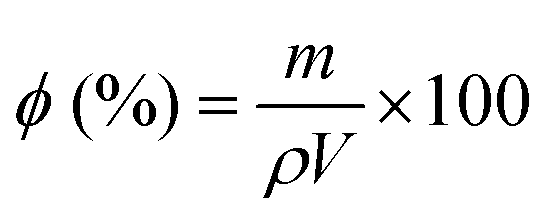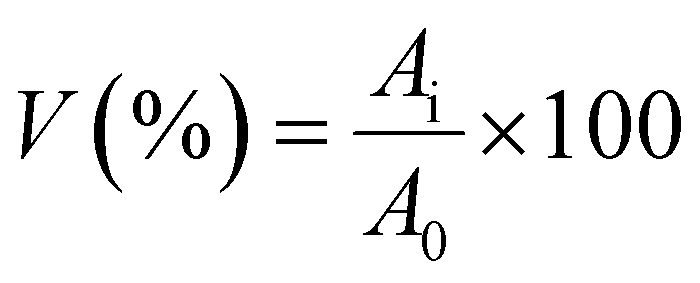Chitosan/hydroxyapatite composite bone tissue engineering scaffolds with dual and decoupled therapeutic ion delivery: copper and strontium
Received
6th May 2019
, Accepted 21st August 2019
First published on 24th September 2019
Abstract
Therapeutic metal ions are a family of metal ions characterized by specific biological properties that could be exploited in bone tissue engineering, avoiding the use of expensive and potentially problematic growth factors and other sensitive biomolecules. In this work, we report the successful preparation and characterization of two material platforms containing therapeutic ions: a copper(II)–chitosan derivative and a strontium-substituted hydroxyapatite. These biomaterials showed ideal ion release profiles, offering burst release of an antibacterial agent together with a more sustained release of strontium in order to achieve long-term osteogenesis. We combined copper(II)–chitosan and strontium–hydroxyapatite into freeze-dried composite scaffolds. These scaffolds were characterized in terms of morphology, mechanical properties and bioactivity, defined here as the ability to trigger the deposition of novel calcium phosphate in contact with biological fluids. In addition, a preliminary biological characterization using cell line osteoblasts was performed. Our results highlighted that the combination of chitosan and hydroxyapatite in conjunction with copper and strontium has great potential in the design of novel scaffolds. Chitosan/HA composites can be an ideal technology for the development of tissue engineering scaffolds that deliver a complex arrays of therapeutic ions in both components of the composite, leading to tailored biological effects, from antibacterial activity, to osteogenesis and angiogenesis.
1 Introduction
Bone repair and regeneration constitute main challenges in reconstructive surgery as many conditions such as tumors, bone trauma, and bone infections cause critical bone defects. Regardless of the etiology, bone disorders are a major health concern with an expected cost by 2020 of $2.5 billion per year only in the USA.1 Major research efforts are being carried out worldwide in the field of allografts, xenografts and other artificial implants.2 However, to date, the golden standard in orthopedic surgery for the reconstruction and repair of bone defects still remains autologous bone.1–3 This consists of tissue harvested from a healthy anatomical site of the patient and transferred to the defect site. Autografts have two major drawbacks: limited availability and invasive harvesting, which can lead to inflammation, hematomas, donor site morbidity and even nerve damage.2 A possible strategy to avoid the drawbacks of autograft with the potential to revolutionize the current capability of medicine for bone repair and regeneration is bone tissue engineering (BTE).1 BTE combines cells, derived from a patient biopsy or from other sources (e.g. stem cells), with a scaffold, a three-dimensional porous structure made from specific biomaterials. Scaffolds are specifically designed to provide temporary support and able to stimulate cell growth and new tissue development,4 for example by delivery of biological signals, in most cases growth factors and biologically active molecules.3
An effective BTE scaffold must own several key properties: interconnected porosity with adequate pore size distribution (10–400 μm), biocompatibility, biodegradability and/or bioresorbability over longer time periods, adequate mechanical properties in the short-term and physicochemical cues for the cells.4,5 The success of BTE scaffolds in clinical applications for bone repair and regeneration is linked to the fulfillment of this complex array of properties simultaneously, which implies that a composite approach, combining organic and inorganic biomaterials could be of great advantage.6 In fact, countless combinations of organic and inorganic phases are currently under study as composite scaffolds for BTE, exhibiting the unique feature of merging the properties of several materials in a single device. Comprehensive reviews on the topic are available.6–8 In this work the focus is on the use of two popular biomaterials in BTE approaches: chitosan as matrix phase and hydroxyapatite (HA) as filler phase.9
Hydroxyapatite (HA) has been widely studied and used for bone tissue engineering applications.10 The production of 3D HA scaffolds from precursor powders was demonstrated using several methods, including thermal bonding, phase leaching, foam replica and additive manufacturing.11 Inorganic HA scaffolds have optimal morphology and porosity, but are usually characterized by insufficient mechanical properties.11 For this reason, a composite approach aiming at the combination of HA powders with natural polymers was often preferred, proving to be a great candidate method for the development of scaffolds.9,12,13 Composite scaffolds based on HA powder dispersed in collagen, silk fibroin, gelatin and chitosan have been all extensively studied.14 Chitosan/HA scaffolds, in particular, are expected to present competitive biocompatibility, osteoconductivity and biodegradation together with sufficient mechanical strength for orthopedic use.9 HA (Ca10(PO4)6(OH)2) is a natural choice when designing novel bone TE approaches: it is a major component of vertebrate hard tissues and it makes up 60–70% of the mass of bone and 98% of the mass of dental enamel. Owing to its similarity to the natural mineral phase of human bone, artificial hydroxyapatite has been used considerably in biomedical applications for bone repair and bone regeneration, where osteoconductivity is considered a key property.10 Furthermore, the particular crystalline structure of hydroxyapatite allows the synthesis of HA derivatives in which ions in the lattice are substituted with other species of interest (i.e. therapeutic ions), making HA a versatile platform for the development of bioceramics with multifaceted biological properties, such as antibacterial effects, osteoinduction, angiogenesis regulation and others.15 These ions are naturally present in HA,16 but their content within the lattice can be artificially tailored and enhanced using several techniques, with specific focus on wet co-precipitation methods,16,17 as reviewed elsewhere.18 These methods offer high tailorability of the final composition of HA, with biologically relevant substitution of ions. For instance, desired ions can be added to a simulated body fluid (SBF) solution and then precipitated to obtain ion-substituted HA19 with biomimetic potential.20,21 Notably, the work published by Bonfield, Best and collaborators on the synthesis of silicon substituted HA showed improved attachment, growth and production of extracellular matrix by osteoblasts.22,23 Substituted HA has also already been combined with polymeric matrices in previous studies.24
This work focuses on the use of therapeutic metal ions (TMIs)25 as possible alternatives to the use of expensive biomolecules such as bone tissue growth factors (e.g. BMP-2). TMIs are a family of metal ions (e.g. strontium, zinc, magnesium or copper) that have been found to have a key role in the regulation of several physiological molecular pathways. TMIs are a main focus of current biomaterials research as they can positively interact with cells and provide a wide range of beneficial effects depending on the specific selected ion (angiogenesis, osteogenesis, improved proliferation or differentiation, antibacterial properties among others).25,26 Traditional approaches to use TMIs in biomaterials usually aim to combine the beneficial properties of ions embedded in particulate inorganic fillers together with a rather inert polymer matrix by means of composites fabrication. In this pilot study, a further improvement is proposed: the possibility to use TMI-carrying biomaterials in both phases of the composite is explored, effectively increasing the spectrum of properties that the final construct is able to deliver. In particular, the design and development of a chitosan/HA composite scaffold characterized by the delivery of strontium and copper ions by means of both its compositional phases is presented. Specifically, strontium is substituted in the HA lattice while copper is complexed with the chitosan matrix.
Strontium and copper were chosen as model ions to prove the concept of multiple ion delivery owing to their tremendously interesting beneficial properties. Strontium is the focus of a big portion of bone tissue engineering research because of its ability to enhance new bone formation and bone resorption.27 The effect of the addition of strontium on bone repair devices was investigated in previous studies and was linked to the ability of this metal ion to influence the gene expression of osteoclasts, shifting the balance of bone remodeling toward osteoblast-mediated bone formation and remodelling.27 Strontium can be delivered orally, but this mode lacks spatial control of the release.24 To locally stimulate regeneration/healing of a bone defect, strontium should be included directly in the scaffold. There are various materials science strategies for the local and controlled release of strontium in bone tissue engineering, many of which are already tested in vivo and in clinical trials, as systematically reviewed by Neves et al. in a recent publication.28 Investigations to date confirm the safety and efficacy of strontium-containing biomaterials and highlight a principal focus on the inclusion of the element in the structure (i.e. lattice/glass network) of inorganic biomaterials, mainly different forms of calcium phosphates (HA, β-TCP),29,30 silicate31,32 and borate28 bioactive glasses, or silicate bioceramics.31 Among other possible approaches, in this work the use of artificial hydroxyapatite was chosen due to the promising possibility to substitute calcium ions in the HA structure by strontium ions, even up to 100%.33
Copper was selected for its strong antibacterial properties and angiogenic effects.34,35 Copper ions are involved in the formation of reactive oxygen species (ROS) that can have dangerous detrimental effects if accumulated in excessive quantity. While eukaryotic cells have mechanisms to defend themselves from ROS, bacteria have weaker defenses and are quickly damaged. In parallel, copper competes for active sites of enzymes with other essential ions (e.g. calcium, iron), denaturing proteins and possibly damaging DNA.36 Conversely, copper can beneficially stimulate eukaryotic cells by inducing the overexpression of vascular endothelial growth factor (VEGF) and cycline D1 genes, stimulating angiogenesis and cell proliferation.37,38
In this study, we demonstrated for the first time the processability of copper(II)–chitosan into a porous copper-containing 3D matrix prepared by freeze-drying, optimizing previously published protocols.9 Strontium-substituted HA, a well established platform for the release of ions promoting bone formation and remodelling, was chosen as inorganic filler in order to enhance the native properties of the polymer. The presence of ions in the two biomaterials was confirmed and their release profiles were assessed. The fabrication of composite chitosan/HA scaffolds proved itself to be a versatile and reproducible way to prepare constructs with the unique property of dual and decoupled delivery of TMIs, loaded in both components of the composite scaffold. The scaffolds were characterized morphologically and physico-chemically using several techniques. Afterwards, a preliminary characterization of the cell response was also performed.
2 Materials and methods
2.1 Production of copper(II)–chitosan
2.1.1 Copper(II)–chitosan preparation and confirmation of complexation.
Medium molecular weight chitosan (Sigma-Aldrich, Germany) with a degree of deacetylation (DDA) of 75–85%, molecular weight (MW) of 190–310 kDa and viscosity 200–800 cP was used to prepare copper(II)–chitosan complexes following a previously published protocol.39 Briefly, chitosan was dissolved (2% w/v) in a 2% v/v water and acetic acid solution (glacial acetic acid, Sigma-Aldrich, Germany). Subsequently, a copper source (CuCl2, Sigma-Aldrich, Germany) was added at different ratios, as previously reported.39 Five different formulations with a range of copper levels corresponding to an estimated saturation of 3 to 18% of the free amino groups of chitosan were prepared (samples named CuChi3 to CuChi18, accordingly). After complete dissolution, mixtures were poured dropwise in a beaker of 0.5 M NaOH (Sigma-Aldrich, Germany) by means of a titration burette. The alkaline environment causes immediate self-assembly of chitosan, leading to the formation of copper(II)–chitosan hydrogel spherical pellets and sodium chloride. This mechanism indicates that copper loading using this technique has very high yield: copper complexes almost completely with chitosan leaving negligible traces of the metal in the aqueous phase.39 The pellets were then carefully washed until complete pH neutralization and reagents removal, as previously demonstrated,39 and finally dried at 37 °C. The final product comprised small pellets (circa 500 μm) that could be easily redissolved and processed as desired. The presence of copper and absence of impurities in the final materials were verified by energy dispersive X-ray spectroscopy (EDX), as previously reported.39 In parallel, Fourier-transform Infrared (FTIR) spectroscopy was used to verify successful complexation between copper and chitosan. In addition to the previously reported analyses,39 the complexation was further confirmed by X-ray diffraction (XRD) using an X’Pert Pro diffractometer with Cu-Kα radiation (40 mV and 40 mA, 2θ = 10–80°, step size 0.03 s−1, Philips).
2.1.2 Copper release measurement by capillary electrophoresis.
To verify and measure the release of Cu2+ from copper(II)–chitosan, films of the different formulations were prepared by solvent casting. Pellets of copper(II)–chitosan were dissolved (2% w/v) in 3% v/v water solution of glacial acetic acid, dried at room temperature in Petri dishes and finally crosslinked with 0.1 M NaOH. The Cu2+ release from the films was studied in phosphate buffer (10 mM, pH 7.4) at 37 °C according to a technique previously reported.40 Aliquots were withdrawn from the media at regular intervals (1, 3 and 6 hours and 1, 3, 7 and 14 days) and the films were re-immersed in fresh buffer at each collection time. Quantification was made by capillary electrophoresis:40 this method combines the in situ complexation of free copper ions with ethylenediaminetetraacetic acid (EDTA) in the electrophoresis column (reverse polarity, acidic conditions) to the subsequent detection and quantification of the complexes by UV radiation.40 This method is specifically designed for complex samples such as composite biomaterials for tissue engineering.40
2.2 Production of hydroxyapatite
2.2.1 Synthesis of hydroxyapatite and strontium substituted hydroxyapatite.
Hydroxyapatite (HA) and strontium substituted HA with a (Sr/Ca + Sr) ratio of 0.2 (Sr–HA) were synthesized by a wet-precipitation method at 40 °C.41 The reagents used for the synthesis were ammonium phosphate dibasic ((NH4)2HPO4, ACS reagent ≥98%, Sigma Aldrich, UK), calcium nitrate tetrahydrate (Ca(NO3)2·4H2O ≥ 99.0%, Sigma Aldrich, UK) and strontium nitrate hexahydrate (Sr(NO3)2·6H2O, ACS reagent ≥99.0%, Sigma Aldrich, UK) (the latter only in the case of Sr–HA synthesis). In a typical synthesis of Sr–HA, 250 mL of two separate solutions (A and B) were prepared. Solution A was prepared by dissolving Ca(NO3)2·4H2O and Sr(NO3)2·6H2O in deionized water in appropriate amounts to reach a total molarity of 0.835 M. Solution B was a 0.5 M solution of (NH4)2HPO4 in deionized water. The amount of reagents was calculated in order to keep the (Ca + Sr/P) ratio constant at 1.667. The pH values of solutions A and B were set at 11 and 9.5, respectively, by adding 1 M ammonium hydroxide (NH4OH, ACS reagent, 28.0–30% NH3 basis, VWR International, UK) solution. Then, solution B was added dropwise to solution A. After mixing the solutions, the pH value was adjusted to 10.75 by adding NH4OH and the final solution was stirred for 16 hours at 400 rpm, followed by aging of the precipitates for 24 hours. The obtained precipitates were washed with deionized water three times and then centrifuged. Samples were dried at 80 °C in an oven and sintered at 900 °C for 3 hours (at the heating rate of 5 °C min−1). Pure hydroxyapatite was also prepared through the same route, but without the addition of Sr(NO3)2·6H2O.
2.2.2 Structural and chemical analysis.
XRD analysis was performed on a Bruker D8 Advance using Bragg–Brentano parafocusing geometry. Data were collected for the 2θ range from 5° to 70° (step size of 0.01° and count time 1 second per step). Collected data were qualitatively examined using the Bruker diffract+ and EVA search-match software (search matched against the ICDD database). The functional groups of HA were analyzed by ATR-FTIR using a Shimadzu IRAffinity-1S (Shimadzu Corp, Japan) equipped with a Quest ATR GS10801-B single bounce diamond accessory (Specac Ltd, England). Data were acquired with 40 scans at a resolution of 4 cm−1 and processed using the LabSolution IR software by Shimadzu. In addition, compositional analysis of HA and Sr–HA samples was performed by X-ray fluorescence (XRF). The test was executed in accordance to ISO standard 12677. Samples were ignited at 1200 °C for 1 hour to examine the loss on ignition. Then, approximately 1.5 g of powdered sample was mixed with 7.5 g of lithium tetraborate (Li2B4O7) and heated in a platinum crucible at 1025 °C for 20 min first and then at 1200 °C for 5 min. The resulting glass melt was cast to create a glass bead. The bead was analyzed using an Axios’ Panalytical XRF spectrometer.
2.2.3 Particle size distribution.
Before the scaffold production, HA and Sr–HA powders were milled to achieve a consistent particle size. Planetary milling was performed using a Pulverisette 7 premium line (Fritsch GmbH, Germany) equipped with 5 mm ZrO2 beads. 3 cycles of 10 min each at 700 rpm were performed and then the particle size was measured using a laser particle size analyzer (Mastersizer 3000, Malvern Instruments Ltd, United Kingdom).
2.2.4 Ion-release profile.
To investigate strontium release from prepared Sr–HA, powder samples were dispersed in simulated body fluid (SBF) at a ratio of 1.5 g L−1 and held in a shaker incubator at 37 °C at 200 rpm for different time intervals (1, 3, 7, 14 and 21 days). SBF was prepared according to the well-established protocol published by Kokubo and Takadama.42 Briefly, 8.0756 g L−1 of NaCl, 0.3532 g L−1 of NaHCO3, 0.2250 g L−1 of KCl, 0.2310 g L−1 of K2HPO4·3H2O, 0.3033 g L−1 of MgCl2·6H2O, 0.3638 g L−1 of CaCl2·6H2O and 0.0716 g L−1 of Na2SO4 were dissolved in deionized water. The solution was buffered at 36.5 °C and pH 7.4 with the addition of 6.0658 g L−1 of tris(hydroxymethyl)aminomethane (Tris buffer, (CH2OH)3CNH2) to reach a final concentration of Tris in SBF of 50 mM, and of 1 M HCl according to need (0–5 mL). Strontium release in SBF under dynamic conditions was measured using ICP-OES (Inductively coupled plasma/optical emission spectroscopy) from IRIS Advantage, Thermo Jarrell Ash. Approximately 1 g of the sample was dissolved in an appropriate amount of 5% HNO3 and heated gently to ensure complete dissolution. The solution was made up to 50 mL volumetrically and was analyzed by ICP-OES (IRIS Advantage, Thermo Jarrell Ash) against a calibration traceable under ISO: 17025 guidance.
2.3 Fabrication of HA–chitosan composite scaffolds
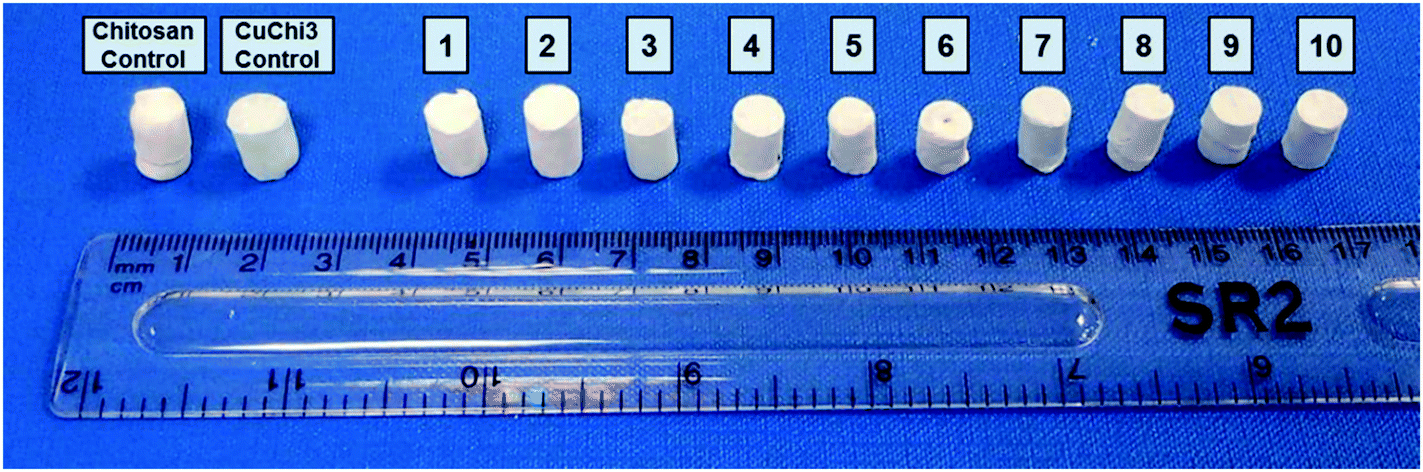
Freeze-drying with previously optimized parameters was performed for the production of HA–chitosan composite scaffolds.9 Copper(II)–chitosan with a 3% saturation of the amino groups (CuChi3, equivalent to 1 wt% copper) was chosen, as it guarantees the lowest cytotoxicity compared to other formulations.39 Chitosan or CuChi3 were dissolved in diluted acetic acid water solutions (3% v/v) at a concentration of 2% w/v. The solution was then cast in Eppendorf tubes used as negative molds and quickly frozen using liquid nitrogen (−196 °C). The rapid freezing encourages the formation of small and homogeneous water crystals, which will result in more reproducible morphologies. A design-of-experiment approach was adopted in order to screen the effect of the following variables on the properties of the scaffolds: (i) type of hydroxyapatite (either undoped or 3% strontium substituted), copper loading (from 0% to 3%) and filler-to-matrix ratio (33% to 80% wt). Table 1 summarizes the samples prepared in this study and the parameters used to produce them. The scaffolds are showed in Fig. 1. Cu1.5 samples were prepared mixing the same amount of undoped chitosan and CuChi3.
Table 1 List of composite scaffold samples prepared for the factorial experimental design (FED)
| # |
Sample name |
Cu loading (wt%) |
Type of HA |
Filler-to-matrix ratio (%) |
| 1 |
Cu0 HA33 |
0 |
Undoped |
33 |
| 2 |
Cu0 HA80 |
0 |
Undoped |
80 |
| 3 |
Cu3 HA33 |
3 |
Undoped |
33 |
| 4 |
Cu3 HA80 |
3 |
Undoped |
80 |
| 5 |
Cu0 SrHA33 |
0 |
3% strontium |
33 |
| 6 |
Cu0 SrHA80 |
0 |
3% strontium |
80 |
| 7 |
Cu3 SrHA33 |
3 |
3% strontium |
33 |
| 8 |
Cu3 SrHA80 |
3 |
3% strontium |
80 |
| 9 |
Cu1.5 HA56.5 |
1.5 |
Undoped |
56.5 |
| 10 |
Cu1.5 SrHA56.5 |
1.5 |
3% strontium |
56.5 |
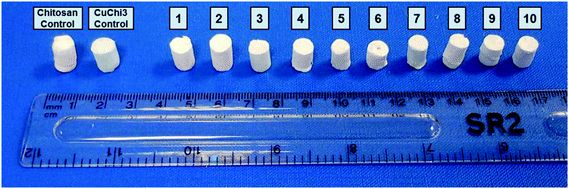 |
| | Fig. 1 Samples of all ten formulations of the composite scaffolds produced by freeze-drying in this study. | |
2.4 Physical characterization of the scaffolds
2.4.1 Scaffold morphology and porosity.
A qualitative assessment of the morphology, pore shape and pore size of the scaffolds was carried out by scanning electron microscopy (SEM, LEO 435 VP, LEO Electron Microscopy Ltd, Cambridge, UK and Ultra Plus, Zeiss, Jena, Germany). Specimen preparation before testing included sputter-coating using a Q150T S equipped with a gold target (Quorum Technologies, United Kingdom). Together with SEM, energy dispersive X-ray (EDX) analysis was performed. The total porosity of the scaffolds was measured by gravimetry.43 Briefly, the mass (m) and dimensions (diameter = ∅ and thickness = h) of several scaffold samples were measured and the percentage porosity (ϕ) was calculated as follows, knowing the theoretical density of the composite material (ρ), which is calculated as a weighted average of the density of the two components of the composite per their weight ratio:| | 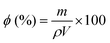 | (1) |
2.4.2 Mechanical properties.
The variations in mechanical properties of the composite scaffolds as a function of their composition were assessed by non-confined compressive testing. The analysis was carried out using a Tinius Olsen H5KS equipped with a 5 kN load cell (strain ramp at 5 mm per min gage speed). The compressive moduli were computed as the slope of the linear region of the stress–strain curve (ε = 0–10%). Tests were performed using 8 replicates per sample type.
2.5 Biological characterization of the scaffolds
2.5.1 Acellular bioactivity test.
Hydroxyapatite is known to be bioactive, that is of triggering the deposition of calcium phosphate, more specifically of hydroxycarbonate apatite (HCA), when in contact with body fluids. To assess whether or not the HA filler embedded in the chitosan matrix was bioactive, an acellular bioactivity test was performed. Simulated body fluid (SBF) was prepared following the protocol by Kokubo and Takadama,42 as previously described. Scaffolds were immersed in SBF using a ratio of 1.5 g L−1, according to the unified in vitro evaluation protocol for apatite-forming ability published by Maçon et al.44,45 The samples were stored in polypropylene flasks and placed in an orbital shaker (80 rpm) at 37 °C for 1, 3 and 7 days. The solution was renewed during soaking time every two days at day 1, 3, 5 and 7 to better simulate the physiological environment. At the end of each incubation time, the scaffolds were washed with deionized water, dried at 50 °C and stored for further characterizations.
The deposition of novel HCA was qualitatively assessed by SEM using the same set-up previously described. Fourier transform infrared spectroscopy (FTIR) using a Shimadzu IRAffinity-1S (Shimadzu Corp, Japan) equipped with a Quest ATR GS10801-B single bounce diamond accessory (Specac Ltd, England) was also performed. Data were acquired with 40 scans at a resolution of 4 cm−1 and processed using the LabSolution IR software by Shimadzu. Native chitosan scaffolds were used as controls in both analyses.
2.5.2 Cell culture.
Homo sapiens osteosarcoma bone cell line (MG-63) was chosen for the preliminary evaluation of the cytocompatibility of the bone tissue engineering scaffolds hereby fabricated. Prior to test, cells were cultured in typical cell culture polystyrene flasks using Dulbecco's Modified Eagle Medium (DMEM) with 10% (v/v) of fetal bovine serum (FBS) and 1% (v/v) combined penicillin–streptomycin antibiotic (PenStrep) (all reagents purchased from Gibco®, Germany). A monolayer of cells close to confluence (∼80%) was detached from the flask at passage 15 using a trypsin/EDTA solution (Life Technologies, Germany) in PBS. Upon cell detachment, trypsin was inactivated by adding fresh DMEM at a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 ratio and the cell suspension was counted by trypan blue exclusion method (Sigma-Aldrich, Germany). Cells were seeded in 24-well culture plates at a density of 100000 cells per well and incubated overnight at 37 °C, 95% relative humidity and 5% CO2. In parallel, samples of all ten scaffold varieties were UV-sterilized for one hour and preconditioned in DMEM for 24 hours. Control samples of native chitosan and copper(II)–chitosan without filler were also similarly prepared. On the second day, the samples were added to the wells of cell culture and incubated for another 24 hours. Finally, the samples were removed and cell viability and fluorescent staining were performed.
:3 ratio and the cell suspension was counted by trypan blue exclusion method (Sigma-Aldrich, Germany). Cells were seeded in 24-well culture plates at a density of 100000 cells per well and incubated overnight at 37 °C, 95% relative humidity and 5% CO2. In parallel, samples of all ten scaffold varieties were UV-sterilized for one hour and preconditioned in DMEM for 24 hours. Control samples of native chitosan and copper(II)–chitosan without filler were also similarly prepared. On the second day, the samples were added to the wells of cell culture and incubated for another 24 hours. Finally, the samples were removed and cell viability and fluorescent staining were performed.
2.5.3 Cell viability.
Cell viability as a function of the mitochondrial activity of MG-63 cells was measured after 24 hours of culture in contact with the composite scaffolds. In particular, the test was performed to evaluate the effect of TMIs on eukaryotic cells. The cell viability was measured using CCK-8 assay kit supplied by Sigma-Aldrich. This kit uses 2-(2-methoxy-4-nitrophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfophenyl)-2H-tetrazolium, monosodium salt (WST-8), a compound which is enzymatically converted to formazan in the mitochondria of viable cells. At the end of the culture period, the medium was removed from the wells, samples disposed and the wells with cells were rinsed with PBS. WST-8 containing culture medium (1% v/v) was pipetted in the wells and left to incubate for 3 hours at 37 °C. After incubation, aliquots (100 μL) of the supernatant were collected and transferred in a 96-wells plate for absorbance reading. Spectrophotometry was performed at 450 nm to detect formazan using a micro plate reader (PHOmo Autobio, Labtec Instruments co. Ltd China). The cell viability (V) was then computed from the absorbance values (A) as follows:| | 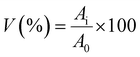 | (2) |
2.5.4 Phenotype expression.
Fluorescent staining of MG63 cells was performed right after WST using both Vybrant® DiI (1,1′-dioctadecyl-3,3,3′3′-tetramethylindocarbocyanine perchlorate, Invitrogen, USA) live staining and DAPI (4′,6-diamidino-2-phenylindole, dilactate, Invitrogen, USA). In particular, DiI is a lipophilic cytoplasm dye that stains living cells entirely. DAPI is a well-known dye to use on fixed samples and which binds to A (Adenine) and T (Thymine) bases within the cell DNA, thus staining nuclei. The staining was carried out following the manuals provided by the supplier. Images of DiI–DAPI stained cells were taken with an Axio Observer D1 fluorescence microscope (Carl Zeiss Microimaging GmbH, Germany).
3 Results
3.1 Copper complexation and release from copper(II)–chitosan
3.1.1 Confirmation of complexation by XRD.
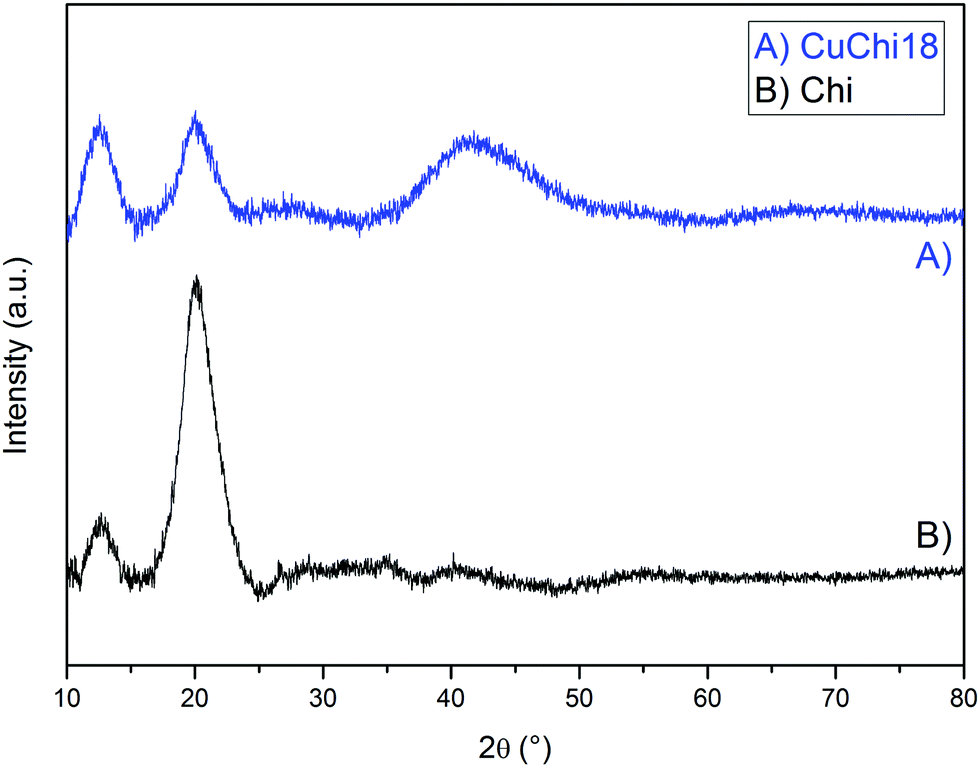
In previous works it was shown by EDX that copper is present in copper(II)–chitosan samples without significant contaminations from other elements.39 Especially, no contamination from reagents (i.e. sodium or chlorine) was detected. Complexation induces clear changes in the characteristic FTIR peaks and bands of chitosan. In particular, changes in relative absorbance were detected for spectral bands relative to the stretching of OH and NH at 3300 cm−1 together with a change in shape of the characteristic peaks of the glycosidic bond around 1100 cm−1, as previously reported.39 In addition, XRD analysis showed the chitosan incurs significant changes in crystalline structure after the addition of copper (Fig. 2).
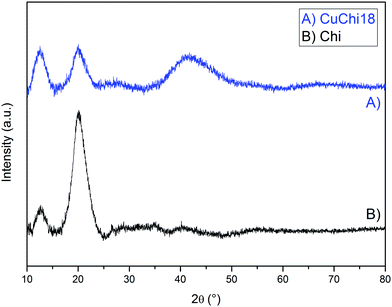 |
| | Fig. 2 Comparison between XRD spectra of native chitosan and CuChi18. All other copper(II)–chitosan formulations behave similarly. | |
The diffractogram of native chitosan showed two broad diffraction peaks at 2θ = 12° and 20°, which are characteristic fingerprints of semi-crystalline chitosan from the selected supplier, as previously reported.46 These peaks are associated to the formation of crystal I and crystal II chitosan structures. After the addition of copper, a decrease in the crystal II peak at 2θ = 20° was observed for all tested formulations, without significant differences between formulations with different levels of copper. No previous literature was found regarding the broad shoulder found at 2θ = 40°. This could be ascribed to the formation of a new crystal conformation of chitosan after the chelation or simply to an increased signal from amorphous domains of the polysaccharide. Further crystallographic studies on the topic should be performed in order to clarify this finding.
3.1.2 Copper release.
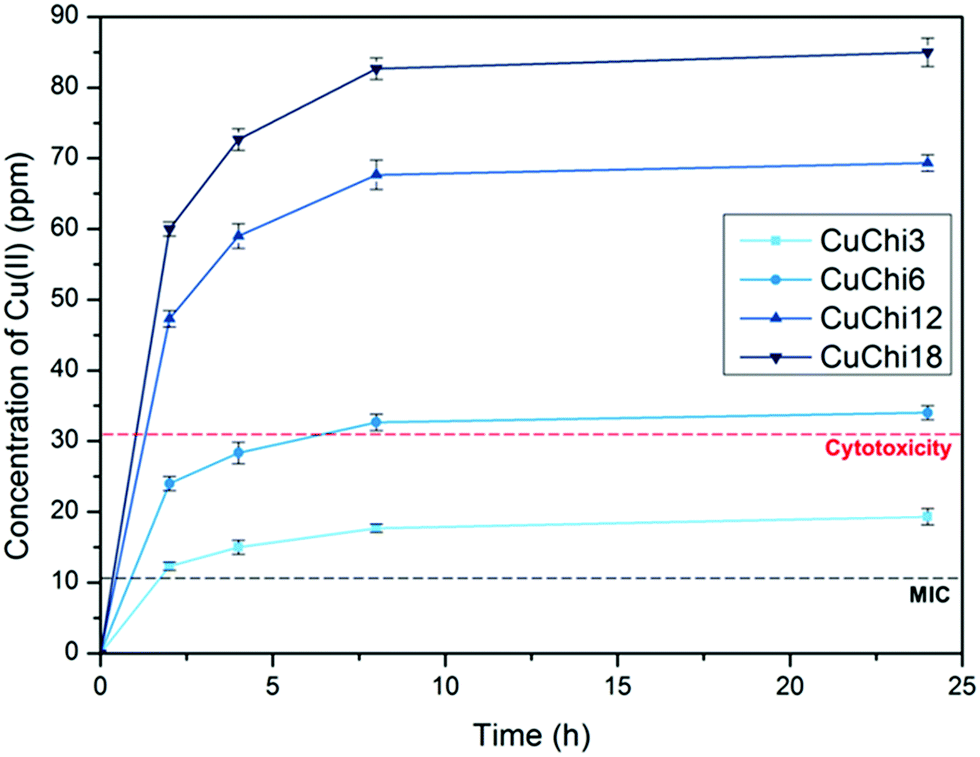
The release of copper from copper(II)–chitosan in PBS was measured by capillary electrophoresis. All samples are characterized by quick copper release within the first 10 hours following exponential profiles with significantly different plateau values (Fig. 3). As the quantity of copper loaded during the preparation of the material increases, the release plateaus vary accordingly. After 24 hours no additional copper is released (Table 2). The values of copper release obtained with this analysis were compared with the reported in vitro cytotoxic concentration (30 ppm) and minimum inhibitory concentration (MIC, 10 ppm) of copper.47,48 Within this window of concentrations (10–30 ppm) it can be anticipated that the material will be antibacterial without being cytotoxic. This behavior was confirmed by a previously reported study in which samples with a ppm release falling within this window were proven to have the desired biological properties.39
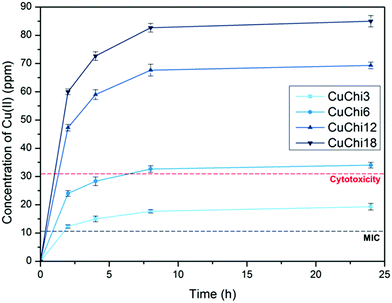 |
| | Fig. 3 Cumulative copper release from all copper(II)–chitosan formulations as a function of time. The release was quantified by capillary electrophoresis. For comparison, the cytotoxic concentration (30 ppm) and minimal inhibitory concentration (MIC, 10–12 ppm) reported in literature for copper are overlaid. | |
Table 2 Quantity of released copper from one day to two weeks. It can be observed that after the first 24 hours no more copper is released from the copper(II)–chitosan samples
|
|
Cumulative amount of copper released (ppm) |
| CuChi3 |
CuChi6 |
CuChi12 |
CuChi15 |
| Days |
1 |
18 |
20 |
20 |
33 |
35 |
34 |
70 |
70 |
68 |
85 |
83 |
87 |
| 7 |
19 |
20 |
20 |
35 |
34 |
34 |
70 |
70 |
69 |
88 |
86 |
84 |
| 14 |
19 |
21 |
19 |
36 |
34 |
36 |
70 |
71 |
70 |
88 |
87 |
88 |
3.2 Strontium substitution and release from hydroxyapatite
3.2.1 Crystallographic analysis by XRD.
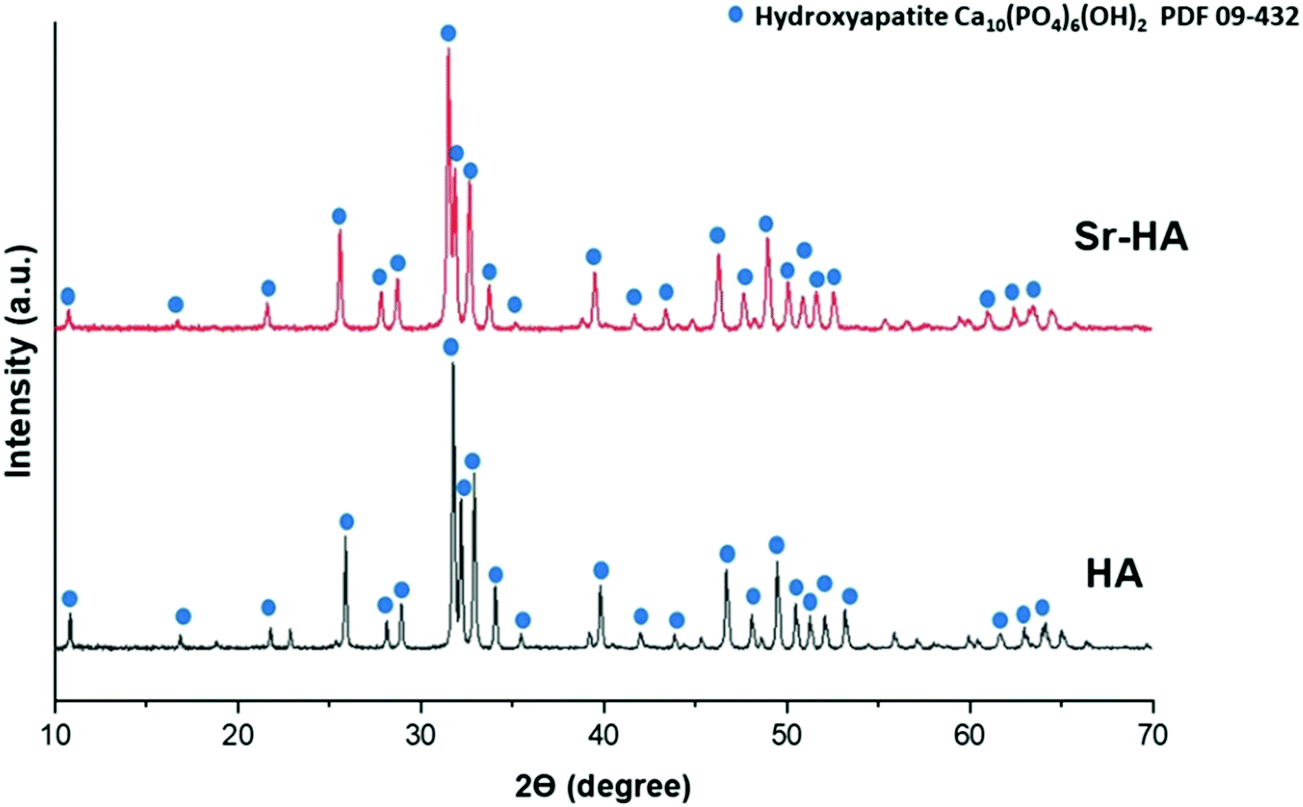
XRD analysis of HA and Sr–HA powders showed highly crystalline diffractograms for both materials (Fig. 4) which matched the standard diffractogram of HA (JCPDS. 09-432). The characteristic diffraction peaks for hexagonal HA were clearly visible in both samples. In addition, the results did not reveal the presence of any additional phases. Broadening in XRD peaks occurs due to distortion of the HA lattice structure. As Sr2+ ions (ionic radius 1.18 Å) are substituted at Ca2+ (0.99 Å) sites in the HA lattice structure, expansion in the unit cell occurs, which consequently results in peak broadening as well as slight shifting in peak position.49 Lattice parameters obtained using Bruker's TOPAS software confirmed this observation. The software showed that the a-axis and c-axis parameters of Sr–HA were increased after the substitution of Sr2+, as presented in Table 3. This is likely to be due to the larger ionic radius of Sr2+ compared to Ca2+, as confirmed by previously reported results.50 In addition, a slight shift in diffraction peaks towards lower angles (2θ), as compared to reference HA peaks, was observed, which can be ascribed to the substitution of calcium ions by strontium ions.49
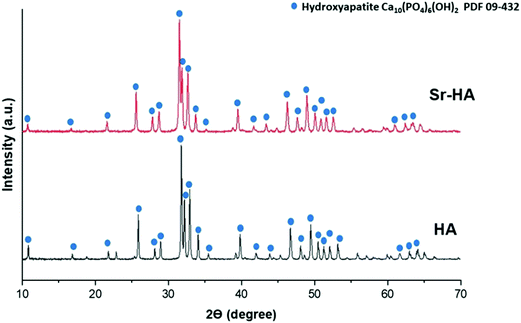 |
| | Fig. 4 X-ray diffractograms of hydroxyapatite (HA) and strontium substituted hydroxyapatite (Sr–HA). | |
Table 3 Variation in lattice parameters a-axis and c-axis of hydroxyapatite lattice structure after the substitution of Sr2+ ions
|
|
a-Axis |
c-Axis |
| HA |
9.4251 |
6.8845 |
| Sr–HA |
9.4845 |
6.9481 |
3.2.2 Functional group analysis by FTIR.
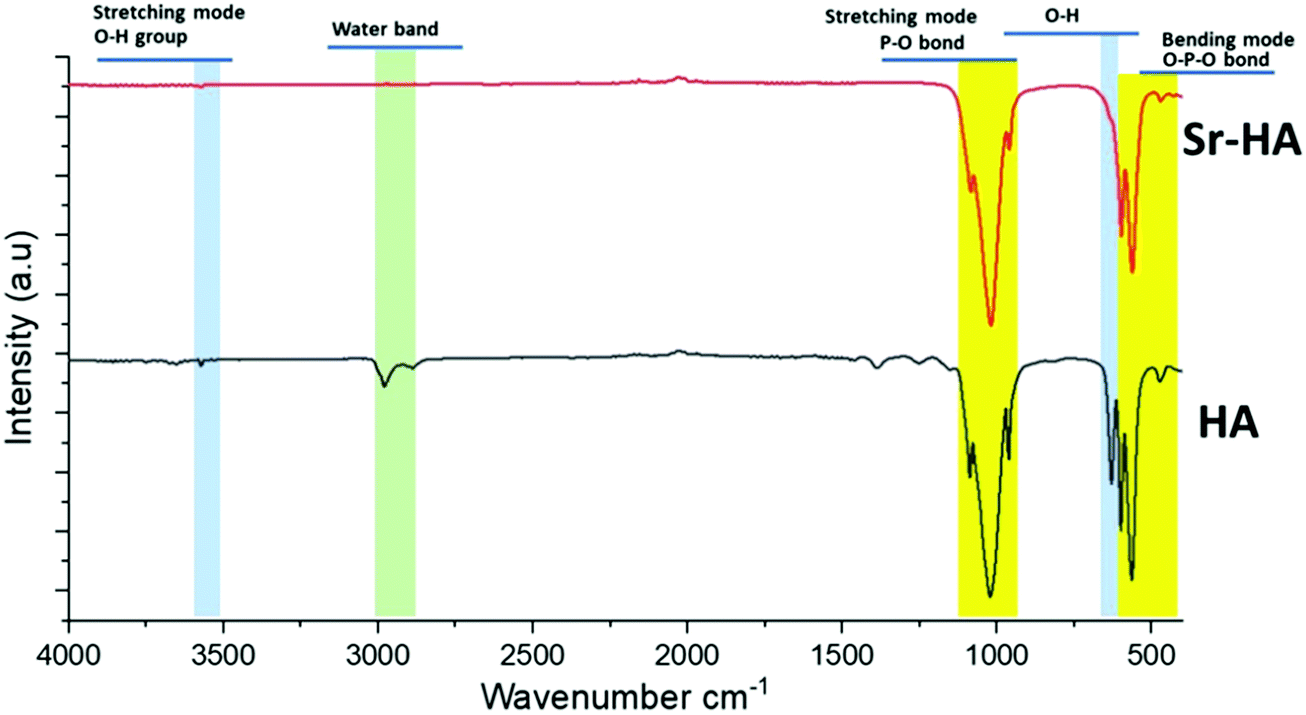
The FTIR spectra of HA and Sr–HA are presented in Fig. 5. The characteristic bands related to phosphate, hydroxyl and adsorbed water can be observed. Notably, all the representative bands that characterize the phosphate (575, 610, 967, and 1014 cm−1) and hydroxyl (3568 and 639 cm−1) groups were still present in FTIR spectra after strontium substitution.51 It was also found that the OH bands in the range of 555–640 cm−1 attributable to molecularly bonded OH are reduced after the substitution of Sr2+ ions.51,52 This might be a consequence of the partial replacement of OH ions by Sr2+ ions.52 Additional bands attributed to carbonates (CO32−) were also present. Furthermore, all relevant FTIR bands corresponding to pure hydroxyapatite functional groups were found at the same position in Sr–HA as they were found in HA.53 Altogether, FTIR determined that both samples show bands typical of pure and Sr-substituted HA composition, with only minor CO23− impurities.
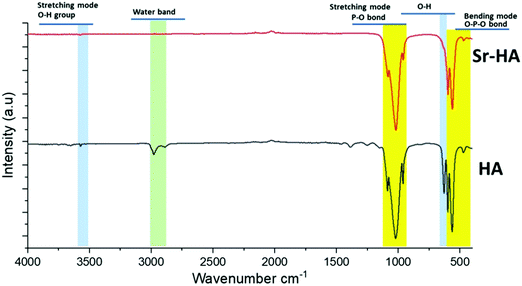 |
| | Fig. 5 FTIR spectra of hydroxyapatite (HA) and strontium substituted hydroxyapatite (Sr–HA). | |
3.2.3 Compositional analysis by XRF.
The compositional analysis of HA and Sr–HA was carried out by semi-quantitative XRF analysis, and the molar ratios were calculated as presented in Table 4. The value of the molar ratios for Ca/P in the case of HA and the molar ratio (Ca + Sr)/P in the case of Sr–HA were very close to the expected value (1.667). The results show that strontium ions added in the reaction successfully substituted calcium within the HA lattice. These compositional results are in good agreement with X-ray diffraction and Rietveld refinement results.
Table 4 XRF results of hydroxyapatite (HA) and strontium substituted hydroxyapatite (Sr–HA)
| Samples |
Calcium (mol%) |
Phosphorus (mol%) |
Strontium (mol%) |
Ca + Sr/P ratio |
| Calculated |
XRF |
| HA |
1.07 |
0.6481 |
— |
1.667 |
1.6509 |
| Sr–HA |
0.8649 |
0.5917 |
0.1254 |
1.667 |
1.6736 |
3.2.4 Particle size distribution.
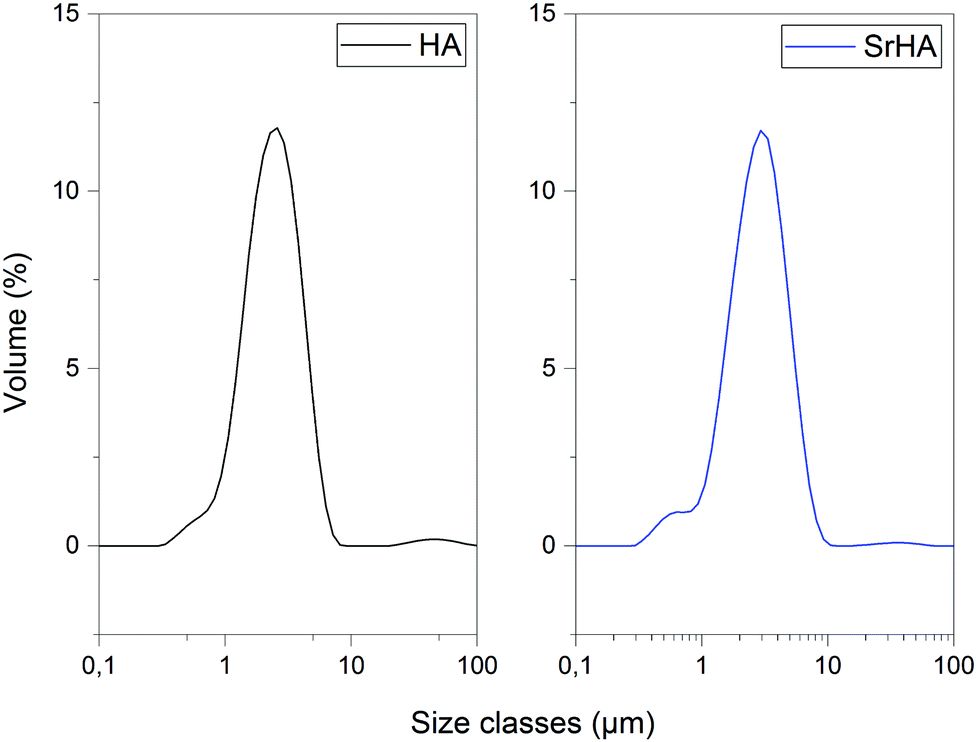
The particle size distribution of HA and Sr–HA after planetary milling was measured by laser particle size analysis suspending the powder in a water medium. Typical distribution profiles for the two powders are shown in Fig. 6. After collection, the distributions were analyzed in terms of size parameters D10, D50 and D90. Results are reported in Table 5. Both HA and Sr–HA are characterized by narrow particle size distributions (within 1–10 μm) and with D50 levels of circa 3 μm. The fineness of the powders produced in this work is ideal for the preparation of scaffolds, as they allow the fabrication of homogeneous and reproducible composite scaffolds.9
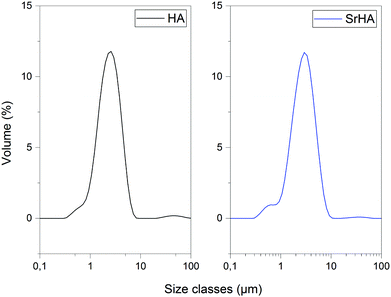 |
| | Fig. 6 Charts showing particle size distributions for HA and Sr–HA. | |
Table 5 Parameters of the particle size distributions of HA and Sr–HA
|
|
Hydroxyapatite |
Strontium substituted hydroxyapatite |
| Dx (10) |
1.25 |
1.32 |
| Dx (50) |
2.41 |
2.78 |
| Dx (90) |
4.37 |
5.13 |
3.2.5 Strontium release.
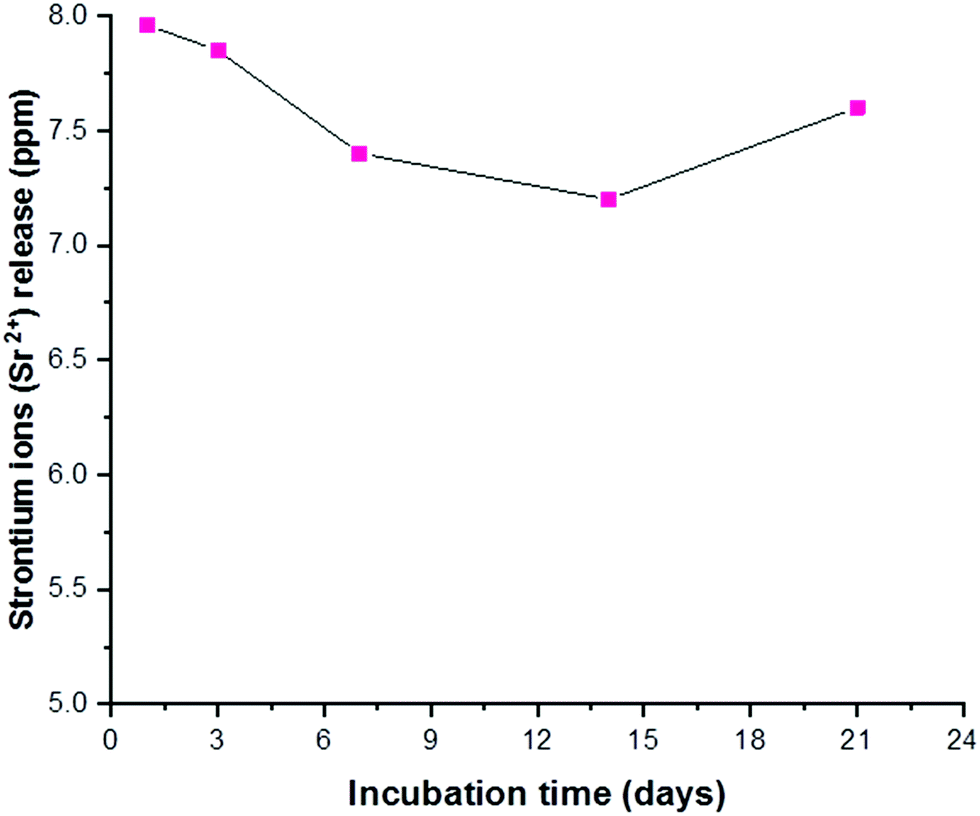
ICP results revealed that there was a steady release of strontium ions in the physiological medium during the 21 days of immersion, as shown in Fig. 7. The concentration of Sr2+ ions released from Sr–HA was within the concentration range of 7–8 ppm at all measured time points. Generally, in such cases, apatite grains are covered by an amorphous layer which is dissolved in the earlier stage after immersion in SBF, consequently exposing less soluble and more stable core.17 As the amorphous grain layer is detached, there is steady release of ions from the bulk of the HA structure. The concentration of released ions did not change significantly with time, which suggests that the majority of the Sr2+ ions was present in the framework of the HA particles and negligible amounts of Sr2+ ions were adsorbed in the amorphous grain layer.
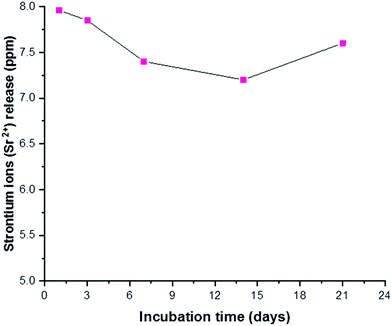 |
| | Fig. 7 ICP results of strontium concentration in ppm as a function of immersion time in SBF. | |
3.3 Characterization of scaffold morphology and porosity
3.3.1 Morphological analysis.
Typical morphological features of chitosan/HA composite scaffolds produced in this study are reported in the SEM micrographs pictured in Fig. 8. Regardless of the type of chitosan and HA used, the scaffolds are characterized by similar and homogeneous morphology and topography, with long and interconnected lamellar pores around 10 μm wide, ideal for cell infiltration and osteoblast differentiation.54 HA particles are evenly distributed within the matrix, well-embedded in the polymer without significant aggregate formation. Furthermore, the qualitative assessment of the particle size from the SEM images confirmed the results of the particle size laser analysis, identifying an average size of the HA particles which sits well below 10 μm.
 |
| | Fig. 8 SEM micrographs of selected samples at two magnifications (1000× and 10000×) showing the typical morphology of freeze-dried chitosan/HA scaffolds. Lamellar structures formed after the sublimation of the solvent crystals can be observed. | |
3.3.2 Scaffold porosity.
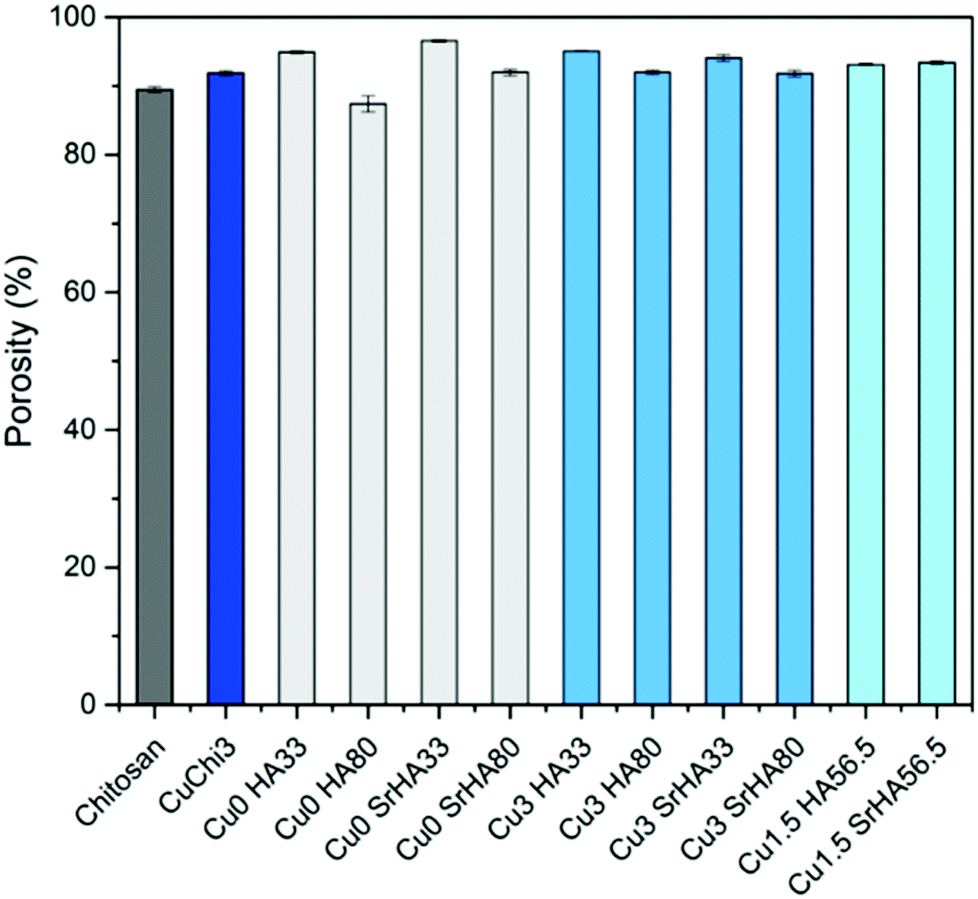
As summarized in Fig. 9, the measurement of porosity by gravimetry performed on the scaffolds showed even levels around 90% regardless of the specific formulation. This is in agreement with unpublished optimization studies performed by our group which identified the concentration of chitosan in acetic acid (which in this study was kept constant) as the determining factor for the final porosity. The lack of variation among samples also indicates that the addition of hydroxyapatite does not significantly affect the porosity. Low standard deviations suggest that this protocol is adequate for the reproducible fabrication of porous composite scaffolds, in terms of morphology and porosity.
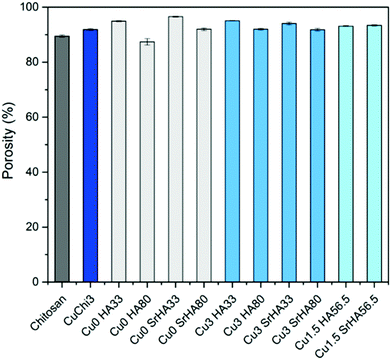 |
| | Fig. 9 Porosity of the scaffolds measured by gravimetry. No significant differences were found: all samples have a porosity of ∼90%. | |
3.3.3 Mechanical properties.
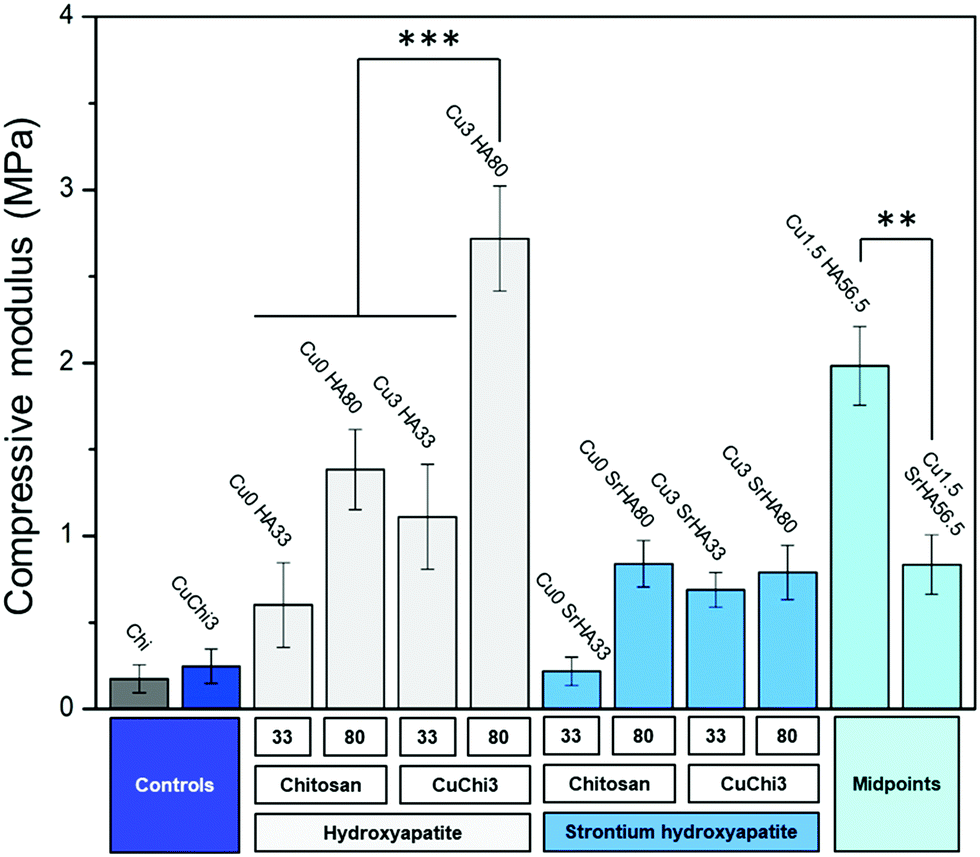
The effect of the hydroxyapatite fillers on the mechanical properties of the chitosan porous scaffolds was investigated by a non-confined compressive test. The compressive modulus of native chitosan and CuChi3 is circa 250 kPa. The addition of HA filler and the use of CuChi3 both led to increases in this value (Fig. 10). The only exception is Cu0SrHA33, which is comparable with the controls. The effect of the composite preparation on the mechanical properties can be divided into three contributions: the effect of copper inside chitosan and the effect of the type and amount of the hydroxyapatite filler. Copper seems to slightly enhance the compressive modulus, although the differences cannot be considered statistically significant, except in the case of Cu0HA80 and Cu3HA80. A more pronounced increase in compressive modulus is due to the addition of the filler: HA significantly increases the mechanical properties for all tested scaffolds, while the improvement due to Sr–HA is lower, even null in the case of Cu0SrHA33. The difference in performance between HA and Sr–HA can be clearly observed when comparing the two midpoints. Finally, the amount of filler seems to have an influence on the mechanical properties a higher filler content results in higher compressive modulus, especially in the case of Cu3HA33/Cu3HA80 and Cu0SrHA33/Cu0SrHA80.
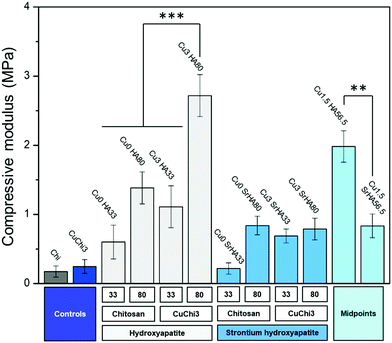 |
| | Fig. 10 Compressive modulus of all ten scaffold formulations plus chitosan and CuChi3 controls. A general increase in compressive mechanical properties after the addition of the filler was assessed. The significance bars highlight the main differences between samples: (i) high HA loading and the presence of copper seem to improve the mechanical properties; (ii) SrHA seems not to have the same beneficial effect as HA (***: p < 0.001, **: p < 0.01). | |
3.4 Biological characterization
3.4.1 Acellular bioactivity test.
The bioactivity of chitosan/HA composite scaffolds was assessed by measuring their ability to trigger the formation of hydroxyl-carbonate apatite (HCA). After 7 days of immersion in SBF, composite scaffolds showed good integrity and stability: they retained their shape and structure, no sign of crumbling or fragmentation was observed for any tested formulation. Upon dehydration, a major structure collapse and shrinkage were observed in HA-free control samples (Chi and CuChi3). However, this effect was strongly reduced for low-HA loaded composite specimens and almost absent in high-HA loadings. SEM inspection showed that the porosity of the scaffolds was reduced as a consequence of the swelling and subsequent drying. The surface of composite scaffolds was covered by a calcium phosphate layer, that was completely absent on control chitosan sponges (Fig. 11).
 |
| | Fig. 11 SEM micrographs of a chitosan control sponge (A) and Cu0HA80 (B). While native chitosan lacks any sign of mineralization after the immersion in SBF, the composite scaffolds are characterized by a deposit that can be ascribed to the precipitation of an amorphous phase of calcium phosphates. | |
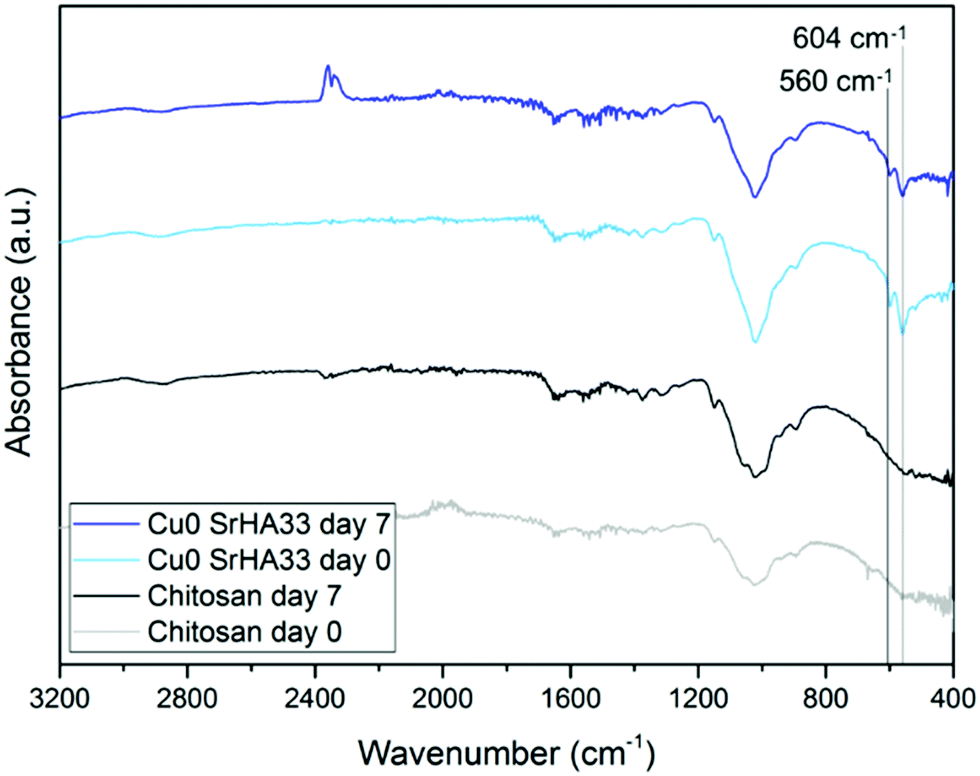
The nature of this layer was unclear from the SEM imaging: the shape of the crystal suggested the formation of HCA, but the cauliflower structure typical of HCA grown in SBF could not be detected.43 This suggests that this layer could be a deposit of amorphous calcium phosphates, precursors of HCA.42 The spectroscopic analysis was not effective in the identification of the nature of this layer as FTIR spectra before and after immersion did not show significant variations. This occurred because specimens were either not bioactive (that is the case of the control samples of chitosan and CuChi3) or already rich in HA. The presence of hydroxyapatite prior to testing, in the case of the composite samples, resulted in the shielding of possible variations in characteristic peaks of the P–O bending at 560 and 604 cm−1 as a consequence of the immersion in SBF.45 As it can be observed in Fig. 12, the spectra of chitosan control and Cu0SrHA33 before and after testing do not show significant alterations.
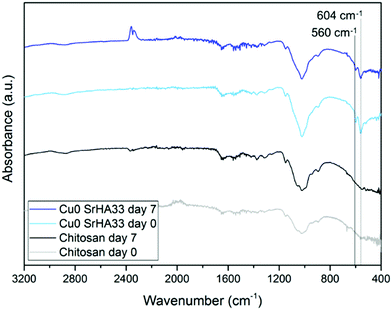 |
| | Fig. 12 FTIR spectra of chitosan and Cu0SrHA33 that show no relevant variation before and after immersion in SBF for 7 days. Cu0SrHA33 was chosen as representative sample, all other specimens behaved similarly. | |
Altogether the results of the acellular bioactivity, or apatite-forming ability, test suggest the deposition of a calcium phosphate layer absent on control samples: HA has a key role in making the scaffolds bioactive; however, further studies will be performed in order to provide conclusive evidence on the nature of the deposit detected in this study. For example, a novel SBF using easily detectable 44Ca isotope of calcium could be a way to better describe the results of the immersion. Otherwise, Raman spectroscopy due to its higher sensitivity could be to accurately identify newly formed hydroxyl-carbonate apatite.
3.4.2 Mitochondrial activity.
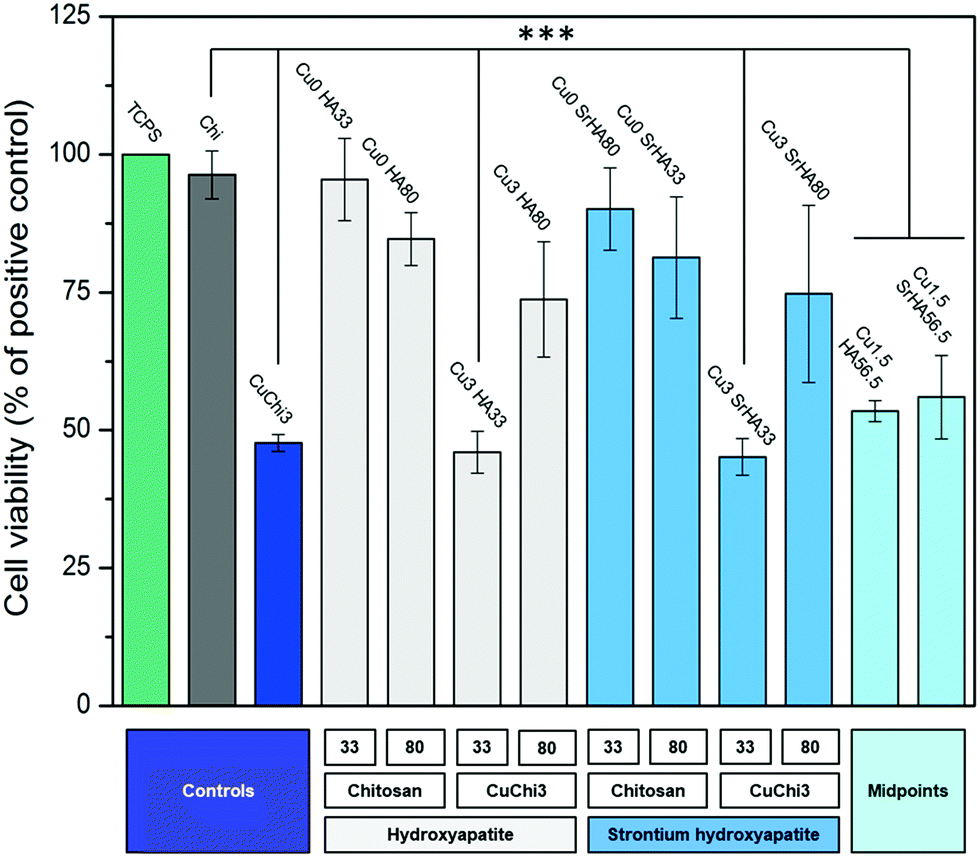
Indirect cell viability was performed on composite scaffolds using MG-63 human osteoblasts cell line. Fig. 13 shows that the cell viability is generally high (i.e. above 70% of the positive control). Copper seems to decrease the viability in some cases, especially for CuChi3, Cu3HA33 and Cu3SrHA33, indicating that higher HA loadings might have an effect in dampening the cytotoxic effect of the metal. In contrast, the presence of strontium did not have a significantly detrimental effect on MG-63 cells.
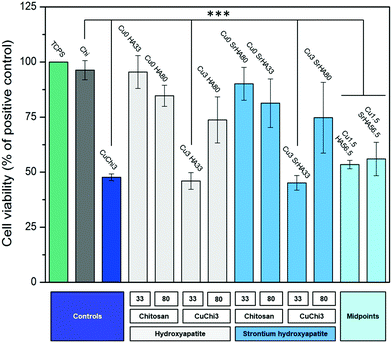 |
| | Fig. 13 Histogram reporting the cell viability of MG-63 cells cultured with all composite scaffold formulations. Tissue culture polystyrene (TCPS) was used as positive control. Chitosan performed similarly. Samples that were significantly different from the controls are highlighted (***: p < 0.001): they are all the copper containing specimens, with exception of samples with high HA content (Cu3HA80 and Cu3SrHA80). | |
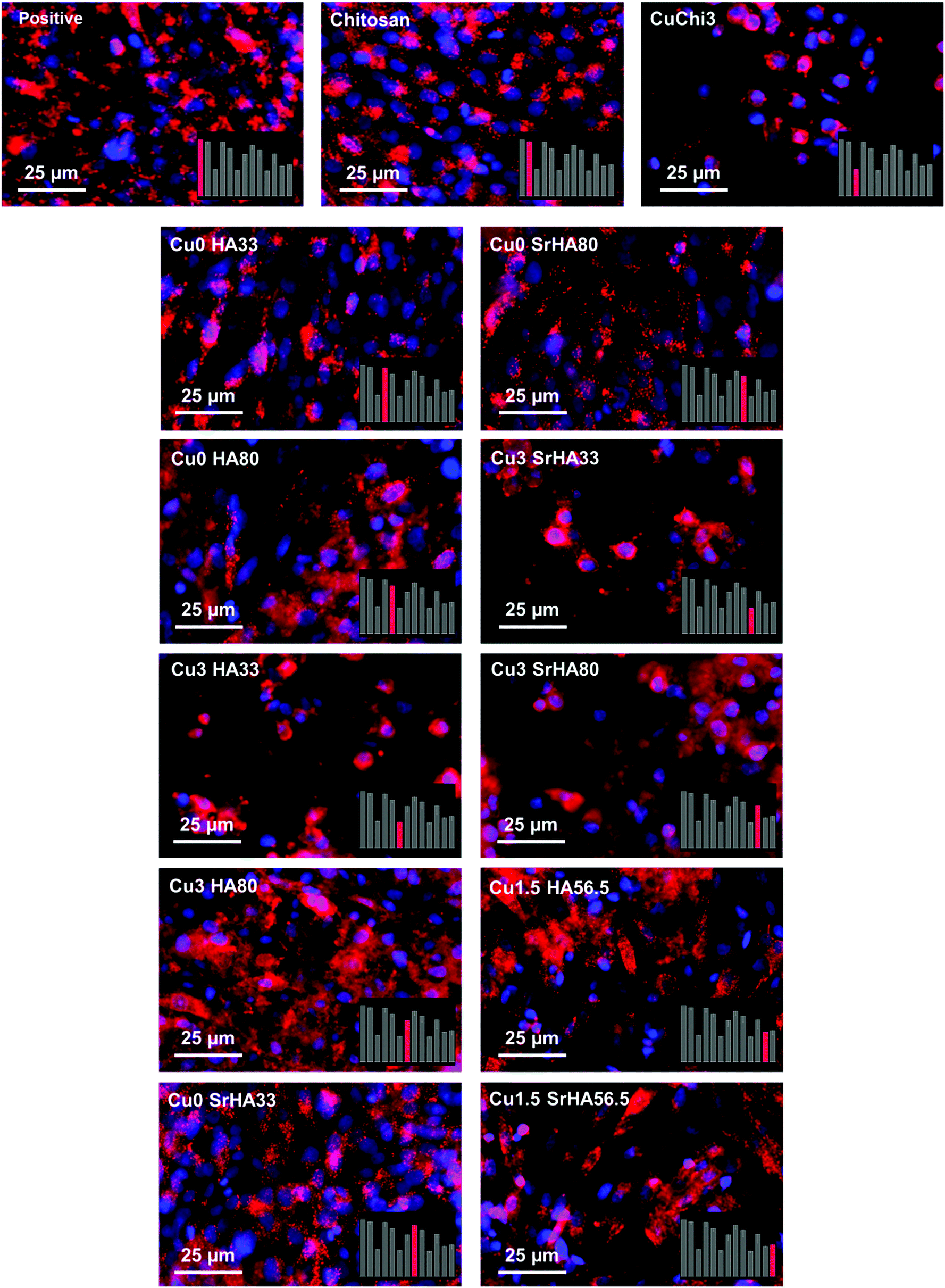
3.4.3 Cell staining.
The staining of the cells with DiI/DAPI was performed to complement the measurements of mitochondrial activity. DiI was found to be partially ineffective, as HA-rich samples showed background noise due to a probable interaction between the dye and HA. In fact, control samples did not show the same issue. Nevertheless, from the fluorescence images it can be observed that cells are numerous, with healthy and well elongated morphology on most samples (Fig. 14). On the basis of a qualitative comparison, the results are also in agreement with cell viability data, since cells are more sparse and round-shaped on the same samples that showed lower viability (i.e. Cu3HA33, Cu3SrHA33 and CuChi3 control).
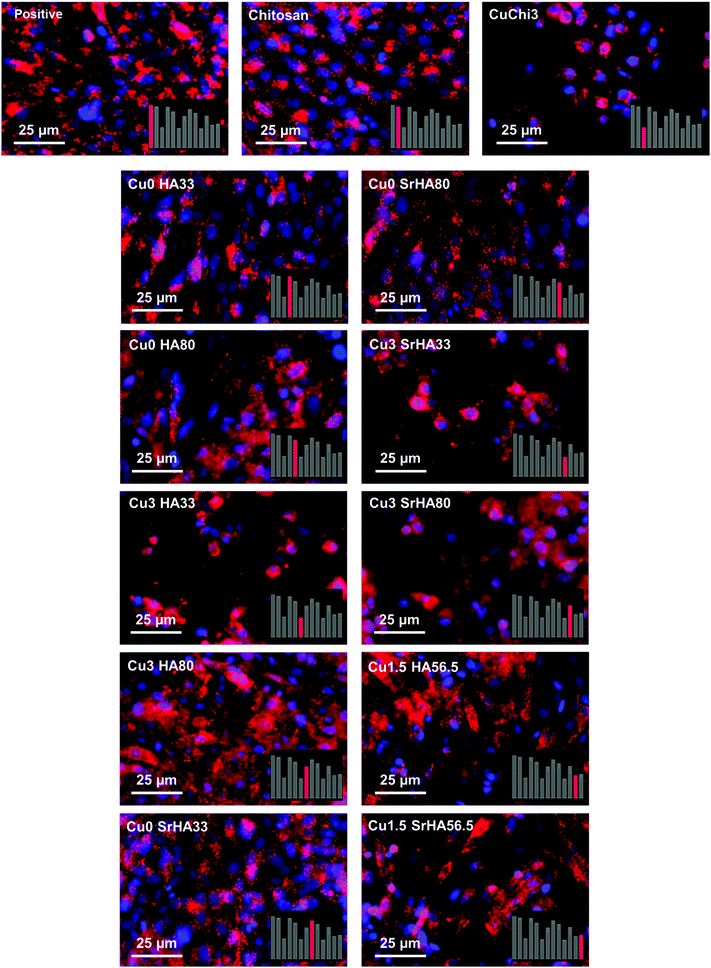 |
| | Fig. 14 Fluorescence images of MG-63 osteoblasts in indirect culture with the different scaffolds stained with DiI and DAPI. The bottom right panels highlight the respective cell viability measurement of each sample: it can be observed that the results of cell viability and cell morphology are qualitatively in accordance. | |
4 Discussion
Copper(II)–chitosan was prepared following the protocol introduced in our previous publication.39 The presence of copper and successful complexation with chitosan were previously confirmed by EDX and FTIR.39 In this work an analysis of the material by XRD was also performed, showing the presence of significant variations in the diffractogram of chitosan following the addition of copper. In particular, the complexation of copper induces a significant decrease in the 2θ = 20° peak of the diffractogram, which is characteristic for the crystal II structure of chitosan.46 This variation was previously reported for a complex of chitosan and iron: the authors suggested that this change in the diffractogram could be ascribed to a decrease in crystallinity of the final material compared to native chitosan.46 The complexation of chitosan with transition metals creates a reduction in binding sites available for hydrogen bonding (NH2 and OH), decreasing the number of inter- and intramolecular bonds between chitosan chains necessary for self-assembly of the polysaccharide.
In parallel, a wet precipitation method was used to successfully produce artificial HA and strontium-substituted HA powders. XRD results showed typical peaks of the hexagonal structure of HA, confirming the high crystallinity of these two materials. Only minor variations were identified as a consequence of the ion substitution and the expansion in the HA unit cell.50 In particular, Sr–HA samples were characterized by general peak broadening and shifts in low 2θ angle peaks, which are typical evidences of ion substitution.49 The analysis by FTIR further confirmed the findings of the XRD analyses: typical peaks and bands of phosphate and hydroxyl vibrations were detected both in HA and Sr–HA. In addition, a minor signal that can be ascribed to carbonate impurities was also identified. Finally, XRF was fundamental in confirming that the calcium phosphate produced was actually hydroxyapatite and to quantify the amount of strontium substituted. In previous studies, authors suggested that XRF is an effective technique for the quantification of substitutions in HA. However, other relevant techniques to perform this measurement could be ICP-MS or atomic absorption spectroscopy (AAS).21 The calculation of the Ca/P molar ratio performed from the results of compositional analysis confirmed a value very close to that characteristic of stoichiometric HA (1.667). These results are in good agreement with previously reported results in literature.51,55–57 Li et al.51 reported that Sr–HA substituted with 0.3 mol% and 1.5 mol% Sr2+ ions showed a monophasic spectrum comparable to pure HA. Kim et al.55 revealed that substitution of Sr2+ over 4 mol% caused transformation in the microstructure from sphere-shaped grains to faceted grains, also β-TCP was formed as secondary phase. The studies by Shen et al.57 about the effect of Sr2+ substitution on compressive strength of calcium phosphate cements (CPC) indicated that addition of 5 wt% Sr–HA whisker in CPC induced higher compressive strength of 2.91 MPa, which is approximately double than that of pure CPC. Kim et al.55 proved that 8 mol% substitution of Sr2+ in Sr–HA caused the increase in Vicker's hardness from 5.2 to 5.5 GPa. Capuccini et al.56 carried out in vitro tests using osteoblast-like MG-63 and human osteoclasts cells. Their results revealed that Sr–HA substituted with 3–7 at% Sr2+ ions considerably stimulated osteoblast proliferation and differentiation, also they reported that 1 at% substitution of Sr2+ ions was significant to reduce the osteoclast proliferation.56
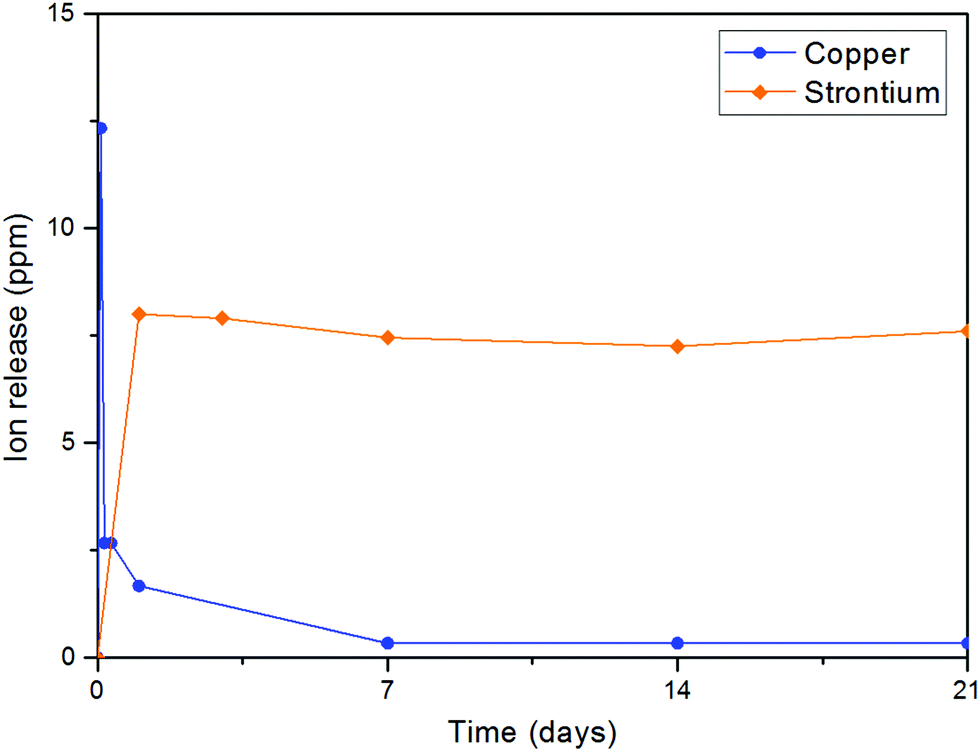
Before using the two synthesized biomaterials for the fabrication of composite scaffolds, ion release tests were also performed. Copper was quantified by a cost-effective capillary electrophoresis method previously developed.40 Results confirmed that copper(II)–chitosan is characterized by a burst release of copper within the first ten hours. The release can be finely tuned by adjusting the amount of copper initially loaded in the polysaccharide. In particular, the formulation CuChi3, used for the production of the scaffolds, releases around 15–20 ppm of copper within the first few hours; this is an ideal release, which can inhibit the colonization by bacteria upon implantation, without excessive cytotoxic effects. It is important to notice that after the burst release a significant quantity of copper is still embedded in the chitosan and will be released only upon degradation of the polymer, which does not occur in saline solutions (i.e. PBS).58 Ion release tests after enzymatic degradation should be performed in order to quantify longer term release of copper from copper(II)–chitosan, which could still be helpful in impeding later stage bacterial colonization and enhancing angiogenesis.38 The release of strontium from Sr–HA was simultaneously determined by ICP-OES and follows a very different trend compared to the one of copper: release reached equilibrium at around 8 ppm per each time point. Strontium release at this concentration sits within the therapeutic window (2–45 ppm) and it is expected to help osteogenesis throughout the first months of regeneration in vivo.59 The different nature of the TMI loading in the two materials highlights the convenience of the present strategy to obtain very different release profiles depending on the desired ion, tailoring the uniformity of release according to need independently of the intrinsic scaffold degradation kinetics (Fig. 15).
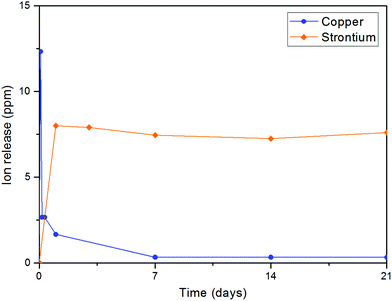 |
| | Fig. 15 Release of copper from copper(II)–chitosan and of strontium from Sr–HA particles: the difference in release can be clearly observed. While copper is characterized by burst release, the release of strontium is uniform over time. | |
After the characterization of the biomaterials and the investigation of ion release, copper(II)–chitosan and strontium substituted hydroxyapatite were successfully combined and fabricated into composite scaffolds for bone tissue engineering by liquid nitrogen freeze-drying, using chitosan as the matrix and HA as the particulate filler phase. In comparison to previously reported systems, in this work a novel concept for the preparation of bone TE scaffolds is introduced, which offers the possibility to dope both the matrix and the filler of the composite with TMIs. In this work, copper and strontium were used. However, chitosan and hydroxyapatite are very versatile material platforms that could allow the design of more complex arrays of ion doping and release. At the same time, the fabrication of TE scaffolds by freeze-drying as reported in this work can be adapted to other derivatives of chitosan and HA. This allows the production of constructs able to deliver several ions with different release profile in time, depending on the specific need. In fact, different ions are needed at different times in order to effectively guide cell adhesion and proliferation as well as, at a later stage, tissue ingrowth and vascularization.25 Having the possibility to engineer the release of TMIs to follow these complex profiles can allow precise engineering of the several stages of inflammation, healing and tissue regeneration. In parallel, TMIs offer also the advantage of enhancing the antibacterial properties of scaffolds with little risk of resistance development. In fact, the antibacterial activity of TMIs is known to be quick and multifaceted: such rapid action inhibits the intrinsic ability of bacteria to develop resistance.60
Liquid nitrogen freeze-drying is a robust and reproducible fabrication technique, especially in terms of morphology and porosity. Chitosan/HA composite scaffolds prepared in this study are characterized by tailorable, hierarchical and interconnected porosity in the ideal range for cell infiltration and correct differentiation.54 The macroscale pore size and distribution can be finely tailored through the variation of the processing parameters, especially by controlling the freezing process and the composition of the HA/chitosan suspensions.9 In particular, rapid freezing with liquid nitrogen is an effective way to decreases the average size of the water crystal formed and thus of the pores. In comparison with previously reported chitosan-based scaffolds,61–63 this approach is an important improvement in order to achieve better control of scaffold morphology. Indeed, in previously developed protocols for the preparation of chitosan/hydroxyapatite scaffolds nitrogen freezing has rarely been used.61–63 Madihally et al.,64 for instance, proposed to use liquid nitrogen for chitosan scaffolds as a solution to achieve pore size and morphology control and bring them closer to physiologically relevant levels. Our results confirm this hypothesis, as chitosan/HA scaffolds prepared in this work have a fairly homogeneous distribution of interconnected pores with a size in the range of 10–100 μm,65 which is known to be ideal for cell infiltration.54,66 Tailoring pore morphology is a key challenge to correctly regulate cell colonization rates and engineer the organization of new tissue into scaffolds.54 In addition to this, pore size, morphology and interconnectivity are known to be key variables in controlling degradation kinetics, mechanical properties and angiogenesis.4,64,67
The introduction of hydroxyapatite in the chitosan matrix was expected to give the composite several beneficial properties that native chitosan lacks. In terms of mechanical properties, the inorganic filler is a key factor in increasing the compressive modulus of the scaffolds. This finding is in agreement with previously reported composite scaffolds and in general with theories on composite materials.9,68 In particular, the scaffold formulations that showed the best mechanical performance are Cu0HA80 and Cu1.5HA56.5. These samples reached values of compressive modulus of around 2–3 MPa, which are approaching the lower end of the modulus of trabecular bone,67 but should be further improved. Interestingly, HA was found to perform better than Sr–HA. Strontium-substituted hydroxyapatite resulted in moduli which barely reached 1 MPa even for high filler loadings. An explanation for this behavior could be a consequence of poorer interface properties: possible leaching of strontium ions from the HA, even in trace amounts, could have been absorbed by the chitosan matrix, inducing the formation of complexes at the interface and resulting in embrittlement and reduction in compliance.69 This local cross-linking of chitosan as a consequence of the complexation could have determined a reduction in the ability of chitosan to deform under compression, increasing the formation of interface voids. In future, interfacial phenomena between Sr–HA and chitosan should be characterized and the interface itself improved by, for example, surface modification of the HA filler. In addition to improving mechanical properties, HA and Sr–HA containing scaffolds were effective in triggering the deposition of a calcium phosphate layer after 7 days in SBF. In contrast, no deposition was assessed on control scaffolds (i.e. chitosan and copper(II)–chitosan). In these samples, the only consequence of the immersion in SBF is the collapse of most of the porosity upon dehydration, which highlights the poor integrity of native chitosan scaffolds and the need for a reinforcing filler to increase mechanical properties, as previously discussed. However, shielding of the signal of this layer by HA initially present in the scaffolds made FTIR analysis inconclusive. Longer incubation times (e.g. 21 or 28 days) in SBF would probably show clearer results. Avoiding the use of Tris buffer is also an important aspect for future optimization of the test: the addition of this buffer in SBF was in fact linked to a possible distortion in the apatite-forming ability of bioactive materials.28 Tris seems to increase the dissolution rate of bioactive glasses (particularly Bioglass® 45S5) and therefore overexpress their bioactivity. Although HA is more stable and less susceptible to possible chemical attack of Tris,28 the possible occurrence of this phenomenon should be verified. Further investigation is needed to clarify the actual extent of scaffold bioactivity and the nature of the deposited phase.
Finally, a preliminary cell culture study was performed using the MG-63 human osteoblast-like cell line. The results of cell viability, measured by WST-8, showed that the ions release had mildly cytotoxic effects only in those samples with little to no HA phase. Fluorescence images of cell morphology are in agreement with cell viability results. In particular, it appears that the presence of HA reduces the cytotoxic character of copper. This is likely due to a protective effect of ions leaching from hydroxyapatite, which can establish biomolecular competition for copper-binding sites.70,71 The lower viability of the CuChi3 sample is somehow in contrast with previous results on the same material39 and could be due to the increase in surface area of the porous samples compared to bulk ones, highlighting the need to perform ion release tests on the full scaffolds.72 These preliminary findings foster further studies to optimize and expand the possible fabrication routes of scaffolds loaded with multiple ions and to characterize the physicochemical and biological performance of these promising tissue engineering platforms. Specifically, in-depth investigations should be carried out in order to explore more combinations of ions and to unravel the specific responses that these ions elicit on cells when released simultaneously and in a controlled manner.
5 Concluding remarks
Two candidate platforms for the delivery of TMIs were prepared: chitosan complexed with copper and strontium substituted hydroxyapatite. The effective loading and release of ions from the two materials was characterized and their use in the production of novel bone TE composite scaffolds with dual ion delivery was demonstrated. The release of copper and strontium follows significantly different profiles due to the different nature of the loading. In particular, copper experiences a burst release from chitosan allowing it to quickly stop possible bacterial colonization, while strontium is released uniformly in order to help and support the regeneration process over a longer period. The materials can be prepared into scaffolds using an optimized and reproducible freeze drying protocol, using chitosan as matrix and a fine, homogeneous particulate HA phase as filler. The resulting scaffolds are ideal for cell infiltration, cell migration and neovascularization thanks to their reproducible pore morphology. They are characterized by interconnected porosity and increased mechanical properties depending on the filler content. Cell cultures performed on the composite scaffolds showed that the materials are also cytocompatible. Compared to existing technologies, chitosan/HA scaffolds produced in this study exhibit the novel feature of carrying therapeutic ions both in the matrix and the filler of the composite. In this study, copper and strontium were loaded respectively in chitosan and hydroxyapatite. However, these two biomaterials are highly versatile and can chelate (chitosan) and include (hydroxyapatite) several different ions in their structure. The combination of chitosan and hydroxyapatite has the potential to design scaffolds with the capability to deliver TMIs depending on the specific need in terms of tissue regeneration properties. The two materials are versatile carriers of biologically relevant ions. In the future, the possibility to have more complex arrays of therapeutic ions in both components of the composite, targeting different cells and tissues, should be explored.
Conflicts of interest
There are no conflicts of interest to declare.
Acknowledgements
This work has received funding from the European Union's Horizon 2020 Research and Innovation Programme under the Marie Sklodowska-Curie (HyMedPoly project, Grant Agreement No. 643050). We acknowledge the important support of the whole HyMedPoly consortium. We would also like to thank Mr Mike Salt for its technical guidance with freeze-drying and of Mr Lee Bullock for mechanical testing consultancy.
References
- A. R. Amini, C. T. Laurencin and S. P. Nukavarapu, CRC Crit. Rev. Bioeng., 2012, 40, 363–408 Search PubMed.
- R. A. Bhatt and T. D. Rozental, Handb. Clin. Neurol., 2012, 28, 457–468 Search PubMed.
- M. Jafari, Z. Paknejad, M. R. Rad, S. R. Motamedian, M. J. Eghbal, N. Nadjmi and A. Khojasteh, J. Biomed. Mater. Res., Part B, 2017, 105, 431–459 CrossRef CAS PubMed.
- F. J. O’Brien, Mater. Today, 2011, 14, 88–95 CrossRef.
- L. Gritsch, D. Meng and A. R. Boccaccini, Biomed. Compos., 2017, 501–542 Search PubMed.
- K. Rezwan, Q. Z. Chen, J. J. Blaker and A. R. Boccaccini, Biomaterials, 2006, 27, 3413–3431 CrossRef CAS PubMed.
- N. Goonoo, A. Bhaw-Luximon, P. Passanha, S. R. Esteves and D. Jhurry, J. Biomed. Mater. Res., Part B, 2017, 105, 1667–1684 CrossRef CAS PubMed.
- A. R. Boccaccini, M. Erol, W. J. Stark, D. Mohn, Z. Hong and J. F. Mano, Compos. Sci. Technol., 2010, 70, 1764–1776 CrossRef CAS.
- L. Pighinelli and M. Kucharska, Carbohydr. Polym., 2013, 93, 256–262 CrossRef CAS PubMed.
- F. Baino, G. Novajra and C. Vitale-Brovarone, Front. Bioeng. Biotechnol., 2015, 3, 202 Search PubMed.
- S. Deville, E. Saiz, R. K. Nalla and A. P. Tomsia, Adv. Sci. Technol., 2006, 49, 148–152 CAS.
- V. Brun, C. Guillaume, S. Mechiche Alami, J. Josse, J. Jing, F. Draux, S. Bouthors, D. Laurent-Maquin, S. C. Gangloff, H. Kerdjoudj and F. Velard, Biomed. Mater. Eng., 2014, 24, 63–73 CAS.
- H. Li, C. Hu, H. Yu and C. Chen, RSC Adv., 2018, 8, 3736–3749 RSC.
- H. Siddiqui, K. Pickering and M. Mucalo, Materials, 2018, 11, 1813 CrossRef PubMed.
- J. T. B. Ratnayake, M. Mucalo and G. J. Dias, J. Biomed. Mater. Res., Part B, 2017, 105, 1285–1299 CrossRef CAS PubMed.
-
I. Cacciotti, Handbook of Bioceramics and Biocomposites, 2016, pp. 145–211 Search PubMed.
- S. Sprio, A. Tampieri, E. Landi, M. Sandri, S. Martorana, G. Celotti and G. Logroscino, Mater. Sci. Eng., C, 2008, 28, 179–187 CrossRef CAS.
- T. Tite, A. C. Popa, L. M. Balescu, I. M. Bogdan, I. Pasuk, J. M. F. Ferreira and G. E. Stan, Materials, 2018, 11, E2081 CrossRef PubMed.
- A. C. Tas, J. Eur. Ceram. Soc., 2000, 20, 2389–2394 CrossRef CAS.
- M. Šupová, Ceram. Int., 2015, 41, 9203–9231 CrossRef.
- G. Graziani, M. Boi and M. Bianchi, Coatings, 2018, 8, 269 CrossRef.
- E. S. Thian, J. Huang, M. E. Vickers, S. M. Best, Z. H. Barber and W. Bonfield, J. Mater. Sci., 2006, 41, 709–717 CrossRef CAS.
- I. R. Gibson, S. M. Best and W. Bonfield, J. Biomed. Mater. Res., 1999, 44, 422–428 CrossRef CAS PubMed.
- Y. Lei, Z. Xu, Q. Ke, W. Yin, Y. Chen, C. Zhang and Y. Guo, Mater. Sci. Eng., C, 2017, 72, 134–142 CrossRef CAS PubMed.
- V. Mouriño, J. P. Cattalini and A. R. Boccaccini, J. R. Soc., Interface, 2012, 9, 401–419 CrossRef PubMed.
- A. Philippart, N. Gómez-Cerezo, D. Arcos, A. J. Salinas, E. Boccardi, M. Vallet-Regi and A. R. Boccaccini, J. Non-Cryst. Solids, 2017, 455, 90–97 CrossRef CAS.
- P. J. Marie, P. Ammann, G. Boivin and C. Rey, Calcif. Tissue Int., 2001, 69, 121–129 CrossRef CAS PubMed.
- Y. Zhang, X. Cui, S. Zhao, H. Wang, M. N. Rahaman, Z. Liu, W. Huang and C. Zhang, ACS Appl. Mater. Interfaces, 2015, 7, 2393–2403 CrossRef CAS PubMed.
- U. Thormann, S. Ray, U. Sommer, T. ElKhassawna, T. Rehling, M. Hundgeburth, A. Henß, M. Rohnke, J. Janek, K. S. Lips, C. Heiss, G. Schlewitz, G. Szalay, M. Schumacher, M. Gelinsky, R. Schnettler and V. Alt, Biomaterials, 2013, 34, 8589–8598 CrossRef CAS PubMed.
- G.-M. Kuang, W. P. Yau, J. Wu, K. W. K. Yeung, H. Pan, W. M. Lam, W. W. Lu and K. Y. Chiu, J. Biomed. Mater. Res., Part A, 2015, 103, 1613–1621 CrossRef PubMed.
- A. A. Gorustovich, T. Steimetz, R. L. Cabrini and J. M. P. López, J. Biomed. Mater. Res., Part A, 2010, 92A, 232–237 CrossRef CAS PubMed.
- G. Molino, A. Bari, F. Baino, S. Fiorilli and C. Vitale-Brovarone, J. Mater. Sci., 2017, 52, 9103–9114 CrossRef CAS.
-
R. Z. LeGeros, Clinical Orthopaedics and Related Research, 2002, pp. 81–98 Search PubMed.
- H. Xie and Y. J. Kang, Curr. Med. Chem., 2009, 16, 1304–1314 CrossRef CAS PubMed.
- G. Grass, C. Rensing and M. Solioz, Appl. Environ. Microbiol., 2011, 77, 1541–1547 CrossRef CAS PubMed.
- R. B. Thurman and C. P. Gerba, Crit. Rev. Environ. Control, 1989, 18, 298–315 Search PubMed.
- M. L. Turski and D. J. Thiele, J. Biol. Chem., 2009, 284, 717–721 CrossRef CAS PubMed.
- L. D. D’Andrea, A. Romanelli, R. Di Stasi and C. Pedone, Dalton Trans., 2010, 39, 7625 RSC.
- L. Gritsch, C. Lovell, W. H. Goldmann and A. R. Boccaccini, Carbohydr. Polym., 2018, 179, 370–378 CrossRef CAS PubMed.
- J. P. Cattalini, V. S. Mouriño and S. E. Lucangioli, Anal. Methods, 2016, 8, 7767–7773 RSC.
- M. R. Saeri, A. Afshar, M. Ghorbani, N. Ehsani and C. C. Sorrell, Mater. Lett., 2003, 57, 4064–4069 CrossRef CAS.
- T. Kokubo and H. Takadama, Biomaterials, 2006, 27, 2907–2915 CrossRef CAS PubMed.
- E. Boccardi, A. Philippart, V. Melli, L. Altomare, L. De Nardo, G. Novajra, C. Vitale-Brovarone, T. Fey and A. R. Boccaccini, Ann. Biomed. Eng., 2016, 44, 1881–1893 CrossRef CAS PubMed.
- A. L. B. Maçon, T. B. Kim, E. M. Valliant, K. Goetschius, R. K. Brow, D. E. Day, A. Hoppe, A. R. Boccaccini, I. Y. Kim, C. Ohtsuki, T. Kokubo, A. Osaka, M. Vallet-Regí, D. Arcos, L. Fraile, A. J. Salinas, A. V. Teixeira, Y. Vueva, R. M. Almeida, M. Miola, C. Vitale-Brovarone, E. Verné, W. Höland and J. R. Jones, J. Mater. Sci.: Mater. Med., 2015, 26, 1–10 CrossRef PubMed.
-
F. Ciraldo, L. Liverani, L. Gritsch, W. Goldmann and A. Boccaccini, Materials, 2018, 11, 692 Search PubMed.
- J. Qu, Q. Hu, K. Shen, K. Zhang, Y. Li, H. Li, Q. Zhang, J. Wang and W. Quan, Carbohydr. Res., 2011, 346, 822–827 CrossRef CAS PubMed.
- M. C. Moolman, Z. Huang, S. T. Krishnan, J. W. J. Kerssemakers and N. H. Dekker, J. Nanobiotechnol., 2013, 11, 12 CrossRef PubMed.
- M. J. Woźniak-Budych, K. Langer, B. Peplińska, Ł. Przysiecka, M. Jarek, M. Jarzębski and S. Jurga, Mater. Chem. Phys., 2016, 179, 242–253 CrossRef.
- K. Ozeki, T. Goto, H. Aoki and T. Masuzawa, Bio-Med. Mater. Eng., 2014, 24, 1447–1456 CAS.
- W. Zhang, N. Cao, Y. Chai, X. Xu and Y. Wang, Ceram. Int., 2014, 40, 16061–16064 CrossRef CAS.
- Z. Y. Li, W. M. Lam, C. Yang, B. Xu, G. X. Ni, S. A. Abbah, K. M. C. Cheung, K. D. K. Luk and W. W. Lu, Biomaterials, 2007, 28, 1452–1460 CrossRef CAS PubMed.
- C. M. Mardziah, I. Sopyan, M. Hamdi and S. Ramesh, Med. J. Malays., 2008, 63(suppl. A), 79–80 Search PubMed.
- Z. H. Cheng, A. Yasukawa, K. Kandori and T. Ishikawa, Langmuir, 1998, 14, 6681–6686 CrossRef CAS.
- Q. L. Loh and C. Choong, Tissue Eng., Part B, 2013, 19, 485–502 CrossRef CAS PubMed.
- H. W. Kim, Y. H. Koh, Y. M. Kong, J. G. Kang and H. E. Kim, J. Mater. Sci.: Mater. Med., 2004, 15, 1129–1134 CrossRef CAS PubMed.
- C. Capuccini, P. Torricelli, E. Boanini, M. Gazzano, R. Giardino and A. Bigi, J. Biomed. Mater. Res., Part A, 2009, 89, 594–600 CrossRef CAS PubMed.
- Y. Shen, J. Liu, K. Lin and W. Zhang, Mater. Lett., 2012, 70, 76–79 CrossRef CAS.
- M. Periayah, A. Halim and A. M. Saad, Pharmacogn. Rev., 2016, 10, 39 CrossRef CAS PubMed.
- A. Aimaiti, A. Maimaitiyiming, X. Boyong, K. Aji, C. Li and L. Cui, Stem Cell Res. Ther., 2017, 8, 282 CrossRef PubMed.
- C. Walsh, Nature, 2000, 406, 775–781 CrossRef CAS PubMed.
- I. Adekogbe and A. Ghanem, Biomaterials, 2005, 26, 7241–7250 CrossRef CAS PubMed.
- T. Garg, A. Chanana, R. Joshi and S. G. L. Bihani, IOSR J. Pharm., 2012, 2, 72–73 Search PubMed.
- G. Pereira, M. Amaral, P. Cesar, M. Bottino, C. Kleverlaan and L. Valandro, J. Mech. Behav. Biomed. Mater., 2015, 45, 183–192 CrossRef CAS PubMed.
- S. V. Madihally and H. W. T. Matthew, Biomaterials, 1999, 20, 1133–1142 CrossRef CAS PubMed.
- D. Narayan and S. S. Venkatraman, J. Biomed. Mater. Res., Part A, 2008, 87, 710–718 CrossRef CAS PubMed.
-
L. Gritsch, D. Meng and A. R. R. Boccaccini, Biomedical Composites, Elsevier, 2017, pp. 501–542 Search PubMed.
- E. Boccardi, F. E. Ciraldo and A. R. Boccaccini, MRS Bull., 2017, 42, 226–232 CrossRef CAS.
- Y. Y. Zheng, Y. Y. Zheng and R. Ning, Mater. Lett., 2003, 57, 2940–2944 CrossRef CAS.
- L. Lupa, R. Voda and A. Popa, Sep. Sci. Technol., 2018, 53, 1107–1115 CrossRef CAS.
- T. Dudev and C. Lim, Chem. Rev., 2014, 114, 538–556 CrossRef CAS PubMed.
- T. Yamazaki, M. Kobayashi, K. Hirano, H. Onuki, J. Shimada, A. Yamazaki, Y. Hibino, H. Nakajima, Y. Yokote, S. Takemoto, Y. Oda and H. Sakagami, In Vivo, 2012, 26, 651–656 CAS.
- M. Manzano and M. Vallet-Regí, J. Mater. Chem., 2010, 20, 5593–5604 RSC.
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  abe,
Muhammad
Maqbool
ab,
Viviana
Mouriño
cd,
Francesca E.
Ciraldo
a,
Mark
Cresswell
b,
Philip R.
Jackson
b,
Christopher
Lovell
b and
Aldo R.
Boccaccini
abe,
Muhammad
Maqbool
ab,
Viviana
Mouriño
cd,
Francesca E.
Ciraldo
a,
Mark
Cresswell
b,
Philip R.
Jackson
b,
Christopher
Lovell
b and
Aldo R.
Boccaccini