyl-chitosan | CpG | TLR9 | OVA | 304 | Encapsulation | Electrostatic interaction | 213 |
| mPEG–chitosan–poly(L-lysine) | CpG | TLR9 | OVA | 203 | Encapsulation | Electrostatic interaction | 196 |
| Pluronis–PSS | CpG | TLR9 | OVA | 25–30 | Surface | Pyridyl disulfide-thiophosphate coupling | 99 and 201 |
| Nanogels | |||||||
| Hyaluronic acid | (Dual-)agonist imidazoquinoline | TLR7; TLR8; TLR7/8 | Diphteria toxoid peptide (CRM197) | — | Encapsulation | Amine–carboxylic acid coupling | 146 |
| mTEGMA-b-PFPMA | Dual-agonist imidazoquinoline | TLR7/8 | Mycobacterium tuberculosis antigen (PPE44) | — | Encapsulation | Amine–ester coupling | 139 |
| mTEGMA-b-PFPMA | Dual-agonist imidazoquinoline | TLR7/8 | Human respiratory syncytial virus protein (SHe) | 40–50 | Encapsulation | Amine–ester coupling | 140 |
| MEO3MA-b-PFPMA | CpG | TLR9 | MUC1 glycopeptide; tetanus toxoid (TT) peptide | 64 | Encapsulation | Electrostatic interaction | 145 |
| β-Glucan SPG | CpG | TLR9 | OVA | 150 | Encapsulation | Hydrogen bonding | 143 |
| β-Glucan SPG | CpG | TLR9 | — | 150 | Encapsulation | Hydrogen bonding | 141 |
| CH-CA-DEAE | CpG | TLR9 | — | 198 | Encapsulation | Electrostatic interaction | 144 |
| Polymersomes | |||||||
| PEG-b-PPS | Gardiquimod; resiquimod | TLR7; TLR7/8 | OVA | 100 | Encapsulation | Hydrophobic interaction | 154 |
| PEG-b-PPS | CL075 | TLR8 | Ag85B peptide | 120 | Encapsulation | Hydrophobic interaction | 156 |
| PGlu(OBn)-b-Psar | CpG | TLR9 | OVA peptide | 39 | Encapsulation | Hydrophobic interaction | 102 |
| Dendrimers | |||||||
| Tris(2-aminoethyl)amine | Dual-agonist imidazoquinoline (dendrimer core) | TLR7/8 | — | — | Surface | Azide–alkyne coupling | 161 |
| Cyanuric chloride (dendrimer core) | Loxoribine; CpG | TLR7; TLR9 | — | 27–106 | Surface | Thiol–maleimide coupling (CpG); azide–alkyne coupling (loxoribine) | 163 |
| PAMAM | CpG | TLR9 | — | 40 | Surface | Electrostatic interaction | 166 and 214 |
| DNA-based platforms | |||||||
| Y-shaped | CpG | TLR9 | — | 7 | Surface | Structure-integrated | 175 |
| Dendrimer | CpG | TLR9 | — | 20–36 | Surface | Structure-integrated | 176 |
| Dendrimer | CpG | TLR9 | — | 16–20 | Surface | Structure-integrated | 177 |
| Nanotube | CpG | TLR9 | — | 40 × 8 | Surface | Structure-integrated | 215 |
| Nanotube | CpG | TLR9 | — | 80 × 20 | Surface or inside | DNA–DNA hybridization | 181 |
| Tetrahedron | CpG | TLR9 | — | 6 | Surface | Structure-integrated | 182 |
| Tetrahedron | CpG | TLR9 | Streptavidin | — | Surface | Structure-integrated | 183 |
| Nanoflowers | CpG | TLR9 | — | 100–300 | Surface/inside | Structure-integrated | 178 |
Similar to PLGA, poly(D,L-lactic-co-hydroxymethyl glycolic acid) (pLHMGA) can also be used to create nanoparticles for drug delivery. Homopolymers such as poly(L-lactic acid) (PLA) and poly(glycolic acid) (PGA) could be used as well, even if unfavorable properties (e.g., slow biodegradation rate) limited their use. Polystyrene (PS) is another synthetic homopolymer used for nanocarrier preparation, which is easy to functionalize but not biodegradable, limiting its safety.118 Poly(propylene sulfide) (PPS) is a biodegradable synthetic homopolymer.107 Due to its oxidation-sensitive properties, ligands can be conjugated to the polymer through reducible linkers to fine-tune the release.119 In Hubbell lab, pluronic-stabilized PPS NPs with thiol-reactive pyridyl disulfide groups were used to create ultrasmall polymeric nanoparticles for the delivery of CpG adjuvant.99In vitro studies showed an enhanced DC maturation, TH 1 profile cytokines secretion and induction of effector CTL and memory compared to the injection of free adjuvant. CpG-loaded NPs reshaped the T helper cell distribution within the tumor towards a TH 1 phenotype, locally increasing antigen-specific effector cells and slowing tumor growth in vivo. Polyethyleneimine (PEI) is a linear or branched synthetic cationic homopolymer. Traditionally used as transfection reagent, it can also be used to create NPs, exploiting its high nucleic acid complexation capacity. Due to its intrinsic buffer capacity, it can induce endosomal escape of cargo ligands by the so-called “proton sponge effect”, useful feature for antigen cross-presentation.120 However, it is not biodegradable and highly toxic, features that limit its clinical use.121 Additionally, natural biopolymers have been exploited to create biocompatible NPs. The most common representative of this class is chitosan, a biodegradable, non-toxic, hydrophilic linear polysaccharide, composed of N-acetyl-D-glucosamine and D-glucosamine units. Chitosan is made by alkaline deacetylation of chitin, a polysaccharide present in insects and crustacean exoskeleton.122 Interestingly, it has been observed that chitosan nanoparticles can have intrinsic adjuvant properties.123 Other references of main studies performed with nucleic acid adjuvant-presenting polymeric nanoparticles can be found in Table 1.
Water-soluble SCNPs have been explored in biomedical applications including drug-delivery and as in vivo imaging reagents, while their use as immune-modulating compounds remains limited to one study.128,129 Nativi and co-workers incorporated the α-Tn antigen mimetic moiety to dextran SCNPs to trigger immune responses similar to immune proteins on human peripheral blood mononuclear cells (PBMCs).130 Since the native antigen has limitations in terms of stability and antibody response, presentation on a SCNP was hypothesized to enhance the immune active potential. Indeed, upregulation of IL-6 and IL-10 was measured, which was absent in non-functionalized SCNPs. Although there seems to be potential for the use of SC-NPs in the immune-engineering domain, no studies using nucleic acid danger signals presenting SCNPs as adjuvants have been performed, which opens opportunities for exciting future developments in this area.
Polyelectrolyte nanogels can readily incorporate oppositely charged biomacromolecules such as DNA and RNA, making them interesting as an adjuvant platform for synthetic vaccine development. The first oligonucleotide-loaded nanogel was presented by Vinogradov et al. and consisted of a cross-linked network of cationic and neutral polymers, e.g., branched polyethylenimine and polyethylene glycol (PEI-cl-PEG).135 Following the work of Vinogradov, research towards the use of nanogels for vaccine adjuvants gained popularity. To stay within the scope of our review, we here only address those nanogel platforms that include oligonucleotide danger signals. For excellent reviews on nanogels in broader pharmaceutical context, the reader is referred to existing literature.134,136–138
De Geest et al. synthesized degradable polymer nanogels that were prepared by self-assembly of amphiphilic block copolymers composed of a hydrophilic, PEG-like polymer block based on methoxy triethylene glycol methacrylate (mTEGMA) and a hydrophobic polymer block based on pentafluorophenyl methacrylate (PFPMA).139 These polymers self-assembled into pH-degradable nanogels that could be covalently functionalized with a small-molecule imidazoquinoline-based TLR7/8 agonist. The resulting system efficiently activated its receptor in vitro, as measured by MHC class II and CD80 expression on BMDCs. More interestingly, the nanogel formulation restricted the immune-activating properties of the molecular adjuvant to the injection site and its draining lymph nodes, as measured by the distribution of IFN-β activation in mice, reducing the systemic inflammatory toxicity associated with the use of soluble TLR agonists. In a follow-up study, the same group demonstrated that the core-crosslinked polymer nanogels massively outperform soluble polymers with exactly the same chemical composition in terms of lymphatic transportation. Additionally, the system was evaluated as potential vaccine adjuvant for human respiratory syncytial virus (RSV) and outperformed a benchmark oil-in-water adjuvant.140 These imiquimod nanogels provoked a robust antibody response of IgG1 and especially TH 1-triggered IgG2a isotypes allowing for viral load reduction in the lungs of mice that received a viral challenge. These promising results pave the way for future translation of nanogels as adjuvants platform in immune engineering.
In the Sakurai lab, nanogels were prepared using the polysaccharide schizophyllan (SPG), a member of β-glucans.141 SPG can function as a delivery carrier of oligonucleotides and the resulting complex can be recognized and subsequently internalized by the Dectin-1 receptor that is present on APCs including macrophages, dendritic cells, monocytes, neutrophils and B cells.142 Crosslinked nanogels were formed consisting of CpG-dA40/SPG and cCpG-dA40/SPG (with the complementary sequence of CpG) complexes through DNA–DNA hybridization. The cross-linked nanoparticle was much larger and showed a higher immune response at significantly low concentrations as measured by IL-6 secretion, than the individual CpG-dA40/SPG, even though some of the immunostimulating CpG portion was hybridized. This initial study was followed up by a larger in-depth evaluation of the adjuvant efficiency in vivo.143 The enhanced immunogenic performance of the cross-linked system was attributed to a larger size of the nanogel and multivalent cluster effect of the β-glucan recognition site in the nanogel structure, resulting in an improved cellular uptake. In the Akiyoshi lab, cationic nanogels consisting of cycloamylose modified with cholesterol and diethylaminoethane, to respectively form hydrophobic cross-linking points and to add positively charged groups, were synthesized.144 They measured that complexes of CpG with the cationic nanogels successfully delivered the CpG danger signal to the macrophage-like cell lines J744A.1 and RAW264.7 and induced a strong IL-12 cytokine production. Zentel and coworkers developed a self-adjuvanting nanogel vaccine platform through the combination of tumor-associated MUC1-glycopeptide B-cell epitope and tetanus toxin T-cell epitope P2 with cationic nanogels.145 The nanogel core consisted of block copolymerized pentafluorophenyl methacrylate (PFPMA) and tri(ethylene glycol)methyl ether methacrylate (MEO3MA), which subsequently reacted with spermine for cross-linking and introduction of cationic moieties. The properties of the nanogel core enabled complexation with the TLR9 ligand CpG, triggering rapid immune activation in bone marrow derived dendritic cells. In vitro studies showed that only the CpG containing nanogels upregulated costimulatory cell-surface markers CD40, CD80 and CD86, necessary for T cell proliferation.
David and coworkers together with de Geest recently published a remarkable study using hyaluronic acid (HA) conjugates of TLR7/8 agonists as targeted adjuvant.146 The synthesized covalent conjugates were completely void of immunostimulatory activity in vitro as demonstrated by a lack of NF-κB induction, though they demonstrated potent adjuvant behavior in animal studies when trafficked to the lymph nodes. They are believed to get “unmasked” by degradation mechanisms resulting in focused immune-stimulation and potent adjuvant effects with negligible systemic exposure.
In the examples presented above, multiple times size has been used to change the in vivo performance of the adjuvant formulations, though a clear correlation with the location of the target needs to be taken into account. For Sakurai and coworkers, the SPG nanogel system performed best when cross-linked, and thus larger (∼100 nm), since their target was located in the endosome.141 On the contrary, a report by the Zentel group demonstrates a preference for smaller nanogels (∼40 nm) when the target is located outside the acidic compartments of the endolysosomal uptake pathway.147 To improve on specificity and to reduce off-target side effects, targeting of nanogels to various tissues or specific cells in lymph nodes has been explored via the addition of ligands on the gels periphery. Hyaluronic acid is known to bind to the CD44 receptor, a single-chain, transmembrane glycoprotein expressed on leukocytes trafficking through the lymphatics,148 as well as to the lymphatic vessel endothelial hyaluronan receptor-1 (LYVE-1), which is expressed almost exclusively on the lymphatic endothelium.149 Therefore, the incorporation of HA in the nanogel formulation has advantageous effects on selective targeting in immune-engineering applications. Additionally, nanogels that use SPG can be recognized and subsequently internalized by the Dectin-1 receptor present on APCs.141 Not discussed here in detail but potentially interesting for future targets is the modification of oligonucleotide-loaded nanogels with transferrin that was shown to facilitate its transport across the blood–brain barrier.150
The first report on the use of polymersomes for immune adjuvants using oligonucleotide danger signals comes from the Hubbell lab, which developed oxidation-sensitive nanoscale polymersomes for both antigen and adjuvant delivery to dendritic cell endosomes.154 Their system consisted of a PEG-b-PPS amphiphile block copolymer that self-assembles into polymersomes under aqueous conditions. Encapsulation of gardiquimod and resiquimod into polymersomes was found to enhance IL-6 and IL-12 cytokine expression significantly. Using the model antigen ovalbumin as a payload, CD8+ T cell cross-priming was demonstrated. Combined, the results demonstrate the utility of an oxidation-sensitive polymersome nanocarriers as potential TLR agonist adjuvant delivery platform for dendritic cells. In a follow-up study, the same lab compared the extent of induced CD4+ or CD8+ T cell responses by oxidation-sensitive polymersomes that presented immunogenic antigens either within the polymersome core or conjugated to the polymersome surface.155 The danger signal CpG 1826 oligonucleotide was used as TLR9 agonist, but it was injected in free form and not as part of the polymersome formulation. It was found that the antigen-loaded polymersomes induced antigen-specific CD4+ T cells in the spleen, lymph nodes and lungs, whereas antigen-conjugated polymersomes induced stronger CD8+ T cell responses. The ability to elicit different T cell immunity by using two separate but very similar particulate-based adjuvant nanosystems opens possibilities for precise vaccine engineering as well as for a better understanding of immune biology in the future.
Levy and coworkers reported a TLR7/8 specific polymersome nanoadjuvant using the small molecule CL075 encapsulated in a block copolymer PEG-b-PPS polymersome.156 CL075 is a potent adjuvant but systemically toxic, therefore a targeted delivery system using polymersomes is beneficial for minimization of systemic toxicity and translational application. Their study focused on a comparison of newborn dendritic cells with adult cells in an attempt to develop an age-specific vaccine formulation better matched to the distinct challenges in newborn immune responses. The in vitro immunostimulatory activities of CL075-containing polymersomes on human newborn and adult monocyte-derived dendritic cells (MoDCs) were benchmarked against conventional adjuvants and human vaccines, including the live attenuated BCG vaccine, which elicits moderate TH 1 immunity in neonates and it is safe and effective at birth.157 The developed TLR7/8 agonist-containing polymersomes matched or exceeded the activating effects of the live attenuated BCG vaccine on human neonatal DCs in vitro, including TH 1-polarizing cytokine IL12p70 and IFN-γ-induction and upregulation of costimulatory molecules.
Polymersomes have also been engineered using peptide building blocks. In this setting, the Barz group produced the first so-called peptosome synthetic vaccine through combination of the SIINFEKL model peptide antigen and TLR9 danger signal CpG 1826 in a peptide-based polymersome.102 The ∼40 nm large peptosomes consisted of a block-copolymer with polysarcosine (PSar) as the hydrophilic block (Xn = 111) and poly(benzyl-glutamic acid) (PGlu(OBn)) as the hydrophobic one (Xn = 46). The loaded peptosomes delivered the antigen and danger signal simultaneously into bone marrow derived dendritic cells. Upon cellular uptake, the cells were stimulated and activated, leading to expression of CD80, CD86 and MHC class II and excretion of pro-inflammatory cytokines (e.g., TNF-α). Furthermore, DC-mediated antigen-specific T-cell proliferation was achieved, thereby demonstrating the combined antigen and adjuvant activity of the developed peptosomes and their overall suitability as potential synthetic vaccine nanoparticles.
Examples of dendrimers as adjuvant platforms using oligonucleotide danger signals are divers, varying in the generation method as well as in the backbone used. David and coworkers reported on the synthesis of a dendrimeric molecule bearing six units of a TLR7/TLR8 dual-agonist imidazoquinoline.161 The work builds on an earlier report on dimeric presentation of the same agonists162 and aimed to explore if multimerization of TLR7/8 would result in altered activity profiles. En route, also the tri-valent variant was tested. The trimeric molecule largely retained both the TLR7- and TLR8-agonistic activities of the monomeric imidazoquinoline, while surprisingly the hexameric dendrimer only displayed TLR7 activity and lost all TLR8 stimulating properties. Fresh human PBMCs were measured to be void of TLR8-driven pro-inflammatory cytokines and IFN-γ, with preservation of TLR7-driven IFN-α induction. An immunization assay performed on rabbits displayed higher titers of high-affinity antibodies to bovine α-lactalbumin in the case of the dendrimers compared to the monomer. While the underlying mechanism(s) on the superior adjuvanticity of the dendrimer remained unknown, the authors hypothesized that a relation to the lack of induction of counter-regulatory IL-10 could be playing parts.
Esser-Kahn and coworkers synthesized dendritic combinations of tri- and di-agonists to understand how specific TLR agonist combinations contribute to the overall immune response.163 They selected the agonists pyrimido[5,4-b]indole, loxoribine and CpG-ODN1826 for respectively TLR4, TLR7 and TLR9 and conjugated these to a triazine-based core via orthogonal coupling chemistries. Once stimulated, each TLR activates a specific immune signaling pathway.27 Therefore, resulting dendrimers were hypothesized to yield a distinct immune response profile, which is helpful to gain a better understanding of TLR synergies. Immune activation was determined by NF-κB activation in RAW264.7 DC-like cells and cytokine transcription levels in bone marrow-derived dendritic cells. Treating immune cells with the linked agonists increased the activation of transcription factor NF-κB and enhanced IL-12 production and gene expression beyond cells treated with an unconjugated mixture of the same three agonists. Through the conjugation of the three agonists in close proximity, a distinctive and more balanced TH 1/TH 2 response and the activation of innate and adaptive immunity were achieved.
Besides functioning as individual adjuvant compounds, dendrimers have also been used to generate an adjuvant coating to magnetic nanoparticles. In the Gunduz group, a PAMAM-dendrimer coated Fe3O4 magnetic nanoparticle (MNP) was developed as a cationic particle to form a complex based on electrostatic interactions with the CpG danger signal.164 The resulting 40 nm particles were screened for cytotoxicity in MDA-MB231 and SKBR3 cancer cells. Individual components were measured to be non-toxic, though the full complexed particle showed significant cell death in the tumor cells. This study builds on the recognition that TLRs exist also in tumor cells besides immune cells.165 The magnetic core of these nanoparticles makes for a promising opportunity for selective drug targeting as they can be concentrated and held in position by means of an external magnetic field. In a follow-up study, the expression levels of some apoptosis-regulating genes in human breast cancer cells treated with the dendrimers-MNP system were investigated.166 The authors observed that treated MDA-MB231 cells showed an increase in Noxa and Bax gene expression levels, whereas the expression level of Survivin decreased. In treated SKBR3 cells, a decline in the c-Flip mRNA level was determined. Treatment with the particles leads to an increase in the release of IL-6 in MDA-MB231 and SKBR3 cells, whereas release of IL-10 and TNF-α did not change significantly. With this study, the mode of cytotoxic action of the dendrimer-based system is believed to involve the expression of apoptosis-related genes and the release of cytokines in the breast cancer cells.
While nucleic acids need transfection agents to penetrate into the cells, it has been shown that DNA nanoparticles are naturally internalized by APCs even without targeting ligands, in a shape- and size-dependent way.168,171 Previous reviews extensively described the delivery of ligands (e.g., siRNA, doxorubicin, aptamers) with DNA nanostructures,172–174 therefore here we only report examples of DNA nanomaterials functionalized with molecules with adjuvant potential. Despite the similarity between DNA sequences of the delivery platform and nucleic acid adjuvants, which makes cargo loading very straightforward via hybridization or chemical modification of nucleic acid sequences, only CpG-containing DNA nanostructures have been developed so far.
The first CpG-containing DNA-based systems were DNA dendrimers. The basic unit is made of three sequences, in which each half of the sequence is complementary to a different part of the other two.175 In 2008, Takakura and co-workers showed that immunostimulatory activity of CpG DNA is dependent on the DNA carrier structure. They demonstrated that Y-shaped oligonucleotides (Y-ODNs) and dendrimer-like DNA (DL-DNA, polypod-like structures) with terminal CpG motifs generate higher levels of cytokines (TNF-α and IL-6) compared with dsODN, showing also greater uptake efficiency.175–177 In particular, DL-DNAs were obtained hybridizing Y-DNA monomers: G2(CpG) and G3(CpG)DL-DNA contained 12 and 24 CpG motifs in one unit respectively and their size is about 19.8 nm and 35.8 nm respectively.176 Interestingly, it has been observed that DL-DNAs have immunostimulatory activity also in absence of CpG motifs, which might be a drawback for the use of these architectures in general cell applications.
Additionally, Mohri et al. studied DL-DNA structures made of three up to eight 26-mer ODNs (called “polypodna”), with size ranges around 15–20 nm and CpG contents between 9 and 36 motifs. Higher size is associated with higher uptake and efficacy, but also reduced stability. In 2015, Zhang et al. developed immune-nanoflowers (NFs), which can be classified as a single chain DNA-based nanomaterial.178 They are self-assembled from a long DNA sequence containing CpG motifs though rolling circle replication, independently from Watson–Crick base pairing, and their size is approximately 300 nm. NFs are nuclease resistant, can be easily uptaken and they induce strong secretion of TNF-α and IL-6 in RAW264.7 DC-like cells. Despite their potential for multivalent CpG presentation, the numbers of CpG copies directly exposed on the surface is not easily determined.
Besides these DNA systems based on a limited number of individual strands, in the past decade, DNA origami CpG-nanodelivery systems have been developed, such as DNA tetrahedrons and DNA nanotubes.179 In the DNA origami technique, a long single-stranded “scaffold” DNA sequence (typically derived from M13 phage DNA) is folded into desired predesigned shape by annealing with a large number of short (typically 15–60 nt) oligonucleotides “staple strands” via selective complementary base pairing.180 2D and 3D structures self-assemble in a one step reaction, where the staples hold the scaffold in specific positions exploiting the programmability of Watson–Crick base pairing.168 To connect DNA double helices and different parts of the scaffold, periodical “cross-overs” are introduced, which are defined as positions based on the Holliday junction structure (four-way junction) in which one sequence starts on one helix and then switches to another one. The architecture of the cross-overs confers stability and rigidity to a DNA origami structure, allowing the creation of a wide variability of shapes of increasing complexity.
In 2011, the first CpG delivery studies with DNA origami structures were published: a 30-helices DNA tube decorated with CpG on the surface designed by Liedl and co-workers181 and a tetrahedron CpG-functionalized on the vertices by Fan and co-workers.182 The nanotube has a length of 80 nm and a diameter of 20 nm, decorated with 62 CpG motifs protruding along each tube. The tetrahedral structure has a size of 6 nm and it has been functionalized with one up to four CpG motifs on the vertices. Both structures showed increased production of cytokines in APCs. In 2012, Liu et al. used a DNA tetrahedron for co-delivery of streptavidin (as model antigen) and CpG motifs, showing that the complex of antigen–CpG–DNA platform can stimulate a stronger immune response in mice compared to a mixture of free antigen and CpG ODNs.183 Of note, however, the limited stability analysis performed in these assays leaves room for future investigations.184
Besides the size, particle shape is another critical parameter affecting circulation, internalization and trafficking of the vaccine carrier.185–188 The local shape, at the points of particle contact, dictate the ability of APCs to phagocytose a particle, a process governed by a combination of cell-particle adhesion through membrane strain energy, cell membrane deformation around the particle and internalization. Given the convenience and versatility in designing DNA nanostructures with controlled shapes and sizes, the DNA origami method offers great opportunity to try various custom-shaped architectures as vaccine vehicles for cellular internalization with a precise and uniform material. Recently, Bastings et al. examined the endocytosis of a diverse library of distinct DNA origami geometries, including round, square, wireframe, solid, hollow, and 3D and 2D forms, of various sizes in different cell lines.171 This study revealed that DCs more preferentially respond to compact shapes with low-aspect-ratio in the size range of 50–80 nm, which are more efficiently internalized compared to wireframe or hollow particles, which mostly remained on the cell surface. These findings highlight the interesting possibility that particle geometry, by mimicking pathogen shapes, can have a large impact in DC uptake and ultimately DC activation.
4 Mode of ligand presentation
As presented previously, targeting vaccines to the lymph node represents an attractive strategy for mounting adaptive immune response. However, besides being successfully accumulated within the lymph nodes, the adjuvant-presenting NPs must also be recognized and internalized by lymphoid tissue residing APCs. Also, it is crucial that the vaccine cargo is released in APCs in a manner that elicits a robust and long-term immune response. In addition, polymeric nanocarriers need to provide long-lasting protection from premature degradation in the biological environment and controllable payload release to the target cells to avoid off-target effects and systemic toxicity. Therefore, rational vaccine design is of great importance in order to fulfill these constraints and guide therapeutic immune outcomes. As reported in Table 1, a wide range of approaches have been developed to enable the complexation of diverse nucleic acid adjuvants to the surface or within the polymer matrix of nanocarriers, improving adjuvant stability and targeted delivery.4.1 Encapsulation vs. surface presentation
The encapsulation of nucleic acid adjuvants increases their half-life by protecting the cargo from enzymatic degradation by endonucleases and allows for a sustained release at the target site, consequently enhancing the exposure duration in DCs. However, it is challenging to ensure that the encapsulated DNA retains its structural integrity throughout the harsh fabrication procedure usually involved in particle fabrication, such as in PLGA-based nanoparticles.189 The use of high-shear forces during emulsification, generated by high-speed homogenization or sonication, and exposure of DNA to organic solvents, especially with double emulsion methods and spray drying techniques, result in loss of DNA activity.100 Moreover, efficient and precise control of adjuvant encapsulation is difficult to replicate with these synthesis methods, usually leading to poor encapsulation efficiency.97 These drawbacks pose a major issue for clinical translation, which requires a precise control of immunostimulatory doses. Alternatively, nucleic acid adjuvants can be located on the surface of the nanoparticle. This method allows for efficient adjuvant complexation without compromising its integrity and bioactivity.190 Although the protection is hampered when exposed to biological milieu after in vivo administration, surface-conjugation of adjuvant potentially allows for easy targeting and interaction with TLRs, boosting immunity.Although both complexation methods have been used for nucleic acid adjuvant delivery, to our knowledge, no comparative studies have yet been conducted to assess the stability in biological fluids and the impact on downstream immune responses of a specific adjuvant. About the antigen, only one study has compared the immune-modulatory properties of antigen encapsulating and surface-presenting PLGA NPs.191 Surface-presentation of the antigen upregulated MHC class II expression level on lymph node-derived DCs in immunized mice, whereas encapsulated antigen increased MHC class I, suggesting a more efficient antigen cross-presentation. In vivo data demonstrated that entrapped antigen drives a more potent antigen-specific T cell response, shown by the activation of both CD4+ and CD8+ T cells, which is critical for the control of tumor and infectious disease. Accordingly, we might hypothesize that the different association of adjuvant to the delivery system (encapsulated or surface-presented) might also elicit qualitatively and/or quantitatively diverse immune responses.
4.2 pH- and reduction-sensitive release in the endosome
Upon particle engulfment by DCs, the nanoparticles are transported within the cell via an endosome, which matures towards lysosomal fusion. During particle processing in the endolysosomal pathway, the intravesicular pH is lowered and the reduction–oxidation state changes from reductive to oxidative in the lysosome. For these reasons, pH- and reduction-sensitive materials have been engineered to release payloads within the endolysosomal compartment. For example, polymers with orthoesters or ketals groups sensitive to acid-hydrolysis have been used to release conjugated payloads into the endosome.108 However, polymers with protonable groups, e.g., polyethylenimine (PEI), poly(amidoamine)s (PAAs), and poly(propylacrylic acid) (PPAA), exhibit a pH buffering capacity that disrupt the endosomal membrane by osmotic imbalance and allows the transport of payloads into the cytoplasm, the so-called “endosomal escape”.120 Although this mechanism is fundamental for antigen cross-presentation in DCs, especially when antigens are co-delivered with adjuvants, it can bring the adjuvant to the cytoplasm, which can activate cytoplasmic sensors and induce excessive immune activation. Therefore, antigen and nucleic acid adjuvant doses should be balanced carefully in vaccine nanocarriers to enable sufficient antigen presentation without excessive toxicity.To tightly control payload release, polymer erosion profile can be tuned via diverse parameters, including polymer composition, size or surface chemistry, but also through changes in physiological parameters, such as temperature, pH or enzymatic activity.192 Detailed mechanisms of general drug release have been extensively reviewed,108,193 here we will focus in systems that have been studied for adjuvants and antigen release. For instance, Kim et al. designed a pH-responsive PLGA formulation for selective endolysosomal delivery of resiquimod.194 PLGA nanoparticle formulation was modified with bicarbonate salt in order to generate carbon dioxide when the particle enters acidic environments. Upon CO2 gas production, the nanoparticle membrane gradually disintegrated, resulting in targeted payload release. Sustained adjuvant release can reduce the need for excessive or repeated vaccine administration. Compared to standard PLGA nanoparticles, this acidic-pH responsive formulation resulted in 33-fold higher encapsulation efficacy of resiquimod and overall the results suggested that anticancer effects in a murine melanoma tumor model improved using the bicarbonate salt incorporation.115
4.3 Ligand complexation/conjugation methods
The strategy enabling the attachment of a therapeutic compound within or onto the surface of the carrier affects specific targeting. The polymer functionality can be customized to achieve the incorporation of drugs with desired properties. Nucleic acid adjuvants can be attached or covalently conjugated to the nanocarrier materials by various methods (Fig. 3B).5 Discussion
5.1 The choice of the right nucleic acid adjuvant
Knowledge about the pattern of TLR cell expression and the choice of the delivery strategy have to be carefully analyzed and coordinated. The expression pattern of TLRs is not equal among different immune cells and this is strictly interconnected with the activity of TLR agonists. As a consequence of this variability, each TLR agonist induces the release of a different cytokine repertoire. For example, despite many TLR ligands activate the production of type I IFN, the induction of TLRs in different cell lines triggers a different IFN subtype and diverse expression ratios.216 Additionally, in preclinical studies the selection of the right cell line and the translation of results from mouse to human should be considered carefully. Contradictory data are present in literature about expression and activity of TLRs in different cell lines31,217 and the expression varies between mouse and human TLRs.218,219 For example, TLR9 expression is limited to B cells and plasmacytoid dendritic cells (pDC), whereas in mouse it has been detected also in monocytes and myeloid dendritic cells.220,221 Consequently, the broader murine expression pattern complicates the extrapolation of mice data to predict human TLR9 activation and possible adverse effects. While so far successful compounds in mice experiments matched with good results in human clinical trials, future studies and designs of adjuvant polymeric architectures should take the potential variabilities explained above in consideration.In addition to receptor expression pattern, species-specific biological microenvironment can affect the metabolism of adjuvants, as it has been verified for poly(I:C), for which serum nuclease activity significantly varies between human and mouse.50 Receptor selectivity and consequent targeted delivery are other parameters to take into account. For TLR7 and TLR9 agonists, delivery to the lymph nodes is necessary to reach the location where pDCs reside. Inversely, for TLR3 ligands, restricted local delivery has to be developed to avoid systemic side effects (overstimulation due to widespread expression of TLR3 also on non-immune cells). To avoid the systemic upregulation of MHC class I, local targeted delivery of poly(I:C) to DCs by conjugation of antigen and poly(I:C) to nanoparticles could help to reduce its toxicity. To prevent autoimmunity, materials used for the delivery should not induce endosomal escape, to avoid the translocation of poly(I:C) into the cytoplasm of non-immune cells where cytosolic sensors are present. Even if a selective TLR3 ligand could exhibit a better safety profile avoiding systemic activation of nonimmune cells, MDA5 activation by poly(I:C) seems to be necessary for high production of type I IFN and induction of memory T cells and antibody production.222,223 Targeted delivery through nanocarriers is crucial also for clinical applications of CpG ODNs as vaccine adjuvants, since phosphorothioate modified backbones of ODNs exhibit higher toxicity compared to the natural backbone.
While most TLR ligands induce the activation of just one subset of DCs, either pDCs or mDCs, bispecific TLR7/8 agonists may have high potential as vaccine adjuvant candidates able to target different cell types involved in the immune response in different in vivo locations. Indeed, previous studies showed the importance of stimulating both pDC and cDC to have strong CD8+ T cell responses.115 Additionally, TLR7 and TLR9 in plasmacytoid DCs induce different pathways in different cellular compartments, according to the cellular location of TLR9 in the endolysosomal trafficking. In particular, CpG-activated TLR9 in the early endosome triggers MyD88-dependent NF-κB activation and inflammatory cytokines, whereas it activates MyD88-IRF7-dependent type I IFN production only after trafficking to the lysosome. This can be exploited in the future by creating nanoparticles releasing CpG in different compartments of the endolysosomal pathway to have a differential cytokine and interferon production, modifying the chemistry of the polymer for releasing the cargo in different compartments.224,225
5.2 The synergy of adjuvants: a new trend
Several groups have examined the combinatorial delivery of multiple TLR agonists for APC activation and showed further amplification of the immune response compared to a single system alone.117,198,226,227 Delivering adjuvants within the same particulate system has the ability to mimic the synergistic immune-response of pathogen components. Targeting of multiple TLRs in dendritic cells can trigger a synergetic activation of pro-inflammatory cytokines, which strengthens T cell activity and induces robust immunity. Bali and coworkers delivered PLGA nanoparticles encapsulating TLR4 (MPL) and TLR7 (imiquimod) agonists in mice and found a synergic increase in antigen-specific antibodies levels and T cell response compared to mice immunized with a single ligand only.117 They showed a robust and durable immune response with an antigen-independent enhancement of IgG antibody titers against H5N1-influenza-derived HA. In a similar study, Madan-Lala et al. assessed dual and triple combinations of TLR ligands, including TLR4 (MPL), TLR7 (imiquimod), and TLR9 (CpG) agonists delivered in PLGA nanocarriers.227 Both TLR4/TLR9 and TLR4/TLR7/TLR9 ligand combinations showed stronger lymph node B220+GL7+ activated germinal center B cell and T follicular helper cell response in mice. Immunization with the combination of three adjuvants induced enhanced antigen-specific antibody titer (IgG1 and IgG2c) with balanced TH 1/TH 2 response, whereas the dual combination induced potent TH 1-polarized antibody responses. Combining multiple adjuvants along with a pathogen- or tumor-associated antigen in particulated systems could thus produce better synergetic immunomodulatory responses than any single system alone.5.3 The choice of the right polymer system: stability and toxicity as key parameters
Several features have to be considered for the choice of a polymer in vaccine nanocarriers, such as biocompatibility, manufacturing simplicity, biodegradability, stability, toxicity and possibility of tuneable payload release. Additionally, manufacturing conditions of polymeric nanoparticles have to be carefully optimized to improve loading and to avoid denaturation/degradation of the loaded drugs, especially for protein antigens. Payloads encapsulated in polymeric NPs are protected in physiological environments. In particular, PLGA can help in the co-delivery of antigens and TLR7/8 agonists, which has been exceptionally challenging since, differently from the other nucleic acid adjuvants, synthetic TLR7/8 are hydrophobic. The low solubility of TLR7/8 agonists limited their use just in topical formulations such as cream and gels, while the antigen was injected separately. PLGA NPs can encapsulate both hydrophilic and hydrophobic ligands, therefore they made in vivo delivery of imidazoquinoline-based TLR7/8 agonists in NPs possible.113 However, the hydrophobicity of PLGA makes protein encapsulation challenging and increases the rate of opsonization and elimination by macrophages in the blood. Indeed, hydrophobic and hydrophilic nanoparticles differ drastically in properties involved in immune activation, such as the capacity to trigger the complement or to be opsonized, phenomena often involved in safety concerns. To prolong the circulation time in the blood, PEG coating has to be added. The fast release of the payload from PLGA slowed the translation of this technology as nanocarrier for vaccine delivery, but it has been verified that fine-tuning the polymer properties (e.g., ratio, molecular weight) can overcome the problem. Therefore, PLGA-based vaccine nanodelivery systems show promising characteristics to be developed into a commercially available platform in the following years.For single chain polymer nanoparticles, to date only initial toxicity evaluations have been performed revealing minimal toxicities that are predominantly related to the presence of external contaminants, such as metal ions or primary amines.129 For a future translation of SCNPs as therapeutic biomaterials, detailed studies are required to gain a general understanding of SCNP-related toxicity and biodistribution.
Stability is always a main concern when aiming for in vivo applications since serum proteins and other molecules easily interact with the designed nanosystem. The interaction of nanohydrogel particles with proteins and serum components was studied in the Zentel laboratory via dynamic light scattering in human blood serum as screening method prior to applications in vivo.139,140 The formation of larger aggregates mostly caused by charge interaction with albumin could be suppressed by nanogel loading with oligonucleotides, affording a neutral zeta potential for the complex. Although both carrier and cargo may have limited stability on their own under physiologically relevant conditions, they appear stable in complexed form. Subsequently, the nanohydrogels’ stability in vivo has been assessed using intravital confocal videography. While the nanohydrogel particles alone caused larger aggregates in the bloodstream, the complexed particles continued circulating without any larger aggregate visible. This high serum stability at optimal oligonucleotide loading secures a future potential for the use of nanogels as true vaccine adjuvants platforms. However, it is worthwhile noting that studies like these should be performed for all new nanogels formulations since the outcomes are expected to be largely material and synthesis dependent.
Due to the synthetic freedom of the individual block-copolymers, polymersomes can display a wide variety of functional and stimuli-responsive behavior that makes them a highly flexible and tuneable system toward nanomedical targets.228 Their composition and molecular weight of individual polymers also define their size and membrane thickness. Usually, polymersomes have a relatively thick (>3–4 nm) and robust membrane, which influences their permeability. The overall dimension and size distribution of polymersomes can be influenced by their preparation technique, using various solvents, sequential extrusion or electric fields.152 Depending on the biological target, the final size and thus preparation methods need to be chosen appropriately. As mentioned earlier, long blood circulation times and stealth behavior can be achieved when one of the polymer blocks consists of PEG. However, pegylation has been shown to negatively influence cellular uptake efficiency of polymersomes by 30%.229 Besides PEG, protein adsorption on polymersomes has been shown to be reduced by introducing various natural or synthetic hydrophilic polymers including polysaccharides, poly(amino acid)s and poly(hydroxyethyl methacrylate) (PHMA).152 Taken together, although reports on polymersomes using endosomal TLR danger signals are limited, results are promising. There exists a clear potential for polymersomes to be further explored and developed into new synthetic vaccines and adjuvant formulations.
Dendrimers show a level of control not manageable with most linear polymer synthesis strategies, leading to nearly monodisperse, globular macromolecules with a large number of functional peripheral groups. For the use of functionalized dendrimers as potent immune modulating materials, careful design and evaluation of their biocompatibility, biodistribution and circulation versus degradation times are of obvious importance.160,230 Since the dendrimer interior structure is to a large extent shielded from the surroundings by the outer shell and the surface, the biological properties of a dendrimer are predominantly governed by the characteristics of the surface groups and of the size of the dendrimer. While amino-terminated dendrimers interact strongly with biological membranes as a result of their highly positive charge, when the generation surpasses G3, the high cell-membrane affinity induces destructive cell-lysis and high cytotoxicity. In vivo, generations ![[double bond splayed left]](https://www.rsc.org/images/entities/char_e009.gif) G6 are believed to cause toxicity problems.231 Negatively charged dendrimers have little interaction with most cellular surfaces and show no significant generation dependent cytotoxicity. Neutral dendrimers can have polar or non-polar end groups, which directly influences their membrane interaction. Polar end groups like PEG create a non-toxic, long-circulating particle, while non-polar groups like lipids readily interact with cell membranes and often trigger immune activation due to their resemblance with bacterial surfaces.160 Interestingly, additives may lead to a significant reduction of cytotoxicity when added to toxic cationic dendrimers. Using the human carcinoma (HeLa) cell line, it has been shown that formulations of fetal calf serum with an amino-terminated G6-PAMAM dendrimer partially modified with the fluorophore Oregon Green exhibit lower toxicity in comparison to the dendrimer alone.232 The cytotoxicity of these formulations was reduced further by complexation with oligonucleotides. Also, formulations of dendrimers with ovalbumin show lower cytotoxicity compared to the free dendrimer.233 These results are likely caused by a strong shielding effect of the many positive charges on the dendrimers’ surface. In terms of clearance and organ accumulation, a direct relation with the generation – and thus size – of the dendrimer is observed. A macromolecule should have a molecular weight larger than 20 kDa to stay in circulation for a prolonged period, but a mass smaller than 40 kDa to be able to be secreted through the kidneys to avoid accumulation in the body.234 With the step-wise synthesis of dendrimers, these ranges can be tightly controlled.159 Altogether, dendrimers have optimal characteristics for efficient delivery of immunostimulating adjuvants, as dendrimers can provide molecularly defined multivalent scaffolds to produce highly defined conjugates with small molecule immunostimulators and/or antigens. However, although cationic PAMAM-type dendrimers are used on a routine basis for gene delivery due to their direct electrostatic complexation with negatively charged DNA, there are surprisingly few reports on the use of dendrimers that deliver oligonucleotide adjuvants.
G6 are believed to cause toxicity problems.231 Negatively charged dendrimers have little interaction with most cellular surfaces and show no significant generation dependent cytotoxicity. Neutral dendrimers can have polar or non-polar end groups, which directly influences their membrane interaction. Polar end groups like PEG create a non-toxic, long-circulating particle, while non-polar groups like lipids readily interact with cell membranes and often trigger immune activation due to their resemblance with bacterial surfaces.160 Interestingly, additives may lead to a significant reduction of cytotoxicity when added to toxic cationic dendrimers. Using the human carcinoma (HeLa) cell line, it has been shown that formulations of fetal calf serum with an amino-terminated G6-PAMAM dendrimer partially modified with the fluorophore Oregon Green exhibit lower toxicity in comparison to the dendrimer alone.232 The cytotoxicity of these formulations was reduced further by complexation with oligonucleotides. Also, formulations of dendrimers with ovalbumin show lower cytotoxicity compared to the free dendrimer.233 These results are likely caused by a strong shielding effect of the many positive charges on the dendrimers’ surface. In terms of clearance and organ accumulation, a direct relation with the generation – and thus size – of the dendrimer is observed. A macromolecule should have a molecular weight larger than 20 kDa to stay in circulation for a prolonged period, but a mass smaller than 40 kDa to be able to be secreted through the kidneys to avoid accumulation in the body.234 With the step-wise synthesis of dendrimers, these ranges can be tightly controlled.159 Altogether, dendrimers have optimal characteristics for efficient delivery of immunostimulating adjuvants, as dendrimers can provide molecularly defined multivalent scaffolds to produce highly defined conjugates with small molecule immunostimulators and/or antigens. However, although cationic PAMAM-type dendrimers are used on a routine basis for gene delivery due to their direct electrostatic complexation with negatively charged DNA, there are surprisingly few reports on the use of dendrimers that deliver oligonucleotide adjuvants.
The main advantage of DNA as nanomaterial is the possibility of controlling the morphology of individual nanoparticles at the nanoscale level. Compared to other systems, DNA origami allow better control over size, shape and surface chemistry, reducing polydispersity. This is extremely important to avoid the induction of different uptake mechanisms in the cells, which would have different outcomes and delivery.235,236 Although CpG-loaded DNA origami activity is demonstrated using cell cultures, so far DNA structures were not protected from degradation by nucleases present in the serum of cell culture media and in vivo. Indeed, DNA origami show low stability in cell media with 10% fetal bovine serum (FBS).237 Moreover, the Mg2+ concentration in cell culture media and blood is below 1 mM, so physiological and cell-culture conditions do not match the relatively high cation concentration (typically 5–20 mM MgCl2) required to obtain a stable self-assembled structure and preventing its denaturation. To further improve the potential and the activity of CpG-presenting DNA origami with adjuvant activity, specific stability-improvement strategies should be applied, as recently reviewed by the Bastings lab.184
6 Conclusions and future perspectives
As has become clear through the above-displayed analysis, our immune system is a highly complex network with many players tightly intertwined. Extensive research on new immunostimulatory agents went beyond the limitations of the old generation of adjuvants, providing a potent but safer and more balanced immune response. However, comparative studies are often not comprehensive and they are performed in different conditions and animal models, making difficult to draw a conclusion about the most effective adjuvant system. Moreover, the amount of adjuvant used for comparative studies differs in the literature. Although many details are still missing, a clear trend is the use of multiple adjuvants in a synergistic manner.While many polymeric architectures have been produced and tested, often just a small subset of the immune-activation cascade is examined. With crucial variations between species and cell types, selection of the correct characterization methods and framework is paramount. PLGA-based polymer adjuvants are numerous, while other classes of polymeric architectures have not yet been extensively explored in the immune adjuvant setting. Due to the high amount of study about PLGA-based vaccine, a commercially available system seems to be expected in the following years as a first delivery platform. Future developments using single chain particles, dendrimers and DNA-based systems can likely be expected. Whereas the lacking analysis of SCNP toxicity and biodistribution slows down their clinical translation, the good serum stability of oligonucleotide loaded nanogels and polymersomes is a promising feature. Although limited studies are performed with adjuvant-loaded dendrimers, their extensive use for gene delivery may accelerate their application in vaccines.
Besides backbone choice, the polymer platform offers great flexibility in the chemical design of complexation and release mechanisms of payloads. Here, the amount of possibilities yields a multifactorial challenge in the future design of the optimal adjuvant architecture. Although the combinations are daunting, a properly performed comparison study could be very beneficial for our fundamental understanding of adjuvant function as well as to carve out some crucial design rules for polymer adjuvants. To add to this challenge, the same statement is true for particle size and charge as well. Since all three TLRs discussed here are located inside the endosomal compartment, the polymeric particles need to be properly taken up through the endosomal pathway before displaying their immune-activating task. Their uptake efficiency and intracellular fate has to be carefully analyzed as in any system expected to perform intracellular interactions. These studies are challenging and unfortunately often lacking in literature, though extremely important when translation is envisioned.
Lastly, the immune system is developed to recognize the highly repetitive and symmetric geometry of viral structures. For example, it has been verified that highly repetitive surface patterns crosslink B cell receptor with increased efficiency: 15–20 haptens or 60 epitopes on a nanoparticle at 5–10 nm of distance induce high B cell response.238 Although nucleic acid adjuvants have been successfully incorporated into nanoparticle platforms, little work has been done to evaluate the influence of the surface density of TLR agonists in shaping the immune response. Recently, Noble et al. developed a series of polystyrene-based nanocarriers with controlled surface densities of CpG conjugated via biotin–NeutrAvidin binding.239 However, no conclusive evidence has been adduced to demonstrate that adjuvant dosage is a critical parameter for directing the stimulation level and response type. Additional studies would therefore be relevant to prove this point. Due to the well-defined and highly programmable properties of nucleic acid strands, DNA-based nanostructures would represent an exciting opportunity to gain control over the density, the position and the spatial orientation of adjuvants, making DNA a suitable vaccine platform.179,240 Now that protection strategies have caught up with the development of bio-active DNA origami designs, exciting future developments of this polymer system in the field of immune engineering can be expected.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to thank mr. Hugo J. Rodríguez Franco for fruitful discussions and assistance on the manuscript. Additionally, we are very grateful for the financial support from the Fondation Pierre Mercier pour la Science (AC) and the Human Frontier Science Foundation (RGY0065/2018) (KP).References
- C. J. Melief, T. van Hall, R. Arens, F. Ossendorp and S. H. van der Burg, J. Clin. Invest., 2015, 125, 3401–3412 CrossRef.
- K. F. Bol, E. H. J. G. Aarntzen, J. M. Pots, M. A. M. O. Nordkamp, M. W. M. M. van de Rakt, N. M. Scharenborg, A. J. de Boer, T. G. M. van Oorschot, S. A. J. Croockewit, W. A. M. Blokx, W. J. G. Oyen, O. C. Boerman, R. D. M. Mus, M. M. van Rossum, C. A. A. van der Graaf, C. J. A. Punt, G. J. Adema, C. G. Figdor, I. J. M. de Vries and G. Schreibelt, Cancer Immunol. Immunother., 2016, 65, 327–339 CrossRef CAS PubMed.
- A. Iwasaki and R. Medzhitov, Nat. Immunol., 2015, 16, 343–353 CrossRef CAS PubMed.
- M. Skwarczynski and I. Toth, Chem. Sci., 2016, 7, 842–854 RSC.
- A. D. Pasquale, S. Preiss, F. T. D. Silva and N. Garçon, Vaccines, 2015, 3, 320–343 CrossRef PubMed.
- P. Marrack, A. S. McKee and M. W. Munks, Nat. Rev. Immunol., 2009, 9, 287–293 CrossRef CAS PubMed.
- O. Majer, B. Liu and G. M. Barton, Curr. Opin. Immunol., 2017, 44, 26–33 CrossRef CAS PubMed.
- B. Ranganathan, C. Miller and A. Sinskey, Pharm. Nanotechnol., 2018, 6, 28–37 CrossRef CAS PubMed.
- J. Han, D. Zhao, D. Li, X. Wang, Z. Jin and K. Zhao, Polymers, 2018, 10, 31 CrossRef PubMed.
- D. M. Klinman, T. Sato and T. Shimosato, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2016, 8, 631–637 CAS.
- A. Hajizade, F. Ebrahimi, A.-H. Salmanian, A. Arpanaei and J. Amani, J. Appl. Biotechnol. Rep., 2014, 1, 10 Search PubMed.
- L. Zhao, A. Seth, N. Wibowo, C.-X. Zhao, N. Mitter, C. Yu and A. P. J. Middelberg, Vaccine, 2014, 32, 327–337 CrossRef.
- B. Sun and T. Xia, J. Mater. Chem. B, 2016, 4, 5496–5509 RSC.
- J. Leleux and K. Roy, Adv. Healthcare Mater., 2013, 2, 72–94 CrossRef CAS.
- M. Zhu, R. Wang and G. Nie, Hum. Vaccines Immunother., 2014, 10, 2761–2774 CrossRef.
- S. G. Reed, M. T. Orr and C. B. Fox, Nat. Med., 2013, 19, 1597–1608 CrossRef CAS.
- M. L. Kapsenberg, Nat. Rev. Immunol., 2003, 3, 984–993 CrossRef CAS.
- J. Banchereau, F. Briere, C. Caux, J. Davoust, S. Lebecque, Y.-J. Liu, B. Pulendran and K. Palucka, Annu. Rev. Immunol., 2000, 18, 767–811 CrossRef CAS.
- K. Palucka and J. Banchereau, Nat. Rev. Cancer, 2012, 12, 265–277 CrossRef CAS.
- S. H. E. Kaufmann, Nat. Rev. Microbiol., 2007, 5, 491–504 CrossRef CAS PubMed.
- S. Nierkens, J. Tel, E. Janssen and G. J. Adema, Trends Immunol., 2013, 34, 361–370 CrossRef CAS PubMed.
- A. S. McKee and P. Marrack, Curr. Opin. Immunol., 2017, 47, 44–51 CrossRef CAS PubMed.
- K. Brown, M. Puig, L. Haile, D. Ireland, J. Martucci and D. Verthelyi, Nucleic Acids as Adjuvants, 2018, pp. 411–444 Search PubMed.
- C. J. Hutcheon, J. O. Becker, B. A. Russell, P. A. Bariola, G. J. Peterson and S. D. Stroop, Vaccine, 2006, 24, 7214–7225 CrossRef CAS.
- A. Wittayanukulluk, D. Jiang, F. E. Regnier and S. L. Hem, Vaccine, 2004, 22, 1172–1176 CrossRef CAS PubMed.
- C. J. Desmet and K. J. Ishii, Nat. Rev. Immunol., 2012, 12, 479–491 CrossRef CAS.
- L. A. J. O'Neill, D. Golenbock and A. G. Bowie, Nat. Rev. Immunol., 2013, 13, 453–460 CrossRef.
- N. J. Gay, M. F. Symmons, M. Gangloff and C. E. Bryant, Nat. Rev. Immunol., 2014, 14, 546–558 CrossRef CAS PubMed.
- M. S. Duthie, H. P. Windish, C. B. Fox and S. G. Reed, Immunol. Rev., 2011, 239, 178–196 CrossRef CAS.
- U. Ohto, T. Shibata, H. Tanji, H. Ishida, E. Krayukhina, S. Uchiyama, K. Miyake and T. Shimizu, Nature, 2015, 520, 702–705 CrossRef CAS.
- J. K. Dowling and J. Dellacasagrande, Toll-Like Receptors: Ligands, Cell-Based Models, and Readouts for Receptor Action, 2016, vol. 1390, pp. 3–27 Search PubMed.
- T. Marichal, K. Ohata, D. Bedoret, C. Mesnil, C. Sabatel, K. Kobiyama, P. Lekeux, C. Coban, S. Akira, K. J. Ishii, F. Bureau and C. J. Desmet, Nat. Med., 2011, 17, 996–1002 CrossRef CAS.
- S. Chattopadhyay and G. C. Sen, J. Interferon Cytokine Res., 2014, 34, 427–436 CrossRef CAS.
- S. Akira and K. Takeda, Nat. Rev. Immunol., 2004, 4, 499–511 CrossRef CAS.
- K. B. Gorden, K. S. Gorski, S. J. Gibson, R. M. Kedl, W. C. Kieper, X. Qiu, M. A. Tomai, S. S. Alkan and J. P. Vasilakos, J. Immunol., 2005, 174, 1259–1268 CrossRef CAS.
- J. M. González-Navajas, J. Lee, M. David and E. Raz, Nat. Rev. Immunol., 2012, 12, 125–135 CrossRef PubMed.
- A. L. Bon, N. Etchart, C. Rossmann, M. Ashton, S. Hou, D. Gewert, P. Borrow and D. F. Tough, Nat. Immunol., 2003, 4, 1009 CrossRef.
- O. Schulz, S. S. Diebold, M. Chen, T. I. Näslund, M. A. Nolte, L. Alexopoulou, Y.-T. Azuma, R. A. Flavell, P. Liljeström and C. R. e Sousa, Nature, 2005, 433, 887 CrossRef CAS.
- W. W. Hancock, M. H. Sayegh, X. G. Zheng, R. Peach, P. S. Linsley and L. A. Turka, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 13967–13972 CrossRef CAS.
- A. M. Krieg, Nat. Rev. Drug Discovery, 2006, 5, 471–484 CrossRef CAS.
- B. Temizoz, E. Kuroda and K. J. Ishii, Curr. Opin. Pharmacol., 2018, 41, 104–113 CrossRef CAS.
- J. T. Patton and E. Spencer, Virology, 2000, 277, 217–225 CrossRef CAS.
- L. Liu, I. Botos, Y. Wang, J. N. Leonard, J. Shiloach, D. M. Segal and D. R. Davies, Science, 2008, 320, 379–381 CrossRef CAS.
- H. Park, L. Adamson, T. Ha, K. Mullen, S. I. Hagen, A. Nogueron, A. W. Sylwester, M. K. Axthelm, A. Legasse, M. Piatak, J. D. Lifson, J. M. McElrath, L. J. Picker and R. A. Seder, J. Immunol., 2013, 190, 4103–4115 CrossRef CAS.
- M. P. Longhi, C. Trumpfheller, J. Idoyaga, M. Caskey, I. Matos, C. Kluger, A. M. Salazar, M. Colonna and R. M. Steinman, J. Exp. Med., 2009, 206, 1589–1602 CrossRef CAS.
- C. Stahl-Hennig, M. Eisenblätter, E. Jasny, T. Rzehak, K. Tenner-Racz, C. Trumpfheller, A. M. Salazar, K. Überla, K. Nieto, J. Kleinschmidt, R. Schulte, L. Gissmann, M. Müller, A. Sacher, P. Racz, R. M. Steinman, M. Uguccioni and R. Ignatius, PLoS Pathog., 2009, 5, e1000373 CrossRef.
- K. Tewari, B. J. Flynn, S. B. Boscardin, K. Kastenmueller, A. M. Salazar, C. A. Anderson, V. Soundarapandian, A. Ahumada, T. Keler, S. L. Hoffman, M. C. Nussenzweig, R. M. Steinman and R. A. Seder, Vaccine, 2010, 28, 7256–7266 CrossRef CAS.
- I. Jelinek, J. N. Leonard, G. E. Price, K. N. Brown, A. Meyer-Manlapat, P. K. Goldsmith, Y. Wang, D. Venzon, S. L. Epstein and D. M. Segal, J. Immunol., 2011, 186, 2422–2429 CrossRef CAS.
- R. A. Robinson, V. T. DeVita, H. B. Levy, S. Baron, S. P. Hubbard and A. S. Levine, JNCI, J. Natl. Cancer Inst., 1976, 57, 599–602 CrossRef CAS.
- K. A. Martins, S. Bavari and A. M. Salazar, Expert Rev. Vaccines, 2015, 14, 447–459 CrossRef CAS PubMed.
- L. Gitlin, W. Barchet, S. Gilfillan, M. Cella, B. Beutler, R. A. Flavell, M. S. Diamond and M. Colonna, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 8459–8464 CrossRef CAS.
- K. S. Lang, M. Recher, T. Junt, A. A. Navarini, N. L. Harris, S. Freigang, B. Odermatt, C. Conrad, L. M. Ittner, S. Bauer, S. A. Luther, S. Uematsu, S. Akira, H. Hengartner and R. M. Zinkernagel, Nat. Med., 2005, 11, 138–145 CrossRef CAS.
- V. Gosu, S. Basith, O. P. Kwon and S. Choi, Molecules, 2012, 17, 13503–13529 CrossRef CAS.
- B. Jasani, H. Navabi and M. Adams, Vaccine, 2009, 27, 3401–3404 CrossRef CAS.
- H. Navabi, B. Jasani, A. Reece, A. Clayton, Z. Tabi, C. Donninger, M. Mason and M. Adams, Vaccine, 2009, 27, 107–115 CrossRef CAS.
- B. B. Gowen, M. H. Wong, K. H. Jung, A. B. Sanders, W. M. Mitchell, L. Alexopoulou, R. A. Flavell and R. W. Sidwell, J. Immunol., 2007, 178, 5200–5208 CrossRef CAS.
- P. Sabbatini, T. Tsuji, L. Ferran, E. Ritter, C. Sedrak, K. Tuballes, A. A. Jungbluth, G. Ritter, C. Aghajanian, K. Bell-McGuinn, M. L. Hensley, J. Konner, W. Tew, D. R. Spriggs, E. W. Hoffman, R. Venhaus, L. Pan, A. M. Salazar, C. M. Diefenbach, L. J. Old and S. Gnjatic, Clin. Cancer Res., 2012, 18, 6497–6508 CrossRef CAS.
- P. Larson, T. A. Kucaba, Z. Xiong, M. Olin, T. S. Griffith and D. M. Ferguson, ACS Med. Chem. Lett., 2017, 8, 1148–1152 CrossRef CAS.
- C. E. Schiaffo, C. Shi, Z. Xiong, M. Olin, J. R. Ohlfest, C. C. Aldrich and D. M. Ferguson, J. Med. Chem., 2014, 57, 339–347 CrossRef CAS.
- H. P. Kokatla, D. Sil, S. S. Malladi, R. Balakrishna, A. R. Hermanson, L. M. Fox, X. Wang, A. Dixit and S. A. David, J. Med. Chem., 2013, 56, 6871–6885 CrossRef CAS.
- P. Larson, T. A. Kucaba, Z. Xiong, M. Olin, T. S. Griffith and D. M. Ferguson, ACS Med. Chem. Lett., 2017, 8, 1148–1152 CrossRef CAS.
- M. Jurk, F. Heil, J. Vollmer, C. Schetter, A. M. Krieg, H. Wagner, G. Lipford and S. Bauer, Nat. Immunol., 2002, 3, 499 CrossRef CAS.
- C. L. Ahonen, S. J. Gibson, R. M. Smith, L. K. Pederson, J. M. Lindh, M. A. Tomai and J. P. Vasilakos, Cell. Immunol., 1999, 197, 62–72 CrossRef CAS.
- M. A. Tomai and J. P. Vasilakos, TLR7/8 Agonists as Vaccine Adjuvants, 2013, pp. 3–18 Search PubMed.
- J. P. Vasilakos and M. A. Tomai, Expert Rev. Vaccines, 2013, 12, 809–819 CrossRef CAS.
- D. Smirnov, J. J. Schmidt, J. T. Capecchi and P. D. Wightman, Vaccine, 2011, 29, 5434–5442 CrossRef CAS.
- S. Bauer, C. J. Kirschning, H. Hacker, V. Redecke, S. Hausmann, S. Akira, H. Wagner and G. B. Lipford, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 9237–9242 CrossRef CAS.
- J. A. Law and S. E. Jacobsen, Nat. Rev. Genet., 2010, 11, 204–220 CrossRef CAS.
- S. Rakoff-Nahoum and R. Medzhitov, Nat. Rev. Cancer, 2009, 9, 57–63 CrossRef CAS.
- R. D. Weeratna, S. R. Makinen, M. J. McCluskie and H. L. Davis, Vaccine, 2005, 23, 5263–5270 CrossRef CAS.
- T. Sugai, M. Mori, M. Nakazawa, M. Ichino, T. Naruto, N. Kobayashi, Y. Kobayashi, M. Minami and S. Yokota, Vaccine, 2005, 23, 5450–5456 CrossRef CAS.
- R. K. Kuan, R. Janssen, W. Heyward, S. Bennett and R. Nordyke, Vaccine, 2013, 31, 4024–4032 CrossRef.
- C. Cooper and D. Mackie, Expert Rev. Vaccines, 2011, 10, 417–427 CrossRef CAS.
- R. J. Hopkins, N. F. Daczkowski, P. E. Kaptur, D. Muse, E. Sheldon, C. LaForce, S. Sari, T. L. Rudge and E. Bernton, Vaccine, 2013, 31, 3051–3058 CrossRef CAS.
- C. J. A. Duncan, S. H. Sheehy, K. J. Ewer, A. D. Douglas, K. A. Collins, F. D. Halstead, S. C. Elias, P. J. Lillie, K. Rausch, J. Aebig, K. Miura, N. J. Edwards, I. D. Poulton, A. Hunt-Cooke, D. W. Porter, F. M. Thompson, R. Rowland, S. J. Draper, S. C. Gilbert, M. P. Fay, C. A. Long, D. Zhu, Y. Wu, L. B. Martin, C. F. Anderson, A. M. Lawrie, A. V. S. Hill and R. D. Ellis, PLoS One, 2011, 6, e22271 CrossRef CAS.
- R. D. Ellis, G. E. Mullen, M. Pierce, L. B. Martin, K. Miura, M. P. Fay, C. A. Long, D. Shaffer, A. Saul, H. Miller and A. P. Durbin, Vaccine, 2009, 27, 4104–4109 CrossRef CAS.
- L. M. Splawn, C. A. Bailey, J. P. Medina and J. C. Cho, Drugs Today, 2018, 54, 399 CrossRef CAS.
- D. E. Speiser, D. Liénard, N. Rufer, V. Rubio-Godoy, D. Rimoldi, F. Lejeune, A. M. Krieg, J.-C. Cerottini and P. Romero, J. Clin. Invest., 2005, 115, 739–746 CrossRef CAS PubMed.
- W. H. Kruit, S. Suciu, B. Dreno, L. Mortier, C. Robert, V. Chiarion-Sileni, M. Maio, A. Testori, T. Dorval, J.-J. Grob, J. C. Becker, A. Spatz, A. M. Eggermont, J. Louahed, F. F. Lehmann, V. G. Brichard and U. Keilholz, J. Clin. Oncol., 2013, 31, 2413–2420 CrossRef CAS.
- D. Valmori, N. E. Souleimanian, V. Tosello, N. Bhardwaj, S. Adams, D. O'Neill, A. Pavlick, J. B. Escalon, C. M. Cruz, A. Angiulli, F. Angiulli, G. Mears, S. M. Vogel, L. Pan, A. A. Jungbluth, E. W. Hoffmann, R. Venhaus, G. Ritter, L. J. Old and M. Ayyoub, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 8947–8952 CrossRef CAS PubMed.
- D. Valmori, N. E. Souleimanian, V. Tosello, N. Bhardwaj, S. Adams, D. O'Neill, A. Pavlick, J. B. Escalon, C. M. Cruz, A. Angiulli, F. Angiulli, G. Mears, S. M. Vogel, L. Pan, A. A. Jungbluth, E. W. Hoffmann, R. Venhaus, G. Ritter, L. J. Old and M. Ayyoub, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 8947–8952 CrossRef CAS.
- X. Wang, M. Bao, M. Wan, H. Wei, L. Wang, H. Yu, X. Zhang, Y. Yu and L. Wang, Vaccine, 2008, 26, 1893–1901 CrossRef CAS.
- R. S. Corral and P. B. Petray, Vaccine, 2000, 19, 234–242 CrossRef CAS.
- V. A. Conforti, D. M. de Avila, N. S. Cummings, K. J. Wells, H. Ülker and J. J. Reeves, Vaccine, 2007, 25, 6537–6543 CrossRef CAS.
- M. Kwissa, H. I. Nakaya, H. Oluoch and B. Pulendran, Blood, 2012, 119, 2044–2055 CrossRef CAS.
- T. K. Ghosh, D. J. Mickelson, J. Fink, J. C. Solberg, J. R. Inglefield, D. Hook, S. K. Gupta, S. Gibson and S. S. Alkan, Cell. Immunol., 2006, 243, 48–57 CrossRef CAS.
- J. Li, C. Fan, H. Pei, J. Shi and Q. Huang, Adv. Mater., 2013, 25, 4386–4396 CrossRef CAS.
- F.-S. Du, Y. Wang, R. Zhang and Z.-C. Li, Soft Matter, 2010, 6, 835–848 RSC.
- E. B. Harrison, S. H. Azam and C. V. Pecot, Front. Pharmacol., 2018, 9, 307 CrossRef PubMed.
- S. Y. Tzeng and J. J. Green, Curr. Opin. Biomed. Eng., 2018, 7, 42–50 CrossRef PubMed.
- S. T. Schmidt, C. Foged, K. S. Korsholm, T. Rades and D. Christensen, Pharmaceutics, 2018, 7, 42–50 Search PubMed.
- K. T. Mody, A. Popat, D. Mahony, A. S. Cavallaro, C. Yu and N. Mitter, Nanoscale, 2013, 5, 5167–5179 RSC.
- D. A. Scheinberg, M. R. McDevitt, T. Dao, J. J. Mulvey, E. Feinberg and S. Alidori, Adv. Drug Delivery Rev., 2013, 65, 2016–2022 CrossRef CAS.
- L. M. M. Neto, A. Kipnis and A. P. Junqueira-Kipnis, Front. Immunol., 2017, 8, 239 Search PubMed.
- J. K. Patra, G. Das, L. F. Fraceto, E. V. R. Campos, M. del Pilar Rodriguez-Torres, L. S. Acosta-Torres, L. A. Diaz-Torres, R. Grillo, M. K. Swamy, S. Sharma, S. Habtemariam and H.-S. Shin, J. Nanobiotechnol., 2018, 16, 71 CrossRef.
- S. Singha, K. Shao, K. K. Ellestad, Y. Yang and P. Santamaria, ACS Nano, 2018, 12, 10621–10635 CrossRef CAS.
- D. J. Irvine, M. A. Swartz and G. L. Szeto, Nat. Mater., 2013, 12, 978–990 CrossRef CAS.
- R. Pati, M. Shevtsov and A. Sonawane, Front. Immunol., 2018, 9, 2224 CrossRef.
- A. de Titta, M. Ballester, Z. Julier, C. Nembrini, L. Jeanbart, A. J. van der Vlies, M. A. Swartz and J. A. Hubbell, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 19902–19907 CrossRef CAS.
- A. Singh, P. Pradhan and K. Roy, Immunobioengineering Approaches Towards Combinatorial Delivery of Immune-Modulators and Antigens, 2013, pp. 161–181 Search PubMed.
- N. I. Ho, L. G. M. H. in't Veld, T. K. Raaijmakers and G. J. Adema, Front. Immunol., 2018, 9, 2874 CrossRef CAS.
- B. Weber, C. Kappel, M. Scherer, M. Helm, M. Bros, S. Grabbe and M. Barz, Macromol. Biosci., 2017, 17, 1700061 CrossRef.
- R. Yang, J. Xu, L. Xu, X. Sun, Q. Chen, Y. Zhao, R. Peng and Z. Liu, ACS Nano, 2018, 12, 5121–5129 CrossRef CAS.
- L. Yang, W. Li, M. Kirberger, W. Liao and J. Ren, Biomater. Sci., 2016, 4, 785–802 RSC.
- M. F. Bachmann and G. T. Jennings, Nat. Rev. Immunol., 2010, 10, 787–796 CrossRef CAS.
- H. Jiang, Q. Wang and X. Sun, J. Controlled Release, 2017, 267, 47–56 CrossRef CAS PubMed.
- S. T. Reddy, M. A. Swartz and J. A. Hubbell, Trends Immunol., 2006, 27, 573–579 CrossRef CAS.
- J. A. Hubbell, S. N. Thomas and M. A. Swartz, Nature, 2009, 462, 449–460 CrossRef CAS PubMed.
- W. Zhang, M. An, J. Xi and H. Liu, Bioconjugate Chem., 2017, 28, 1993–2000 CrossRef CAS.
- M. A. Swartz and A. W. Lund, Nat. Rev. Cancer, 2012, 12, 210–219 CrossRef CAS.
- N. L. Trevaskis, L. M. Kaminskas and C. J. H. Porter, Nat. Rev. Drug Discovery, 2015, 14, 781–803 CrossRef CAS PubMed.
- A. Kumari, S. K. Yadav and S. C. Yadav, Colloids Surf., B, 2010, 75, 1–18 CrossRef CAS.
- H. K. Makadia and S. J. Siegel, Polymers, 2011, 3, 1377–1397 CrossRef CAS.
- F. Danhier, E. Ansorena, J. M. Silva, R. Coco and A. L. Bre, ton and V. Préat, J. Controlled Release, 2012, 161, 505–522 CrossRef CAS.
- H. Kim, L. Niu, P. Larson, T. A. Kucaba and K. A. Mur, phy, B. R. James, D. M. Ferguson, T. S. Griffith and J. Panyam, Biomaterials, 2018, 164, 38–53 CrossRef CAS.
- L. J. Cruz, P. J. Tacken, C. Eich, F. Rueda, R. Torensma and C. G. Figdor, Nanomedicine, 2017, 12, 491–510 CrossRef CAS.
- S. P. Kasturi, I. Skountzou, R. A. Albrecht and D. Koutso, nanos, T. Hua, H. I. Nakaya, R. Ravindran, S. Stewart, M. Alam, M. Kwissa, F. Villinger, N. Murthy, J. Steel, J. Jacob, R. J. Hogan, A. García-Sastre, R. Compans and B. Pulendran, Nature, 2011, 470, 543–547 CrossRef CAS.
- C. Loos, T. Syrovets, A. Musyanovych, V. Mailänder, K. Landfester, G. U. Nienhaus and T. Simmet, Beilstein J. Nanotechnol., 2014, 5, 2403–2412 CrossRef.
- S. Hirosue, I. C. Kourtis, A. J. van der Vlies, J. A. Hubbell and M. A. Swartz, Vaccine, 2010, 28, 7897–7906 CrossRef CAS.
- A. K. Varkouhi, M. Scholte, G. Storm and H. J. Haisma, J. Controlled Release, 2011, 151, 220–228 CrossRef CAS.
- H. Lv, S. Zhang, B. Wang, S. Cui and J. Yan, J. Controlled Release, 2006, 114, 100–109 CrossRef CAS.
- H. S. Kas, J. Microencapsulation, 1997, 14, 689–711 CrossRef CAS.
- B. Slütter, L. Plapied, V. Fievez, M. A. Sande, A. des Rieux, Y.-J. Schneider, E. V. Riet, W. Jiskoot and V. Préat, J. Controlled Release, 2009, 138, 113–121 CrossRef.
- M. K. Aiertza, I. Odriozola, G. Cabañero, H.-J. Grande and I. Loinaz, Cell. Mol. Life Sci., 2012, 69, 337–346 CrossRef CAS.
- S.-D. Li and L. Huang, Mol. Pharmaceutics, 2008, 5, 496–504 CrossRef CAS.
- M. A. Gauthier, M. I. Gibson and H.-A. Klok, Angew. Chem., Int. Ed., 2009, 48, 48–58 CrossRef CAS.
- D. E. Whitaker, C. S. Mahon and D. A. Fulton, Angew. Chem., Int. Ed., 2013, 52, 956–959 CrossRef CAS.
- A. P. P. Kröger and J. M. J. Paulusse, J. Controlled Release, 2018, 286, 326–347 CrossRef.
- A. B. Benito, M. K. Aiertza, M. Marradi, L. Gil-Iceta, T. S. Zahavi, B. Szczupak, M. Jiménez-González, T. Reese, E. Scanziani, L. Passoni, M. Matteoli, M. D. Maglie, A. Orenstein, M. Oron-Herman, G. Kostenich, L. Buzhansky, E. Gazit, H.-J. Grande, V. Gómez-Vallejo, J. Llop and I. Loinaz, Biomacromolecules, 2016, 17, 3213–3221 CrossRef CAS.
- R. Gracia, M. Marradi, G. Salerno, R. Pérez-Nicado, A. P.-S. Vicente, D. Dupin, J. Rodriguez, I. Loinaz, F. Chiodo and C. Nativi, ACS Macro Lett., 2018, 7, 196–200 CrossRef CAS.
- K. S. Soni, S. S. Desale and T. K. Bronich, J. Controlled Release, 2016, 240, 109–126 CrossRef CAS.
- F. R. Kersey, T. J. Merkel, J. L. Perry, M. E. Napier and J. M. DeSimone, Langmuir, 2012, 28, 8773–8781 CrossRef CAS.
- J. P. Rolland, B. W. Maynor, L. E. Euliss, A. E. Exner, G. M. Denison and J. M. DeSimone, J. Am. Chem. Soc., 2005, 127, 10096–10100 CrossRef CAS PubMed.
- J. Ramos, J. Forcada and R. Hidalgo-Alvarez, Chem. Rev., 2014, 114, 367–428 CrossRef CAS PubMed.
- S. Vinogradov, E. Batrakova and A. Kabanov, Colloids Surf., B, 1999, 16, 291–304 CrossRef CAS.
- Y. Tahara and K. Akiyoshi, Adv. Drug Delivery Rev., 2015, 95, 65–76 CrossRef CAS.
- A. V. Kabanov and S. V. Vinogradov, Angew. Chem., Int. Ed., 2009, 48, 5418–5429 CrossRef CAS.
- S. A. Ferreira, F. M. Gama and M. Vilanova, Nanomedicine, 2013, 9, 159–173 CrossRef CAS.
- L. Nuhn, N. Vanparijs, A. D. Beuckelaer, L. Lybaert, G. Verstraete, K. Deswarte, S. Lienenklaus, N. M. Shukla, A. C. D. Salyer, B. N. Lambrecht, J. Grooten, S. A. David, S. D. Koker and B. G. D. Geest, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 8098–8103 CrossRef CAS PubMed.
- L. Nuhn, L. V. Hoecke, K. Deswarte, B. Schepens, Y. Li, B. N. Lambrecht, S. D. Koker, S. A. David, X. Saelens and B. G. D. Geest, Biomaterials, 2018, 178, 643–651 CrossRef CAS PubMed.
- N. Miyamoto, S. Mochizuki and K. Sakurai, Chem. Lett., 2014, 43, 991–993 CrossRef CAS.
- G. D. Brown and S. Gordon, Nature, 2001, 413, 36 CrossRef CAS.
- N. Miyamoto, S. Mochizuki, S. Fujii, K. Yoshida and K. Sakurai, Bioconjugate Chem., 2017, 28, 565–573 CrossRef CAS.
- Y. Tahara, J. Yasuoka, S. Sawada, Y. Sasaki and K. Akiyoshi, Biomater. Sci., 2015, 3, 256–264 RSC.
- S. Hartmann, L. Nuhn, B. Palitzsch, M. Glaffig, N. Stergiou, B. Gerlitzki, E. Schmitt, H. Kunz and R. Zentel, Adv. Healthcare Mater., 2015, 4, 522–527 CrossRef CAS.
- E. Yoo, A. C. D. Salyer, M. J. H. Brush, Y. Li, K. L. Trautman, N. M. Shukla, A. D. Beuckelaer, S. Lienenklaus, K. Deswarte, B. N. Lambrecht, B. G. D. Geest and S. A. David, Bioconjugate Chem., 2018, 29, 2741–2754 CrossRef CAS.
- L. Nuhn, S. Tomcin, K. Miyata, V. Mailänder, K. Landfester, K. Kataoka and R. Zentel, Biomacromolecules, 2014, 15, 4111–4121 CrossRef CAS.
- S. Misra, P. Heldin, V. C. Hascall, N. K. Karamanos, S. S. Skandalis, R. R. Markwald and S. Ghatak, FEBS J., 2011, 278, 1429–1443 CrossRef CAS.
- D. G. Jackson, Immunol. Rev., 2009, 230, 216–231 CrossRef CAS.
- S. V. Vinogradov, E. V. Batrakova and A. V. Kabanov, Bioconjugate Chem., 2004, 15, 50–60 CrossRef CAS.
- B. M. Discher, Y.-Y. Won, D. S. Ege, J. C.-M. Lee, F. S. Bates, D. E. Discher and D. A. Hammer, Science, 1999, 284, 1143–1146 CrossRef CAS.
- J. S. Lee and J. Feijen, J. Controlled Release, 2012, 161, 473–483 CrossRef CAS PubMed.
- X. Y. Zhang and P. Y. Zhang, Curr. Med. Chem., 2017, 13, 124–129 CAS.
- E. A. Scott, A. Stano, M. Gillard, A. C. Maio-Liu, M. A. Swartz and J. A. Hubbell, Biomaterials, 2012, 33, 6211–6219 CrossRef CAS PubMed.
- A. Stano, E. A. Scott, K. Y. Dane, M. A. Swartz and J. A. Hubbell, Biomaterials, 2013, 34, 4339–4346 CrossRef CAS.
- D. J. Dowling, E. A. Scott, A. Scheid, I. Bergelson, S. Joshi, C. Pietrasanta, S. Brightman, G. Sanchez-Schmitz, S. D. V. Haren, J. Ninković, D. Kats, C. Guiducci, A. de Titta, D. K. Bonner, S. Hirosue, M. A. Swartz, J. A. Hubbell and O. Levy, J. Allergy Clin. Immunol., 2017, 140, 1339–1350 CrossRef CAS.
- A. Demirjian and O. Levy, Eur. J. Immunol., 2009, 39, 36–46 CrossRef CAS.
- D. A. Tomalia and J. M. J. Fréchet, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 2719–2728 CrossRef CAS.
- E. Abbasi, S. F. Aval, A. Akbarzadeh, M. Milani, H. T. Nasrabadi, S. W. Joo, Y. Hanifehpour, K. Nejati-Koshki and R. Pashaei-Asl, Nanoscale Res. Lett., 2014, 9, 247 CrossRef.
- P. M. H. Heegaard, U. Boas and N. S. Sorensen, Bioconjugate Chem., 2010, 21, 405–418 CrossRef CAS PubMed.
- N. M. Shukla, D. B. Salunke, R. Balakrishna, C. A. Mutz, S. S. Malladi and S. A. David, PLoS One, 2012, 7, e43612 CrossRef CAS PubMed.
- N. M. Shukla, C. A. Mutz, S. S. Malladi, H. J. War-shakoon, R. Balakrishna and S. A. David, J. Med. Chem., 2012, 55, 1106–1116 CrossRef CAS PubMed.
- J. K. Tom, E. Y. Dotsey, H. Y. Wong, L. Stutts, T. Moore, D. H. Davies, P. L. Felgner and A. P. Esser-Kahn, ACS Cent. Sci., 2015, 1, 439–448 CrossRef CAS.
- N. T. Pourianazar and U. Gunduz, Biomed. Pharmacother., 2016, 78, 81–91 CrossRef.
- Y. Sato, Y. Goto, N. Narita and D. S. Hoon, Cancer Microenviron., 2009, 2, 205–214 CrossRef.
- N. T. Pourianazar and U. Gunduz, Int. J. Pharm., 2016, 515, 11–19 CrossRef.
- N. C. Seeman and H. F. Sleiman, Nat. Rev. Mater., 2017, 3, 17068 CrossRef.
- P. Wang, G. Chatterjee, H. Yan, T. H. LaBean, A. J. Turberfield, C. E. Castro, G. Seelig and Y. Ke, MRS Bull., 2017, 42, 889–896 CrossRef CAS.
- T. J. Bandy, A. Brewer, J. R. Burns, G. Marth, T. Nguyen and E. Stulz, Chem. Soc. Rev., 2011, 40, 138–148 RSC.
- F. Praetorius, B. Kick, K. L. Behler, M. N. Honemann, D. Weuster-Botz and H. Dietz, Nature, 2017, 552, 84–87 CrossRef CAS PubMed.
- M. M. C. Bastings, F. M. Anastassacos, N. Ponnuswamy, F. G. Leifer, G. Cuneo, C. Lin, D. E. Ingber, J. H. Ryu and W. M. Shih, Nano Lett., 2018, 18, 3557–3564 CrossRef CAS PubMed.
- J. Li, C. Fan, H. Pei, J. Shi and Q. Huang, Adv. Mater., 2013, 25, 4386–4396 CrossRef CAS PubMed.
- K. Mohri, M. Nishikawa, Y. Takahashi and Y. Takakura, Eur. J. Pharm. Sci., 2014, 58, 26–33 CrossRef CAS PubMed.
- Y. Zhang, J. Tu, D. Wang, H. Zhu, S. K. Maity, X. Qu, B. Bogaert, H. Pei and H. Zhang, Adv. Mater., 2018, 1703658 CrossRef.
- M. Nishikawa, M. Matono, S. Rattanakiat, N. Matsuoka and Y. Takakura, Immunology, 2008, 124, 247–255 CrossRef CAS.
- S. Rattanakiat, M. Nishikawa, H. Funabashi, D. Luo and Y. Takakura, Biomaterials, 2009, 30, 5701–5706 CrossRef CAS.
- K. Mohri, E. Kusuki, S. Ohtsuki, N. Takahashi, M. Endo, K. Hidaka, H. Sugiyama, Y. Takahashi, Y. Takakura and M. Nishikawa, Biomacromolecules, 2015, 16, 1095–1101 CrossRef CAS PubMed.
- L. Zhang, G. Zhu, L. Mei, C. Wu, L. Qiu, C. Cui, Y. Liu, I.-T. Teng and W. Tan, ACS Appl. Mater. Interfaces, 2015, 7, 24069–24074 CrossRef CAS PubMed.
- F. Hong, F. Zhang, Y. Liu and H. Yan, Chem. Rev., 2017, 117, 12584–12640 CrossRef CAS PubMed.
- V. J. Schüller, S. Heidegger, N. Sandholzer, P. C. Nickels, N. A. Suhartha, S. Endres, C. Bourquin and T. Liedl, ACS Nano, 2011, 5, 9696–9702 CrossRef PubMed.
- V. J. Schüller, S. Heidegger, N. Sandholzer, P. C. Nickels, N. A. Suhartha, S. Endres, C. Bourquin and T. Liedl, ACS Nano, 2011, 5, 9696–9702 CrossRef.
- J. Li, H. Pei, B. Zhu, L. Liang, M. Wei, Y. He, N. Chen, D. Li, Q. Huang and C. Fan, ACS Nano, 2011, 5, 8783–8789 CrossRef CAS PubMed.
- X. Liu, Y. Xu, T. Yu, C. Clifford, Y. Liu, H. Yan and Y. Chang, Nano Lett., 2012, 12, 4254–4259 CrossRef CAS PubMed.
- H. Bila, E. E. Kurisinkal and M. M. C. Bastings, Biomater. Sci., 2019, 7, 532–541 RSC.
- S. Dasgupta, T. Auth and G. Gompper, Nano Lett., 2014, 14, 687–693 CrossRef CAS PubMed.
- J. C. Sunshine, K. Perica, J. P. Schneck and J. J. Green, Biomaterials, 2014, 35, 269–277 CrossRef CAS PubMed.
- S. Kumar, A. C. Anselmo, A. Banerjee, M. Zakrewsky and S. Mitragotri, J. Controlled Release, 2015, 220, 141–148 CrossRef CAS.
- R. Agarwal, V. Singh, P. Jurney, L. Shi, S. V. Sreenivasan and K. Roy, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 17247–17252 CrossRef CAS.
- A. Z. Wilczewska, K. Niemirowicz, K. H. Markiewicz and H. Car, Pharmacol. Rep., 2012, 64, 1020–1037 CrossRef CAS PubMed.
- A. Singh, P. Pradhan and K. Roy, Immunobioengineering Approaches Towards Combinatorial Delivery of Immune-Modulators and Antigens, 2013, pp. 161–181 Search PubMed.
- E. Zupančič, C. Curato, M. Paisana, C. Rodrigues, Z. Porat, A. S. Viana, C. A. M. Afonso, J. Pinto, R. Gaspar, J. N. Moreira, R. Satchi-Fainaro, S. Jung and H. F. Florindo, J. Controlled Release, 2017, 258, 182–195 CrossRef PubMed.
- S. Senapati, A. K. Mahanta, S. Kumar and P. Maiti, Signal Transduction Targeted Ther., 2018, 3, 7 CrossRef PubMed.
- P. T. Wong and S. K. Choi, Chem. Rev., 2015, 115, 3388–3432 CrossRef CAS PubMed.
- H. Kim, D. Sehgal, T. A. Kucaba, D. M. Ferguson, T. S. Griffith and J. Panyam, Nanoscale, 2018, 10, 20851–20862 RSC.
- M. T. Speth, U. Repnik, E. Müller, J. Spanier, U. Kalinke, A. Corthay and G. Griffiths, Mol. Pharmaceutics, 2017, 14, 4098–4112 CrossRef CAS PubMed.
- Y. Han, Q. Duan, Y. Li and J. Tian, RSC Adv., 2017, 7, 39962–39969 RSC.
- T. I. Croll, A. J. O'Connor, G. W. Stevens and J. J. Cooper-White, Biomacromolecules, 2004, 5, 463–473 CrossRef CAS PubMed.
- M. Ebrahimian, M. Hashemi, M. Maleki, G. Hashemitabar, K. Abnous, M. Ramezani and A. Haghparast, Front. Immunol., 2017, 8, 1077 CrossRef PubMed.
- A. Napoli, H. Bermudez and J. A. Hubbell, Langmuir, 2005, 21, 9149–9153 CrossRef CAS PubMed.
- C. Thauvin, J. Widmer, I. Mottas, S. Hocevar, E. Allémann, C. Bourquin and F. Delie, Eur. J. Pharm. Biopharm., 2019, 139, 253–261 CrossRef CAS PubMed.
- S. N. Thomas, E. Vokali, A. W. Lund, J. A. Hubbell and M. A. Swartz, Biomaterials, 2014, 35, 814–824 CrossRef CAS PubMed.
- S. L. Demento, N. Bonafé, W. Cui, S. M. Kaech, M. J. Caplan, E. Fikrig, M. Ledizet and T. M. Fahmy, J. Immunol., 2010, 185, 2989–2997 CrossRef CAS PubMed.
- R. A. Rosalia, L. J. Cruz, S. van Duikeren, A. T. Tromp, A. L. Silva, W. Jiskoot, T. de Gruijl, C. Löwik, J. Oostendorp, S. H. van der Burg and F. Ossendorp, Biomaterials, 2015, 40, 88–97 CrossRef CAS PubMed.
- Q. Chen, J. Chen, Z. Yang, J. Xu, L. Xu, C. Liang, X. Han and Z. Liu, Adv. Mater., 2019, 31, e1802228 CrossRef PubMed.
- A. Seth, H. Lee, M. Y. Cho, C. Park, S. Korm, J.-Y. Lee, I. Choi, Y. T. Lim and K. S. Hong, Oncotarget, 2016, 8, 5371–5381 Search PubMed.
- L. J. Cruz, P. J. Tacken, C. Eich, F. Rueda, R. Torensma and C. G. Figdor, Nanomedicine, 2017, 12, 491–510 CrossRef CAS PubMed.
- S. Rahimian, M. F. Fransen, J. W. Kleinovink, J. R. Christensen, M. Amidi, W. E. Hennink and F. Os-sendorp, J. Controlled Release, 2015, 203, 16–22 CrossRef CAS PubMed.
- D. Schmid, C. G. Park, C. A. Hartl, N. Subedi, A. N. Cartwright, R. B. Puerto, Y. Zheng, J. Maiarana, G. J. Freeman, K. W. Wucherpfennig, D. J. Irvine and M. S. Goldberg, Nat. Commun., 2017, 8, 1747 CrossRef PubMed.
- J. M. Silva, E. Zupancic, G. Vandermeulen, V. G. Oliveira, A. Salgado, M. Videira, M. Gaspar, L. Graca, V. Préat and H. F. Florindo, J. Controlled Release, 2015, 198, 91–103 CrossRef CAS PubMed.
- J. Widmer, C. Thauvin, I. Mottas, V. N. Nguyen, F. Delie, E. Allémann and C. Bourquin, Int. J. Pharm., 2018, 535, 444–451 CrossRef CAS PubMed.
- H. D. Han, Y. Byeon, J.-H. Jang, H. N. Jeon, G. H. Kim, M. G. Kim, C.-G. Pack, T. H. Kang, I. D. Jung, Y. T. Lim, Y. J. Lee, J.-W. Lee, B. C. Shin, H. J. Ahn, A. K. Sood and Y.-M. Park, Sci. Rep., 2016, 6, 38348 CrossRef CAS PubMed.
- A. Malik, M. Gupta, R. Mani, H. Gogoi and R. Bhatna-gar, Front. Immunol., 2018, 9, 562 CrossRef PubMed.
- B. Slütter and W. Jiskoot, J. Controlled Release, 2010, 148, 117–121 CrossRef PubMed.
- N. T. Pourianazar and U. Gunduz, Biomed. Pharmacother., 2016, 78, 81–91 CrossRef PubMed.
- S. Sellner, S. Kocabey, K. Nekolla, F. Krombach, T. Liedl and M. Rehberg, Biomaterials, 2015, 53, 453–463 CrossRef CAS PubMed.
- P. Hillyer, V. P. Mane, L. M. Schramm, M. Puig, D. Verthelyi, A. Chen, Z. Zhao, M. B. Navarro, K. D. Kirschman, S. Bykadi, R. G. Jubin and R. L. Rabin, Immunol. Cell Biol., 2012, 90, 774–783 CrossRef CAS PubMed.
- S. E. Applequist, Int. Immunol., 2002, 14, 1065–1074 CrossRef CAS PubMed.
- J. Mestas and C. C. W. Hughes, J. Immunol., 2004, 172, 2731–2738 CrossRef CAS PubMed.
- M. Rehli, Trends Immunol., 2002, 23, 375–378 CrossRef CAS PubMed.
- V. Hornung, S. Rothenfusser, S. Britsch, A. Krug, B. Jahrsdorfer, T. Giese, S. Endres and G. Hartmann, J. Immunol., 2002, 168, 4531–4537 CrossRef CAS PubMed.
- N. Kadowaki, S. Ho, S. Antonenko, R. de Waal Male-fyt, R. A. Kastelein, F. Bazan and Y.-J. Liu, J. Exp. Med., 2001, 194, 863–870 CrossRef CAS PubMed.
- Y. Wang, M. Cella, S. Gilfillan and M. Colonna, J. Immunol., 2010, 184, 2751–2755 CrossRef CAS PubMed.
- A. M. Hafner, B. Corthésy and H. P. Merkle, Adv. Drug Delivery Rev., 2013, 65, 1386–1399 CrossRef CAS PubMed.
- T. Kawasaki and T. Kawai, Front. Immunol., 2014, 5, 461 Search PubMed.
- K. Honda, H. Yanai, H. Negishi, M. Asagiri, M. Sato, T. Mizutani, N. Shimada, Y. Ohba, A. Takaoka, N. Yoshida and T. Taniguchi, Nature, 2005, 434, 772–777 CrossRef CAS PubMed.
- C. Liu, X. Chu, M. Yan, J. Qi, H. Liu, F. Gao, R. Gao, G. Ma and Y. Ma, Int. J. Pharm., 2018, 553, 327–337 CrossRef CAS PubMed.
- R. Madan-Lala, P. Pradhan and K. Roy, Sci. Rep., 2017, 7, 2530 CrossRef PubMed.
- F. Meng, Z. Zhong and J. Feijen, Biomacromolecules, 2009, 10, 197–209 CrossRef CAS PubMed.
- A. Vertut-Doï, H. Ishiwata and K. Miyajima, Biochim. Biophys. Acta, 1996, 1278, 19–28 CrossRef.
- S. Svenson and D. A. Tomalia, Adv. Drug Delivery Rev., 2005, 57, 2106–2129 CrossRef CAS PubMed.
- N. Malik, R. Wiwattanapatapee, R. Klopsch, K. Lorenz, H. Frey, J. W. Weener, E. W. Meijer, W. Paulus and R. Duncan, J. Controlled Release, 2000, 65, 133–148 CrossRef CAS PubMed.
- H. Yoo and R. L. Juliano, Nucleic Acids Res., 2000, 28, 4225–4231 CrossRef CAS PubMed.
- P. Rajananthanan, G. S. Attard, N. A. Sheikh and W. J. Morrow, Vaccine, 1999, 17, 715–730 CrossRef CAS PubMed.
- L. D. Margerum, B. K. Campion, M. Koo, N. Shargill, J.-J. Lai, A. Marumoto and P. C. Sontum, J. Alloys Compd., 1997, 249, 185–190 CrossRef CAS.
- S. Zhang, H. Gao and G. Bao, ACS Nano, 2015, 9, 8655–8671 CrossRef CAS PubMed.
- J. Mercer and U. F. Greber, Trends Microbiol., 2013, 21, 380–388 CrossRef CAS PubMed.
- J. Hahn, S. F. J. Wickham, W. M. Shih and S. D. Per-rault, ACS Nano, 2014, 8, 8765–8775 CrossRef CAS PubMed.
- A. Jegerlehner, T. Storni, G. Lipowsky, M. Schmid, P. Pumpens and M. F. Bachmann, Eur. J. Immunol., 2002, 32, 3305–3314 CrossRef CAS PubMed.
- J. Noble, A. Zimmerman and C. A. Fromen, ACS Biomater. Sci. Eng., 2017, 3, 560–571 CrossRef CAS.
- L. H. Tan, H. Xing and Y. Lu, Acc. Chem. Res., 2014, 47, 1881–1890 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2019 |