
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A muscle mimetic polyelectrolyte–nanoclay organic–inorganic hybrid hydrogel: its self-healing, shape-memory and actuation properties†
Sovan Lal
Banerjee
a,
Thomas
Swift
 b,
Richard
Hoskins
b,
Stephen
Rimmer
*b and
Nikhil K.
Singha
*a
b,
Richard
Hoskins
b,
Stephen
Rimmer
*b and
Nikhil K.
Singha
*a
aRubber Technology Centre, Indian Institute of Technology, Kharagpur, India. E-mail: nks8888@yahoo.com
bSchool of Chemistry and Biosciences, University of Bradford, Bradford, West Yorkshire BD7 1DP, UK. E-mail: S.Rimmer@bradford.ac.uk
First published on 28th January 2019
Abstract
In this investigation, we report a non-covalent (ionic interlocking and hydrogen bonding) strategy of self-healing in a covalently crosslinked organic–inorganic hybrid nanocomposite hydrogel, with specific emphasis on tuning its properties fitting into a muscle mimetic material. The hydrogel was prepared via an in situ free radical polymerization of sodium acrylate (SA) and successive crosslinking in the presence of starch grafted with poly(2-(methacryloyloxy)ethyl trimethyl ammonium chloride) (PMTAC) and montmorillonite modified with cetyl ammonium bromide (OMMT). This hydrogel shows stimuli triggered self-healing following damage in both neutral and acidic solutions (pH = 7.4 and pH = 1.2). This behavior was reported using stress–strain experiments and rheological analyses of the hydrogel segments joined at their fracture points. The hydrogel was also able to display shape memory properties in the presence of water as well as stimuli (salt, acid and electric impulse) driven actuation behavior. It was observed that the ultimate tensile strength (UTS) of the self-healed hydrogel at pH = 7.4 was comparable to the extensor digitorum longus (EDL) muscle of a New Zealand white rabbit and the as synthesized self-healable hydrogel was found to be non-cytotoxic against NIH 3T3 fibroblast cells.
1. Introduction
Muscle is a soft tissue, containing protein filaments capable of undergoing stimuli driven contraction, responsible for most forms of locomotion in animal species. There are several different types of muscle tissues; skeletal, cardiac and smooth. In recent years various types of synthetic materials have been produced to mimic the nature of these muscle types. In this field, polymer gels are of great interest due to their viscoelastic mechanical properties, their capability to hold a large amount of solvents (specifically water) and structural similarity to the components of animal or human tissues.1–4 Usually, the connecting tissue consists of “fibroblast” cells which, in biological tissues, are capable of healing damage to the material substrate. Ideally, to mimic a muscle system, a polymer hydrogel should have good self-healing characteristics, excellent shape memory properties and stimuli driven actuation properties. The self-healing characteristics are important for challenging events, like when the muscles are torn or undergo cleavage under high stress and strain. The material also requires water-based shape memory properties, as the muscle can undergo cramping due to dehydration and/or other physiological conditions. Thus the muscle mimetic polymeric hydrogel should have dynamic self-healing behavior which can restore its mechanical integrity after failure.Self-healable hydrogels can be prepared by using different physical and chemical interactions. Physically interacting self-healable hydrogels involve hydrogen bonding,5,6 metal coordination,7 hydrophobic interactions,8 host–guest interactions,9 π–π stacking,10 electrostatic interactions,11,12 Schiff base reactions,13,14 thiol–yne click reactions15etc., whereas chemically interacting self-healable gels utilize light,16 temperature17etc. to self-heal via the formation of new chemical bonds. The main disadvantages observed in most physically interacting self-healable hydrogels are their poor mechanical stability (i.e. contractility, extensibility and elasticity), which is an important factor to mimic a muscle system. Physically interacting hydrogels have poor elasticity and are unable to dissipate the absorbed energy, so they are unsuitable for use as an artificial muscle, cartilage, tendons etc. They require high load-bearing capacity without permanent shape deformation (shape memory effect) as well as self-healing properties. Therefore, many researchers have focused on the preparation of stronger hydrogels. Possible solutions in this area include different types of nano-filler18 and double network (DN) hydrogels in which both physical and chemical interactions are present.19,20 The formation of intimate interfacial interactions between the filler and the polymer matrix contributes to the enhanced mechanical properties of the hydrogel. MMT clay is one of the most tested components to prepare a nanocomposite hydrogel, due to its high biocompatibility, easy availability and ability to form strong interactions with the polymer matrix (due to the availability of layered structures).21 It is a smectic-type clay which has an expandable 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of aluminosilicate clay mineral, a high platelet aspect ratio, excellent cation exchange capacity, and very high specific surface area and can show high water swelling capability due to the penetration of water into the layered structure.22,23
:1 ratio of aluminosilicate clay mineral, a high platelet aspect ratio, excellent cation exchange capacity, and very high specific surface area and can show high water swelling capability due to the penetration of water into the layered structure.22,23
In this work we have prepared a nanocomposite hydrogel system in which we have used “ionic interaction” as a sacrificial physical bond along with the permanent covalent crosslinks to prepare the hydrogel system. Irreversible crosslinked systems are known to restrict self-healing behavior due to the limited crosslink repair available within the highly restrictive network. In this study, we have incorporated both physical crosslinks and covalent crosslinks to introduce self-healing properties to the hydrogels as well as to improve elasticity. These properties together create a suitable muscle-mimic material. In our previous report, we have described a simple method for the preparation of self-healable nanocomposite hydrogels based on starch and poly(2-(acryloyloxy)ethyltrimethyl)ammonium chloride as a polycation and poly(acrylic acid) as a polyanion. These monomers were polymerized and crosslinked randomly in the presence of cetyl trimethyl ammonium bromide (CTAB) modified MMT clay (OMMT) to give a solid gel mass. However, the main problem with that nanocomposite hydrogel is its lower stretchability due to the random grafting of the (polycationic) and (polyanionic) segments in the starch backbone. For that reason, it showed only 200% elongation on manual stretching before it breaks down.6
This material is a new class of smart self-healable hydrogel that can repair incisions in a wide range of pH, from pH 1.2 (pH inside stomach) to pH 7.4 (physiological pH), and can elongate with a higher extension and heal faster than the previously formulated hydrogels.6 We have also studied the shape memory property of this hydrogel. The healing property was studied by evaluating the tensile property and rheological behaviour. The shape memory property was evaluated by measuring the shape fixity (Rf) and shape recovery ratio (Rr). Along with the shape memory and self-healing properties, the actuation property of the synthesized hydrogel was also monitored in the presence of salt solution, acidic conditions and electrical impulse. Cell viability assay was carried out against a fibroblast cell line to monitor its cytotoxic effect.
2. Materials and methods
2.1. Materials
Soluble starch (SS), analytical grade ammonium persulfate (APS), N,N′-methylene-bis-acrylamide (MBA) (crosslinking agent), 2-(methacryloyloxy)ethyltrimethyl ammonium chloride (MTAC) solution (80% in H2O) and acrylic acid (AA) monomer were purchased from Sigma-Aldrich, USA. Analytical grade ethanol (EtOH) and sodium hydroxide (NaOH) were purchased from Merck, India. Montmorillonite (K-10) (MMT) clay, cetyl trimethyl ammonium bromide (CTAB) and 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) were also purchased from Sigma-Aldrich. Fetal bovine serum (FBS) and Dulbecco's modified Eagle's medium (DMEM) were obtained from HiMEDIA, India. NIH 3T3 fibroblast cells were obtained from the National Centre for Cell Science, Pune, India. Deionized (DI) water was used throughout the work.2.2. Synthesis of starch-graft-poly(2-(methacryloyloxy)ethyltrimethyl ammonium chloride) (SS-g-PMTAC)
In a typical formulation, soluble starch (SS, 4.0 g) was dissolved in deionized water (50 ml) in a three-neck round bottom (RB) flask fitted with a condenser and heated for 1 h (50 °C) to solubilize the starch. Cationic monomer, 2-(methacryloyloxy) ethyltrimethyl ammonium chloride (MTAC) solution (80% in H2O, 12 g, 57.9 mmol, 1:3 wt ratio with SS), was injected into the system with continuous stirring under nitrogen (10 min, 65 °C). Following this, an aqueous solution of the thermal initiator APS (5 ml, 12 mg, 1 wt% of monomer) was added dropwise into the reaction mixture under N2 and the reaction mixture was left to polymerize for 3 h. The reaction mixture was cooled to room temperature and then precipitated via dropwise addition into methanol (500 ml) under stirring. Purification of the graft copolymer was carried out via repeated reprecipitation from the water. The purified polymer was filtered, resolubilized in water and freeze-dried to provide a fluffy white mass. This was characterized by 1H NMR, 1H diffusion-ordered NMR spectroscopy (DOSY), FTIR and XRD analyses as detailed below. For the DOSY analysis, soluble starch was taken as a control. The grafting percentage was determined using the following formula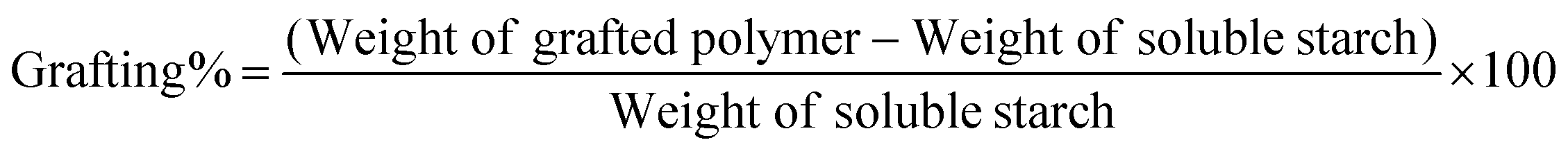
2.3. Synthesis of polyelectrolyte containing nanocomposite hydrogels
In a typical synthesis process, a predetermined amount of OMMT clay (2 wt%, 5 wt% and 7 wt%) was dispersed in deionized water and sonicated for 1 h to form a stable dispersion. CTAB modified montmorillonite (OMMT) clay was prepared by following a previously reported method.24 After that the modified cationic soluble starch (CSS), base neutralized AA (12 g, 6 mol) solution and MBA (0.12 g, 1 wt% of total monomer concentration) were added sequentially to the clay aqueous dispersion taken in a three neck RB flask fitted with a N2 inlet–outlet system under constant stirring at 65 °C and left for 30 min for homogenization. Neutralization of AA was done by adding NaOHaq (25 ml, 4 mol dm−3) solution in AAaq (24 ml, add the molar concentration). After complete dispersion of the OMMT clay (visualized with the naked eye), an aqueous solution of APS (0.12 g, 1 wt% of total monomer concentration) was added into the reaction mixture under an inert atmosphere (N2 atm) and continuous agitation of the reaction mixture. This reaction mixture was allowed to pre-polymerize and after an increase in the viscosity of the reaction mixture the whole mass was quickly transferred to a cylindrical mould having a dimension of 5 cm in length and 1 cm in diameter. The same process was also adapted to prepare rectangular shaped samples having dimensions of 40 mm × 15 mm × 3 mm. The post-polymerization reaction was carried out by dipping the closed cylindrical mould in a hot water bath maintained at a temperature of 65 °C for 2 h to obtain a solid hydrogel mass. During hydrogel preparation, the amount of cationically modified starch was varied to 0 wt%, 10 wt%, 30 wt% and 50 wt%. The sample is denoted as PSASxCy. For example, PSAS30C5 signifies the poly(sodium acrylate) hydrogel having 30 wt% soluble starch and 5 wt% OMMT clay. The as-prepared cylindrical hydrogel was then dipped into an ethanol–water mixture (1:4 volume ratio) and left for 24 h to make the hydrogel free from the unreacted mass. Then all the formulated cylindrical hydrogel samples were kept inside a vacuum oven and allowed to dry for 48 h at 50 °C. Details of the formulation are summarized in Table 1.
| No. | Sample name | Cationic soluble starch (CSS) content (wt%) | OMMT content (wt%) | M c (g mol−1) | ρ c (10−20) |
|---|---|---|---|---|---|
| Total batch volume maintained at 40 ml. APS content = 1 wt% with respect to monomer concentration. MBA content = 1 wt% with respect to monomer concentration. Sodium acrylate content = 12 wt% of total volume. | |||||
| 1 | PSAS0C0 | 0 | 0 | 12106 |
0.56 |
| 2 | PSAS50C0 | 50 | 0 | 10750 |
0.67 |
3. Characterization
Fourier Transform Infrared (FTIR) spectroscopy (Perkin Elmer, model spectrum-2) was used to characterize the OMMT and hydrogel samples (range 500–4000 cm−1). Fine dust samples were mixed with KBr in a 1:10 weight ratio and palletized using a hydraulic press. Proton nuclear magnetic resonance (1H NMR) spectra and diffusion-ordered NMR spectroscopy spectra (DOSY spectra) were recorded on a Bruker Avance III 400 MHz spectrometer equipped with a 5 mm SMART Probe at room temperature (25 ± 1 °C) using deuterated solvents (D2O). In the case of DOSY analysis, the magnetic gradient was increased gradually from 2% to 95% in a sequence of 64 gradient steps. Bipolar rectangular gradients were maintained for a duration of 2 ms, and the gradient recovery delay was 200 ms. The maximum gradient strength, which used was 0.535 T m−1, and the diffusion time was recorded between 0.5 s and 1.0 s. The obtained spectrum was analyzed using Topspin 2.1.6 software (Bruker). The crystalline behavior of the sample was investigated by using X-ray diffraction (XRD) (PANalytical, Netherlands). The data were acquired using a copper X-ray source (1.549 Å) with a standard angle of 0–80 degrees (2θ) and a fixed scan rate of 10° per minute. The morphology of the self-assembled polymer and dispersion of the OMMT filler inside the hydrogel were monitored using a TEM (JEOL, JEM-2000E7) instrument operated at an accelerating voltage of 200 kV. For the solid gel samples before microscopy the samples were cryo-microtomed at a thickness of 50 nm and placed over copper grids having 300 mesh sizes, and for the liquid sample the dilute aqueous solution of the self-assembled polymer was drop cast over the copper grids. For porosity determination of the formed hydrogel Microcomputed Tomography (GE Phoenix Vtomex-s) was used and the images were processed using VG Studio Max 2.2 software.
3.1. Swelling study of the synthesized hydrogel
The swelling ratio of the synthesized hydrogel was determined gravimetrically. To study the swelling behaviour of the respective gels, a preweighed cylindrical shaped hydrogel sample was dipped into the container having deionized water, buffer solution of pH = 1.2 and 7.4 and different types of salt solutions (0.1 mol dm−3) having different valencies [di-valent (Ca2+, CaCl2) and tri valent (Fe3+, FeCl3)] respectively. After that, at a predetermined time interval the swollen hydrogel was taken out and gently pressed with tissue paper to remove surface water before being weighed. The swelling ratio was calculated using the following formula:where Wswell and Wdry are the swollen weight and the dry weight of the hydrogel sample respectively. The equilibrium swelling ratio (Qe) of the polymer was calculated by taking the weight of the swollen hydrogel under equilibrium conditions (We) and using the same equation as mentioned above. Different buffer solutions were prepared by using our previously reported method.6
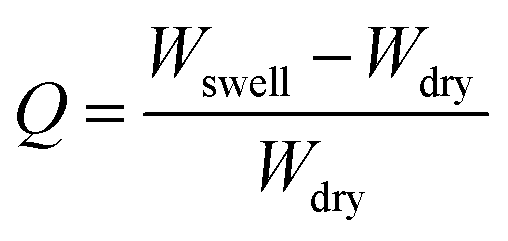
3.2. Determination of the gel structural parameters
The crosslink density (ρc) was determined by using the Flory–Rehner theory. It is reported that the average molar mass of the polymer chains between two crosslinks (Mc) is directly linked to the density of the gel. All the physical and mechanical properties of the hydrogel depend on Mc. According to the theory, Mc can be determined by using the following equation25,26where V1 is the molar volume of the solvent (ml mol−1), Vs is the volume fraction of the swollen hydrogel under equilibrium conditions, χ is the Flory–Huggins solvent–polymer interaction parameter and dp is the density of the hydrogel. Here χ was calculated using the following equation27
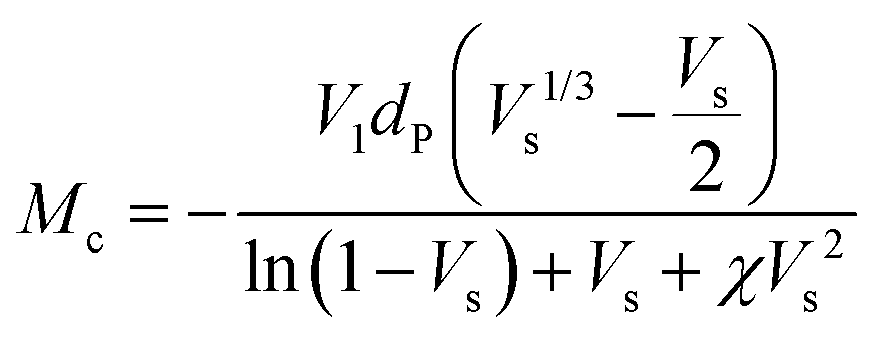
The volume fraction of the polymer (Vs) was calculated using the following equation28
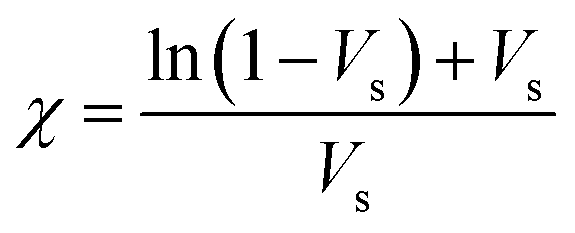
where Ma and Mb are the masses (g) of the swollen and dry hydrogel. dp and ds are the densities of the gel and solvent in g ml−1 respectively. After that the crosslink density (ρc) of the gel was calculated using the equation29
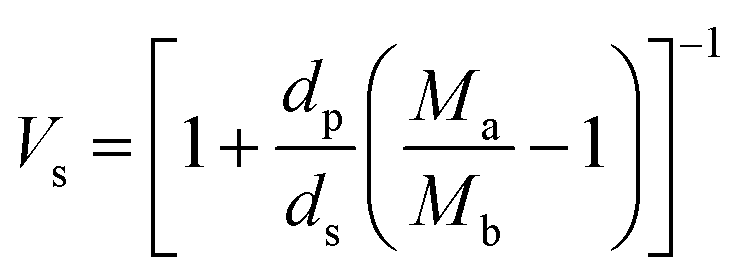
where NA is Avogadro's number having a value of 6.023 × 1023.
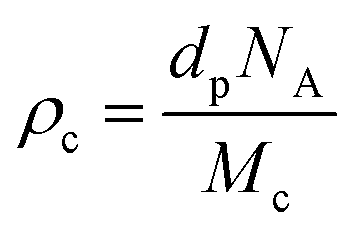
3.3. Tensile and optical rheology analysis
The self-healing study of the hydrogel was carried out using a universal tensile test machine (Hounsfield H10KS) and rheological analysis. Tensile tests of the uncut and self-healed hydrogels were carried out by maintaining a 10 mm min−1 crosshead speed at room temperature using a 500 N load cell. Cylindrical samples of 10 mm diameter and 13 mm length were used to study the tensile strength and elongation at break of the pure and self-healed hydrogels. The self-healing study of the single component (PSAS0C0, control) and the composite hydrogel (PSA50C5, experimental) was also carried out through optical microscopy analysis. For this a notch was created over the hydrogel sample using a sharp knife and after that buffer solution of pH 7.4 was applied over it and the samples were kept for one hour in this condition. After the retention time the hydrogels were dried and healing of the notch was monitored through an optical microscope. For the cyclic tensile study, the water swollen hydrogels were coated with silicon oil to prevent loss of water during the loading–unloading cycles and a strain% of 100% was applied for each cycle which was 7–8 times higher as compared to the maximum strain sustained by the muscle (12–15% strain%).30 We have applied this drastic condition to the hydrogel system to examine its robustness during the fatigue testing.The isothermal rheomicroscopy analysis of the prepared hydrogels in the swollen state was carried out using an Anton Paar (USA), parallel plate optical rheometer by varying the shear strain rate between 0.001 and 10% at a constant angular frequency of 1 Hz at 25 °C temperature. For the self-healing study, the cylindrical shaped hydrogel having a thickness of 0.5 cm was placed between the parallel plates of the rheometer and the shear strain was varied as in the previously mentioned conditions. The variation of the rheological and viscoelastic behavior of the synthesized hydrogel samples was examined. Due to the presence of the optical microscope, the change in the orientation of polymeric macrostructures and the alignment of the filler with a gradual change in the shear strain can be visualized.
3.4. Shape-memory study of the composite hydrogel
To study a preliminary shape memory effect of the hydrogel, the hydrogels (PSAS0C0 and PSAS50C5) were prepared in the form of a strip having a dimension of 40 mm × 15 mm × 3 mm. Then the water swollen hydrogel was bent to 180°, and the programmed “U” type shape was fixed via water evaporation (drying). After that, the shape deformed hydrogel samples were dipped into deionized water, and the shape recovery of the hydrogel system was studied. The following equations were utilized to evaluate the shape fixity and shape recovery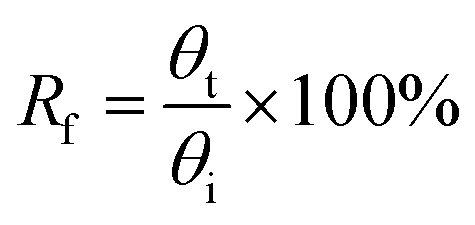
where Rf = shape fixity ratio, Rr = shape recovery ratio, θi = angle provided to the hydrogel, θt = temporarily fixed angle and θf = final angle after recovery.
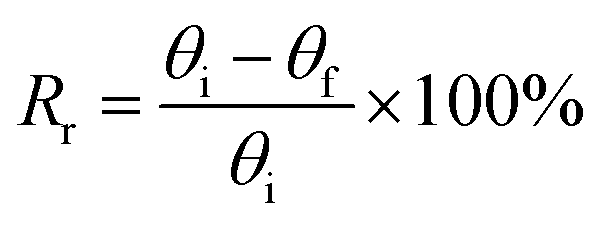
To measure the resistance against shape deformation, bending (180°) and twisting (3 fold) forces were applied over the composite hydrogel sample (PSAS50C5) and the recovery from the deformed state was monitored.
3.5. Study of hydrogel actuation in the presence of external stimuli
To study the acid induced actuation in the polyelectrolyte hydrogel, the hydrogel strip having a dimension of 40 mm × 10 mm × 1 mm was sequentially dipped in the buffer solution having pH 7.4 and pH 1.2 respectively in programmed time intervals. The change in the shape of the hydrogel was observed.A similar experiment was carried out in a salt induced actuation study. A NaCl solution having 2 M concentration was used for the analysis. As with the acid induced actuation, the hydrogel strip was sequentially dipped into the buffer solution having pH 7.4 and salt water solution in programmed time intervals.
For the electric field stimulated actuation property study, a custom built voltameter was used where flat surfaced carbon electrodes having a dimension of 30 mm × 15 mm × 1.5 mm were used. The electrodes were placed 50 mm apart from each other and deionized water having 0.1 M NaCl solution was used as an electrolyte. After that the hydrogel strip having a dimension of 40 mm × 10 mm × 1 mm was immersed into the electrolyte by maintaining a nearly equal distance from two electrodes. It has to be mentioned that before dipping in the electrolyte, the gels were preactivated with the salt solution having the same molarity NaCl solution. A predetermined bias of 18 V was applied to the electrodes for five minutes and as a result, twisting in the hydrogel strip was observed and the respective images with time were captured.
A similar method was applied in the case of the control hydrogel sample (PSAS0C0, anionic hydrogel) to monitor the disparity of it with the composite hydrogel system (PSAS50C5).
3.6. Preliminary in vitro cell cytotoxicity assay
In vitro cell cytotoxicity assay was performed with a fibroblast cell line by using the MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) colorimetric technique. Here the MTT assay was used to study the change in the metabolic activity of the fibroblast cell in the presence of a composite hydrogel, which can be identified by measuring the O.D. of the formed purple colored formazan salt at 590 nm. The salt was produced due to the reduction of the MTT salt by the oxidoreductase enzymes under definite cell conditions. Fibroblast cells at a density of 1 × 104 cells per well in 180 μl DMEM medium containing 10% FBS and 1% antibiotic were seeded in a 96-well plate. Then hydrogel suspension in PBS buffer was added to the medium, and it was cultured for 48 h in a humidified atmosphere in the presence of 5% CO2. Cells without the treatment of the hydrogel suspension in growing medium were taken as a positive control. After that, the culture was washed with PBS solution and MTT solution having a concentration of 1 mg ml−1 was added before incubation for 4 h at 37 °C. Then the supernatant was removed, and the insoluble formazan salt was dissolved in DMSO and the O.D. of the purple solution was measured at a wavelength of 590 nm. This method provides the number of the viable cells present in the medium.31–33 The relative cell viability was calculated using the following equation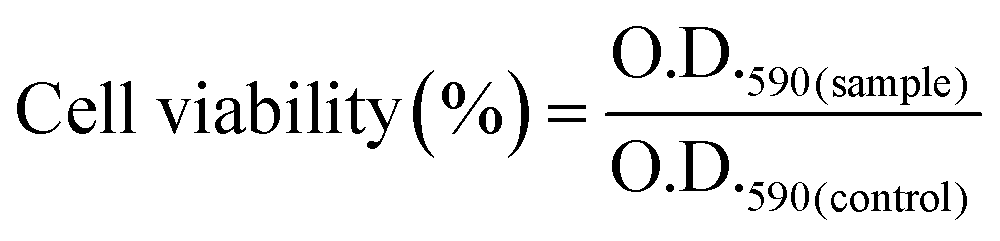
4. Results and discussion
Scheme 1 summarizes the preparation of a self-healable composite hydrogel and its healing study at different pH. In this case, initially, starch was cationically modified via graft copolymerization of MTAC over the starch backbone (grafting percentage = 80%). After that, the composite polyelectrolyte hydrogel was prepared via in situ copolymerization of sodium acrylate in the presence of cationic modified starch and OMMT using APS as a thermal initiator and MBA as a crosslinker.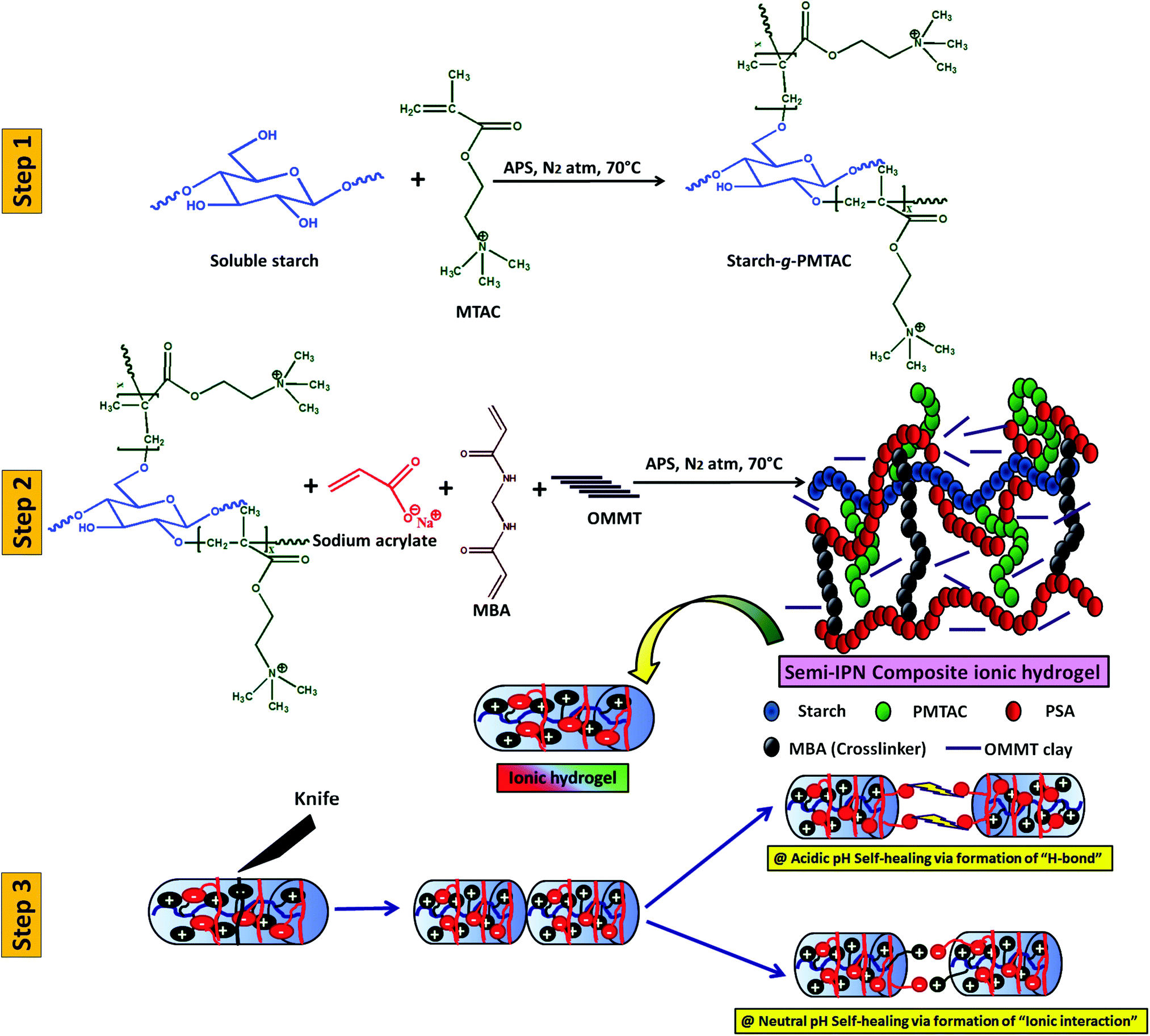 | ||
| Scheme 1 Schematic diagram of the preparation of a three-component self-healable hydrogel and its behaviour in media at different pH. | ||
The formation of starch-g-PMTAC was confirmed by 1H NMR and DOSY NMR analyses. The designation of different protons in starch is shown in Fig. 1a. In the case of starch, all typical glucose ring proton signals (b, c, d, e, and f) appeared at 3.5–3.9 ppm. In the case of a, the proton signal appeared at 5.33 ppm.34 After modification with PMTAC, new characteristic signals appeared at 3.24 ppm for q, 1.04 ppm for n, 4.51 ppm for o, 3.67 ppm for p and at 3.17 ppm for m which are due to the different protons of the PMTAC segment as shown in Fig. 1b.12,35 This confirmed the grafting of PMTAC onto the starch backbone. Successful grafting of PMTAC onto starch was confirmed by utilizing Diffusion Ordered Spectroscopy (DOSY) analysis, a technique that produces 2D correlation maps, representing the chemical shifts on the horizontal axis and diffusion coefficients on the vertical axis. By DOSY analysis (Fig. 1c and d), it was observed that the diffusion value of starch was higher than the graft polymer and the resonances of the protons of the graft copolymer maintained a horizontal line across the diffusion axis, which indicates that they are part of the same graft copolymer macromolecule.36,37 Furthermore, by considering the shift in solvent diffusion as well as viscosity of the sample, accurate hydrodynamic radii were determined.38 In this case, the unmodified soluble starch was found to have a peak hydrodynamic radius (RH) of 10.6 nm increasing to 39 nm following PMTAC modification (eqn (S1), ESI†).
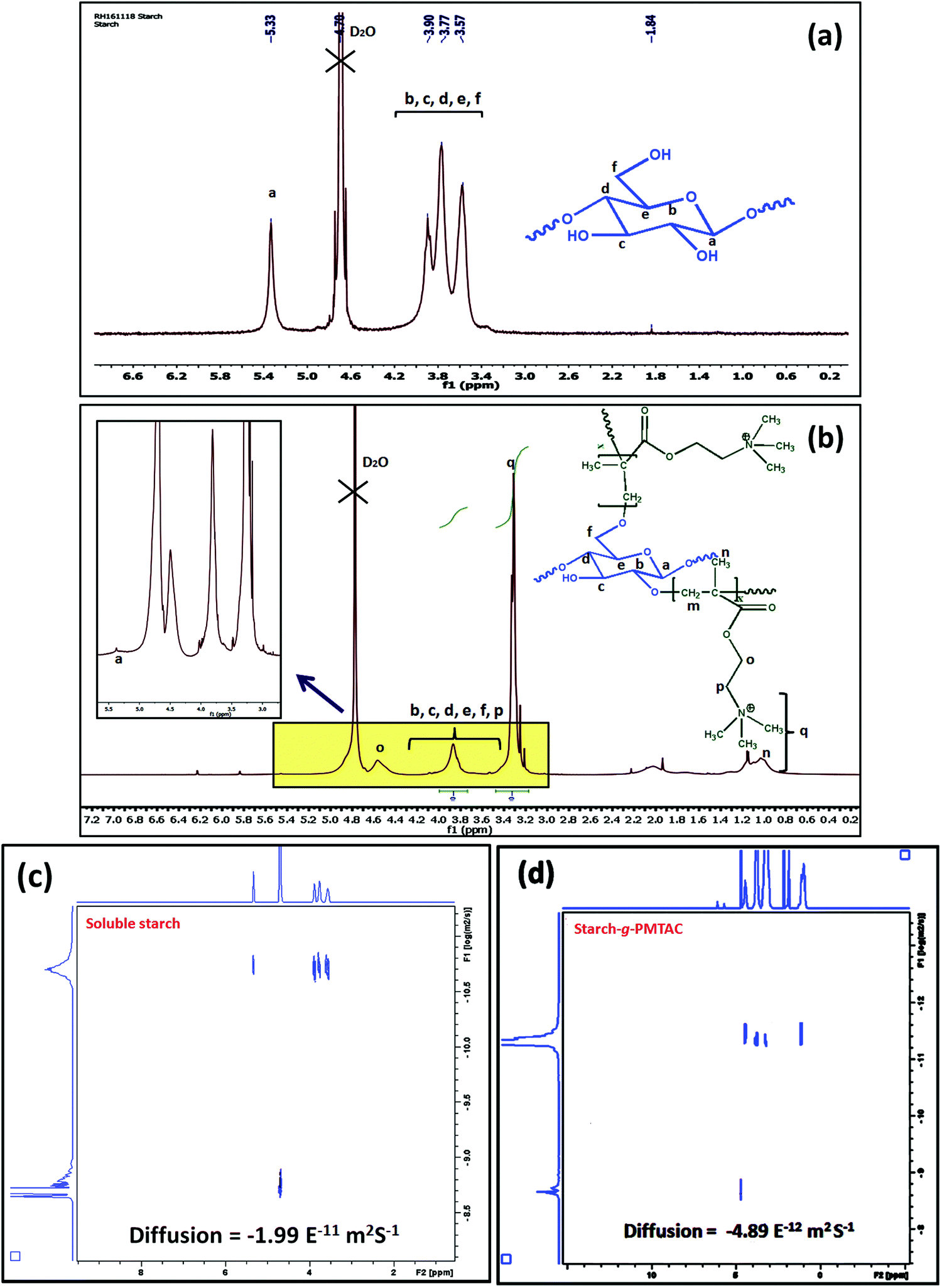 | ||
| Fig. 1 1H NMR analysis of (a) soluble starch and (b) starch-g-PMTAC. DOSY NMR analysis of (c) soluble starch and (d) starch-g-PMTAC. | ||
From FTIR analysis (Fig. S1(a), ESI†) of the hydrogel (PSAS50C0), absorption bands at 1491 cm−1 and 942 cm−1 were observed due to the bending and stretching vibration of the quaternary ammonium group respectively along with the characteristic peaks of starch (3416 cm−1, –OH stretching). The presence of the vibration at 1560 cm−1 delineates the presence of a –COO− ion of poly(sodium acrylate). Details of the obtained results from FTIR have been explained in the ESI.†
The surface modification of MMT clay and the change in crystallinity in starch upon modification with the cationic polymer were studied by XRD analysis. In the case of starch-g-PMTAC, a broad peak was observed at 24° (Fig. S1(b), ESI†) as compared to the strong crystalline peaks of starch at 2θ of 17° and 23° that defines the destruction of the intermolecular H-bonding present among the –OH groups in starch after grafting. A similar kind of observation has also been reported by Fengdan et al.39,40 A shift of the Bragg angle from 6.10° to 5.49° was observed (Fig. S1(c), ESI†) after the modification of the MMT clay with CTAB. This indicates the increase in spacing between the clay layers.41
The TEM image showed the polymeric ionic segments interacting with each other to give an ionically interlocked structure (Fig. 2a). This observation is comparable to the report of Kim et al.42 and Stuparu et al.43 The dark and thin lines observed in Fig. 2b and c indicate the presence of OMMT clay in the bulk of the hydrogel.44,45 It was observed that the extent of intercalation intensified with increasing content of OMMT. The presence of the clay inside the hydrogel was further confirmed by the EDAX analysis of the composite hydrogel, indicating the presence of silica (Fig. 2d). It was observed that the distribution of the OMMT clay inside the hydrogel is much similar to the construction of the muscle as shown in Fig. 2e. Here, it can be assumed that the presence of OMMT clay simulates the activity of the muscle fibre (composed of elastin and actin fibre) that basically imparts the elasticity to the muscle system46,47 as shown latter.
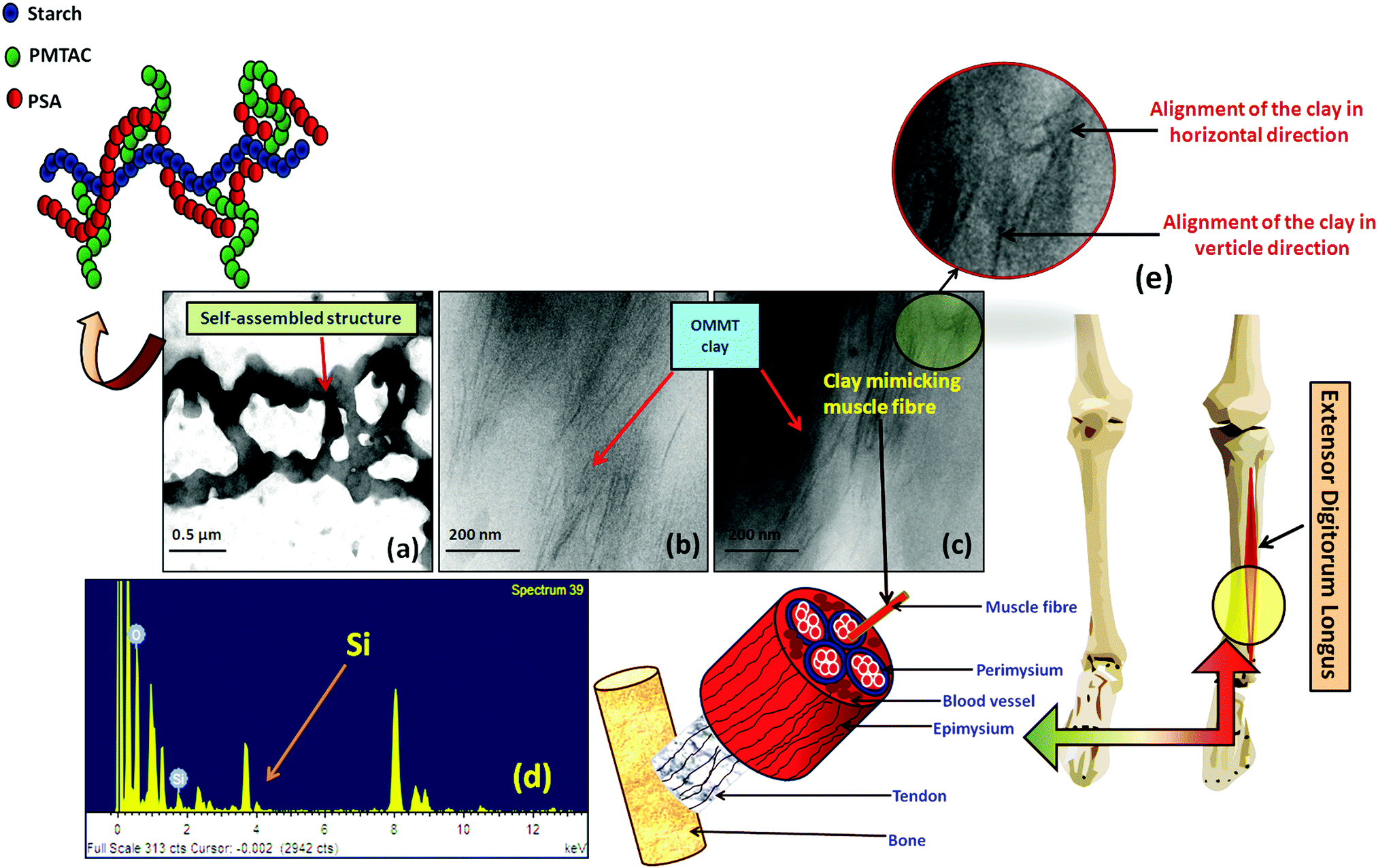 | ||
| Fig. 2 HRTEM images of (a) the self-assembled structure of cationic starch and anionic pol(sodium acrylate); (b and c) composite hydrogel with 5.0 wt% (PSAS50C5) and 7.0 wt% (PSAS50C7) of OMMT; (d) EDAX analysis of the composite hydrogel containing 5 wt% of filler (PSAS50C5) and (e) schematic of the location of EDL muscle present in the human body. | ||
4.1. Swelling behavior and gel parameter analysis of the hydrogel
Swelling is an important phenomenon for the muscle system. Myofibril, which is one of the prime components of the muscle, contains 20% protein and rest of the part is occupied by water. Like in the muscle, the swelling behavior of the hydrogel also depends on the bulk structure of the hydrogel system. The volume fraction of the swollen gel (Vs) (determined by the ratio of the volume of the polymer to the swollen gel) describes the amount of water that can be assimilated into the hydrogel.48Vs is dictated by the molar mass between the two consecutive crosslinking points (Mc) and the Flory–Huggins solvent–polymer interaction parameter (χ). These govern the swelling of the hydrogel due to the generation of the osmotic pressure and capillary action. It was found that, χ in the range of 0.5 in our system signifies a lower polymer solvent interaction and higher polymer to polymer interaction.49 The obtained values of these network parameters are summarized in Table 1. It was observed that an increase in cationic starch content results in an increase in the crosslink density (ρc). This affects the swelling of the hydrogel as the free volume was decreased. This indicates that a lower inter-crosslinked space is available for water to diffuse into the hydrogel structure.Fig. 3a represents the swelling behavior of the composite hydrogel (containing 5 wt% OMMT filler, (PSAS50C5)) with variation in the pH and salinity of the solution. At very low pH (pH 1.2), the minimal swelling was observed. This is due to the protonation of the pendant carboxylic group of poly(sodium acrylate), and this accelerates the formation of the intermolecular polymer–polymer H-bonding. The formation of H-bonding enhances the crosslink density, and consecutively reduces the degree of swelling. According to Hamm the charge over the thin and the thick filament of myofibril is negative at pH > 5 which imparts a repulsive force between the filaments resulting in an enlargement of the lattice due to swelling.50 Whereas an opposite phenomenon (swelling) occurred at lower pH. A complementary effect of electrostatic repulsion between the fibrils and the contraction by the presence of the cross-bridges governs the swelling of the myofibrils.51 Similar like muscle, at pH 7.4 (physiological pH), we observed an increase in the swelling ratio of the hydrogel due to the deprotonation of the carboxylic groups and the ratio of –COO− to –COOH increased.52 However, this increase in the swelling was abruptly decreased at very high pH (pH 10). This is probably due to the screening effect of the Na+ ion over the –COO− group and this nullified the effect of repulsion between the carboxylate moieties. So the Donnan osmotic pressure decreases resulting in a poor swelling at higher concentration of Na+. On the other hand, at very high pH, –N(CH3)3+ converts to –N(CH3)3OH which also restricts the swelling due to the reduction in the repulsion between the positively charged quaternary ammonium ion moieties.53
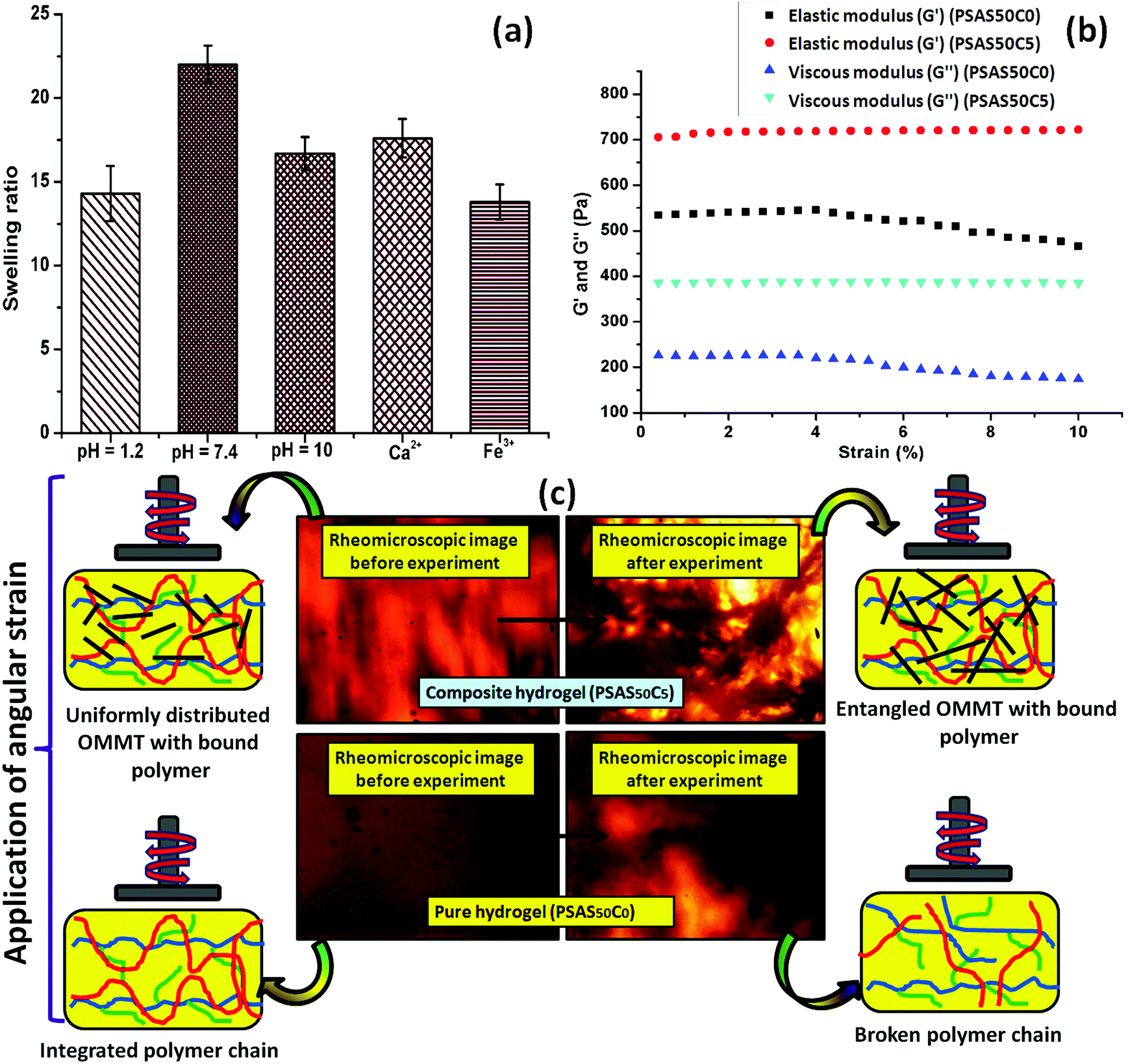 | ||
| Fig. 3 (a) Swelling study of the prepared hydrogel with variation in pH and salt solution of different valence; (b) the rheomicroscopic analysis of the hydrogel with the content of OMMT clay (left: initial, right: final) and (c) respective optical rheomicroscopic images obtained during the experiment. | ||
Along with the pH the presence of the salt solution also imparts a significant finger print over the swelling of the muscle fibres. Hofmeister first experienced the effects of different ions over the proteins and experienced that some ions exhibit a stronger salting-out effect (Hofmeister/lyotropic series).54 Ions like Na+, H+ (monovalent cations), Ca2+, and Mg2+ (divalent cations) acted as kosmotropes (structure-making salts) whereas K+ large monovalent ions like C(NH3)4+ were classified as chaotropes (structure breaking salts). To evaluate the effect of salinity over the swelling behavior of the hydrogel, the composite hydrogel was dipped into CaCl2 and FeCl3 solutions of the same molarity (0.1 M) having a pH of 6.5 and 3 respectively. It was observed that the presence of the counter positive ions of different valencies significantly reduced the swelling of the hydrogel. This effect is mainly due to the increase in the osmotic pressure as the charge increased.55,56 As expected, in the presence of the trivalent metal salt, the extent of swelling was lower compared to that in the presence of divalent metal salt. Like in our system, in muscle the divalent ion like Ca2+ used to bind with the F-actin of myofibril which reduces the swelling.57
4.2. Analysis of the mechanical properties
Fig. 3b shows the rheological properties of the composite hydrogel in the incorporation of OMMT clay. The rheological analysis was carried out by varying the shear strain from 0.001% to 10% at a constant frequency (1 Hz) in compression mode where G′ and G′′ depict the elastic and viscous components. It was observed that the elastic modulus of the composite hydrogels containing 5 wt% OMMT clay (PSAS50C5) was drastically improved compared to the non-filler containing hydrogel (PSAS50C0) (Fig. 3b). This is due to the formation of the network structure by the clay particles in the bulk of the hydrogel as evidenced by the rheo-microscopic image (Fig. 3c).58The change in the distribution of the OMMT clay upon the application of mechanical force was visually observed through the rheomicroscopic analysis as summarized in Fig. 3c. It was observed that upon application of the shearing force, the integrity of the hydrogel sample having no filler gradually reduced and as a result a drop in both the elastic and viscous modulus was observed. Whereas in the case of the composite hydrogel sample, OMMT clay acts as a shock absorber which is also entangled with the polymer during the shear force application as observed from the rheomicroscope image.59,60 This phenomenon protects the polymer chain from breaking under shear stress.
The composite hydrogel has much higher complex viscosity than the single component hydrogel (Fig. 4a). In the case of the poly(sodium acrylate) containing hydrogel, where no positively charged ionic components and filler were present, a drastic drop of the viscous modulus was observed at a strain of 1.5%. The drastic drop in the complex viscosity value can be attributed to the breakage of covalent links at this high percentage of shear strain. The reason behind the loss in the modulus is schematically represented in Fig. 4b. A crossover point of the complex viscosity value was observed in the case of the high filler loaded (7 wt%, PSAS50C7) composite hydrogel system as compared to the complex viscosity trend followed by the hydrogel having a lower loading of filler (PSAS50C5 and PSAS50C2). It can be explained by considering the distribution of the clay in bulk of the hydrogel. In the case of the higher loading of clay, agglomeration of the filler in the bulk of the hydrogel was witnessed from the HRTEM (Fig. 2c) image, which reduces the polymer–filler interactions as schematically represented in Fig. 4c. This agglomeration of the OMMT clay may allow the free movement of the polymer chains. The relaxation of the polymer chain is due to the detachment from the clay cage with an increase in the strain% resulting in a decrease in the resistance against the flow (viscosity). As a result, a significant drop in the complex viscosity value is observed. The fate of the mutual interaction of the OMMT clay and polymer chain for the low (PSAS50C2) and medium (PSAS50C5) filler loading with an increase in the shear strain% is schematically represented in Fig. 4d. In both cases it was observed that during the lower loading of the filler, interaction of the filler to polymer is high which helps in the retention of the viscous modulus at high shear strain%.
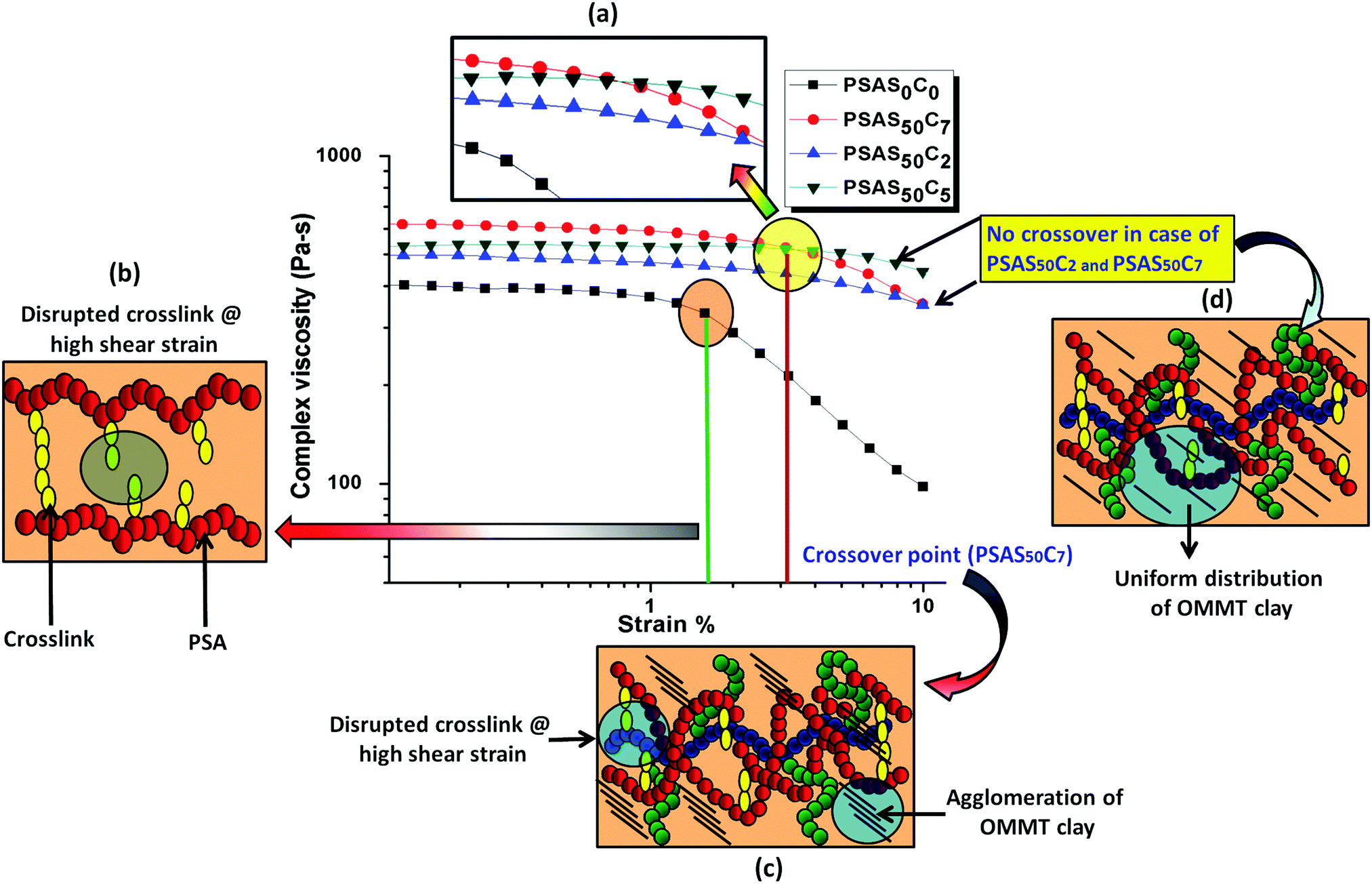 | ||
| Fig. 4 (a) Variation of complex viscosity of the synthesized hydrogel with a change in strain%; schematic representation of the probable behavior of (b) a single component hydrogel (PSAS50C0) under high shear strain% at a crossover point; (c) composite hydrogel with high OMMT loading (PSAS50C7) under high shear strain% at a crossover point and (d) composite hydrogel with low OMMT (PSAS50C2) and moderate OMMT (PSAS50C5) under high shear strain%. | ||
The tensile data (Fig. 5a), important properties for the muscle, showed that the hydrogel with composition of PSAS50C7 had an elongation to break of about (285 ± 10)% which was lower than that of the material containing less OMMT (PSAS50C5) having an elongation to break of (500 ± 10)%. The data showed that upon increasing the filler content the stiffness of the hydrogel was increased. This phenomenon can be explained by considering the extent of dispersion of the filler into the bulk of the hydrogel sample. Fig. 5b shows that as the filler content increased from 0 wt% to 5 wt% to 7 wt%, the transparency of the hydrogel decreased. These data signify that a higher level of filler loading generates aggregation points inside the hydrogel due to the poor distribution of the filler resulting in a weak point and subsequent failure with an increase in strain%.
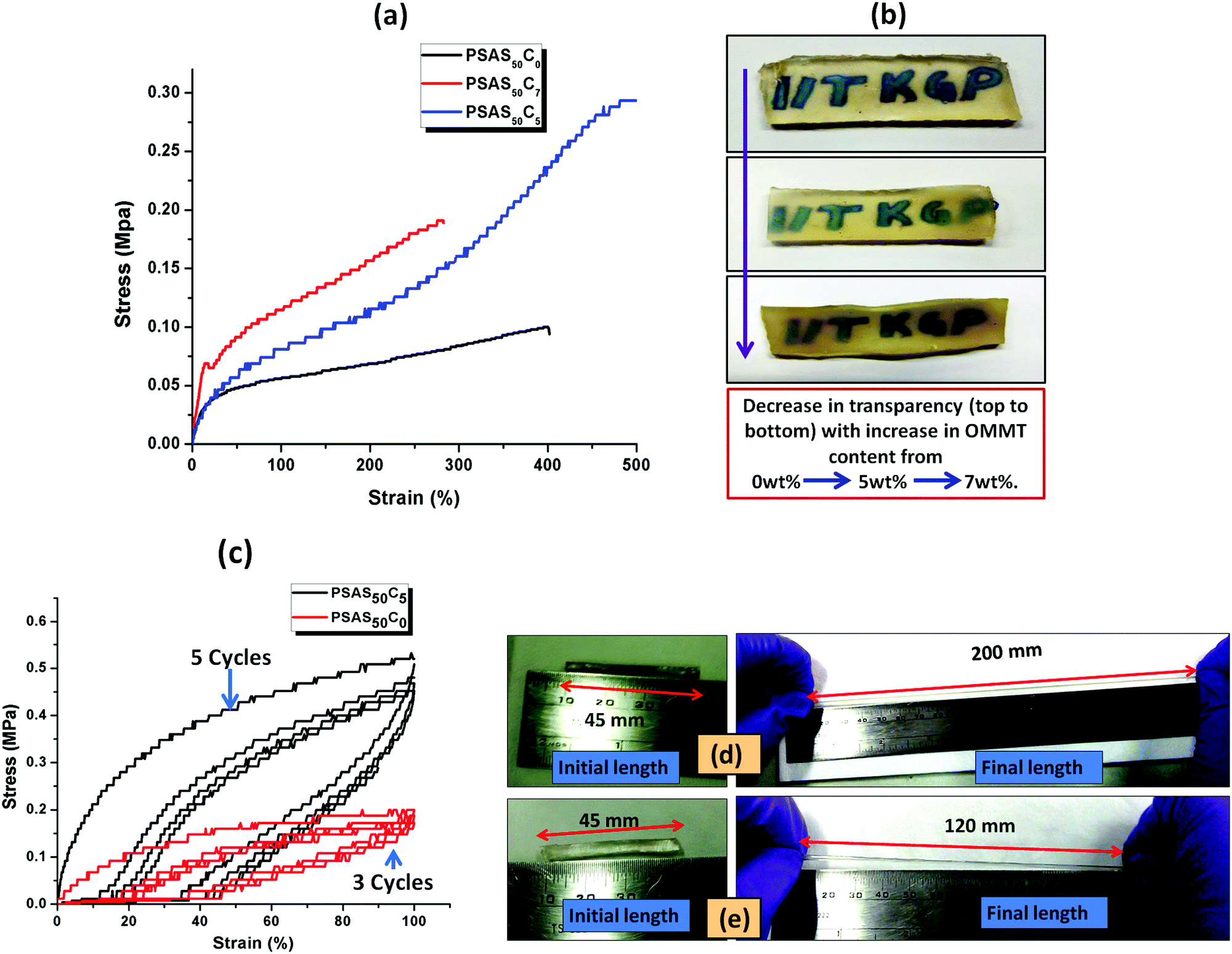 | ||
| Fig. 5 (a) Tensile study with the variation in OMMT content; (b) change in the optical transparency of the synthesized hydrogel with the clay content; (c) cyclic tensile test measurement of a single component hydrogel (PSAS0C0) and a composite hydrogel (PSAS50C5); elongation study of (d) PSAS50C5 and (e) PSAS0C0. | ||
To examine the flex–fatigue property of the composite hydrogel, we have studied the cyclic loading–unloading tensile test (Fig. 5c). It was observed that hydrogels without filler maintained integrity up to 3 cycles before fracture whereas the composite gel (PSAS50C5) did not fracture until 5 cycles under a high shear strain% of 100%, much higher than the strain% tolerated by the muscle system (12%).30 This indicates that it may be used for continuous flexing purposes. It was observed that the composite hydrogel (PSAS50C5) (Fig. 5d) got elongated to a greater extent before fracture compared to the pristine hydrogel (PSAS50C0) (Fig. 5e). Therefore, comparing the rheological, tensile and cyclic loading–unloading tensile studies, it can be concluded that the composite hydrogel having 5 wt% OMMT (PSAS50C5) provides a tougher material as compared to other compositions.
4.3. Self-healing study of the hydrogel
The self-healing ability of the composite hydrogel in environments with different pHs was studied after joining the two cylindrical shaped hydrogel samples having a dimension of 20 mm in length and 10 mm in diameter. Before the self-healing study, two parts of the cylindrical composite gel samples (PSAS50C5) having dimensions of 10 mm in length and 10 mm in diameter were swollen at pH 1.2 and pH 7.4 respectively. After taking them out, the terminal ends of the two hydrogel samples were joined together and pressed in this condition using a weight of 10 g and left for drying at 37 °C (body temperature) (Scheme 1). After complete drying, the hydrogel samples were again swollen, and the tensile test was performed for both the PSAS50C0 and the composite hydrogels, PSAS50C5, at a cross-head speed of 10 mm min−1 using a load cell of 500 N. For visual identification, one part of the hydrogel was colored with rhodamine-B dye (the red part of the cylindrical hydrogel).Fig. 6a shows the image of elongation behavior of the self-healed hydrogel (PSAS50C5) at pH 7.4 over time. The hydrogel at pH 7.4 had an elongation at break of 350% ± 10% whereas the hydrogel at pH 1.2 had an elongation at break of 430% ± 15%. These values are slightly less than that of the hydrogel without a cut, 500% ± 10%. The stress vs. strain plot of the uncut and self-healed hydrogel having a composition of PSAS50C5 is shown in Fig. 6b. The main reason behind the comparative elongation of the self-healed hydrogel [both in acidic (pH of 1.2) and neutral media (pH 7.4)] with the uncut gel is due to the presence of an elastic ionic bond at pH 7.4 that formed between an anionic carboxylate ion (–COO−) and a cationic tertiary ammonium ion (–N(CH3)3+) and due to the formation of hydrogen bonds in an acidic environment (pH 1.2) (shown in Fig. 6c) respectively.
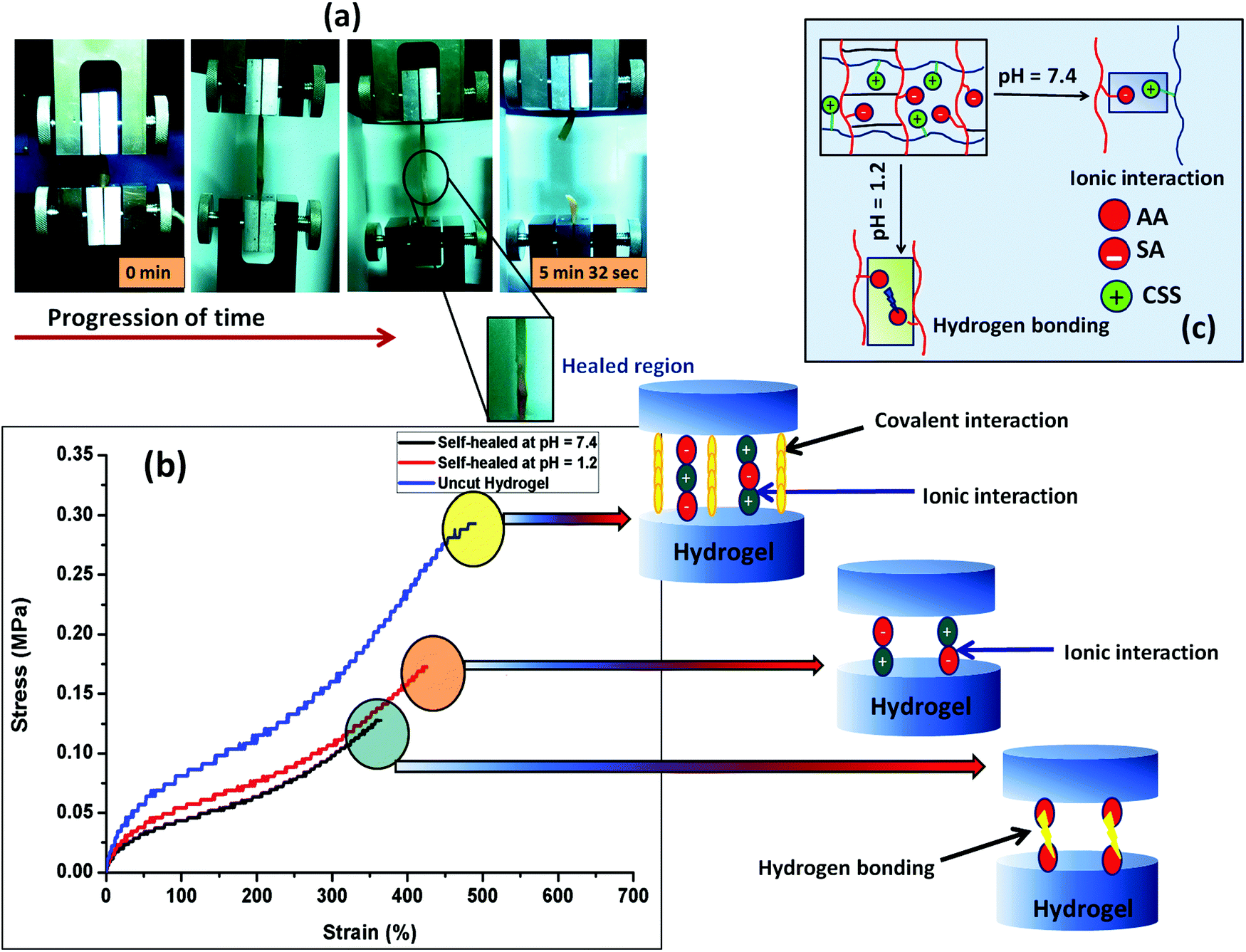 | ||
| Fig. 6 (a) Image of the tensile study of the self-healed hydrogel (at pH 7.4) with time, (b) schematic of the mechanism involved in the self-healing in buffer solution of different pHs, (c) stress–strain curve of the pure hydrogel and the self-healed hydrogel at different pHs. | ||
The fracture stress to break was determined as the self-healing time at pH 7.4 was varied (Fig. S2a, ESI†). It was observed that healing increased with an increase in the healing time but stabilized after 6 h of healing. The self-healing behavior was also studied with the content of the cationic component (Fig. S2b, ESI†). The data showed that as the cationic content increased from 10% to 50%, the extent of healing was increased. We believe that the formation of the highly associated ionic interaction between the positively charged starch molecules and the negatively charged poly(sodium acrylate) counterpart generates an intense ion association energy resulting in a significant increase in the healing effect by the hydrogel. Recently, Liang et al. reported a similar observation, where they have explained a comparative interaction between the polydimethyldiallylammonium chloride and poly(acrylic acid) in a poly(acrylamide) based hydrogel.61 Morrow et al. studied the elongation behavior of the extensor digitorum longus (EDL) muscle (a type of skeletal muscle) of a New Zealand white rabbit and found that it showed an ultimate tensile stress of 163 ± 75.7 kPa in the longitudinal direction.62 In the case of our study, both the uncut hydrogel and self-healed hydrogel show a similar tensile behavior.
The self-healing property of the hydrogel sample was again cross-checked using the rheological analysis. Rheological analysis provides an isothermal mechanical response obtained from the cylindrical shaped hydrogel when placed between the two parallel plates of the rheometer. From Fig. S2c (ESI†), it was observed that the elastic (G′) and the viscous (G′′) modulus values decreased after the self-healing. Both the elastic modulus and viscous modulus decreased but the deviation from the original value (as obtained in the case of the uncut hydrogel) was not so high indicating a very effective healing efficiency in the hydrogel at physiological pH.
To compare the self-healing ability of the single component hydrogel (anionic hydrogel, PSAS0C0) and the composite hydrogel (containing both cationic and anionic segments, PSAS50C5) a “scratch and heal” test was performed over both the hydrogel samples. From Fig. S3 (ESI†), it was observed that the single component hydrogel was unable to heal in the presence of buffer solution due to the repulsion existing between the anionic –COO− groups (Fig. S3a and b, ESI†). Whereas the composite hydrogel containing both the ionic segments (cationic and anionic) efficiently healed the notch that was created over it via the ionic interaction formed between –COO− and –N(CH3)3+ ions (Fig. S3c and d, ESI†).
4.4. Study of the shape memory behavior of the hydrogel
For an effective muscle mimetic hydrogel, both the self-healing properties and the shape memory properties are important. To study the shape memory ability, the hydrogels having a composition of PSAS0C0 and PSAS50C5 were prepared in the strip form having a dimension of 40 mm × 15 mm × 3 mm. As described in the experimental section, a pre-programmed angle of 180° was given to the water-swollen hydrogel, and the “U” shape was locked via drying of the hydrogels. After that, the shape recovery of the deformed hydrogel samples was monitored by dipping them into water (Fig. 7). The data in Fig. S4a and b (ESI†) showed that the hydrogel could tolerate a large amount of torsional force and bending force respectively. In both cases, we have studied the time taken for the hydrogel to recover from its deformed state after releasing the deformation force. It was noticed that in both cases the composite hydrogel can re-achieve its initial shape within a few seconds (30 s approx.).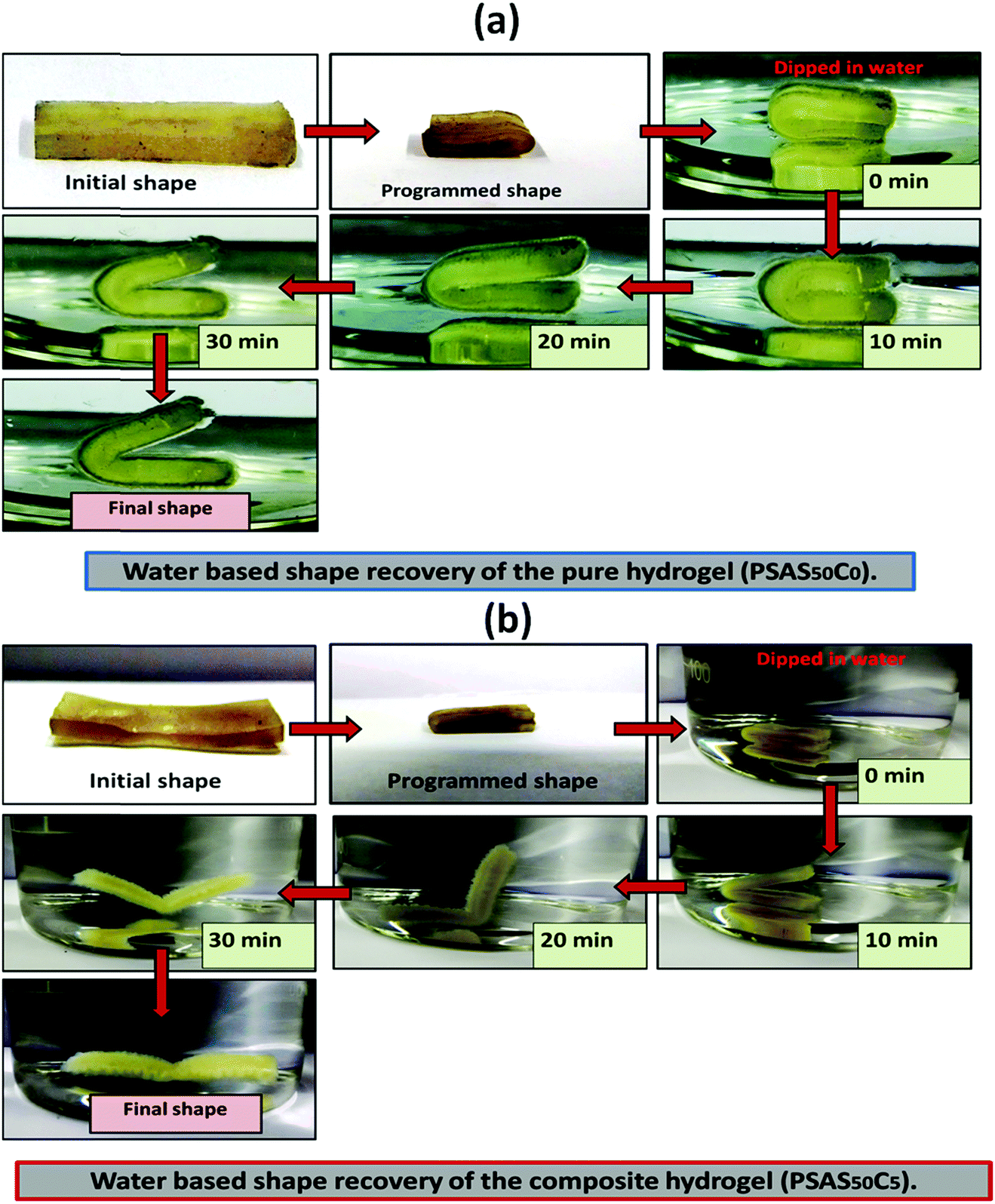 | ||
| Fig. 7 Water-based shape recovery study over the (a) single component hydrogel (PSAS50C0) and (b) composite hydrogel (PSAS50C5). | ||
After that, we have studied the water base shape recovery behavior of the pure hydrogel (Fig. 7a) and composite hydrogel (by varying the cationic starch content). The composite hydrogel consists of both cationic starch (50 wt%) and OMMT (5 wt%) (PSAS50C5) (Fig. 7b) showed 90% shape-recovery when it was dipped into water whereas the hydrogel with 5 wt% OMMT (PSAS0C5) (control sample) showed 75% self-recovery. On the other hand, the non-modified hydrogel (PSAS0C0) (control sample) showed 30% self-recovery. The electrostatic interactions between the chains are key to this shape-recovery behavior. In the pre-fractured material, there are many ionic interactions between chains. Some of these interactions between cationic and anionic segments are broken along the fracture surface, but they can be reformed as the two pieces of the hydrogel are brought back into contact. It was observed that when both the cationic starch and OMMT clay were present in the system (PSAS50C5) it endows an instant reformation of the shape which might be due to the higher positive charge density present inside the hydrogel system as compared to the presence of only OMMT (PSAS0C5). The importance of the segmental electrostatic interactions and the addition of the clay were shown by considering the non-modified hydrogel, which showed poor self-recovery. The results, therefore, show that tuning the segmental electrostatic interactions in these clay–hydrogel composites can provide shape recovery properties to the materials. The water based shape memory study was carried out thrice to get an idea about the repeatability of the shape memory property and it was observed that the same property has been achieved in every cycle. The obtained data have been enlisted in Table S1 (ESI†). The probable reasons behind the recovery of the hydrogel in the presence of water are schematically represented in Scheme 2.
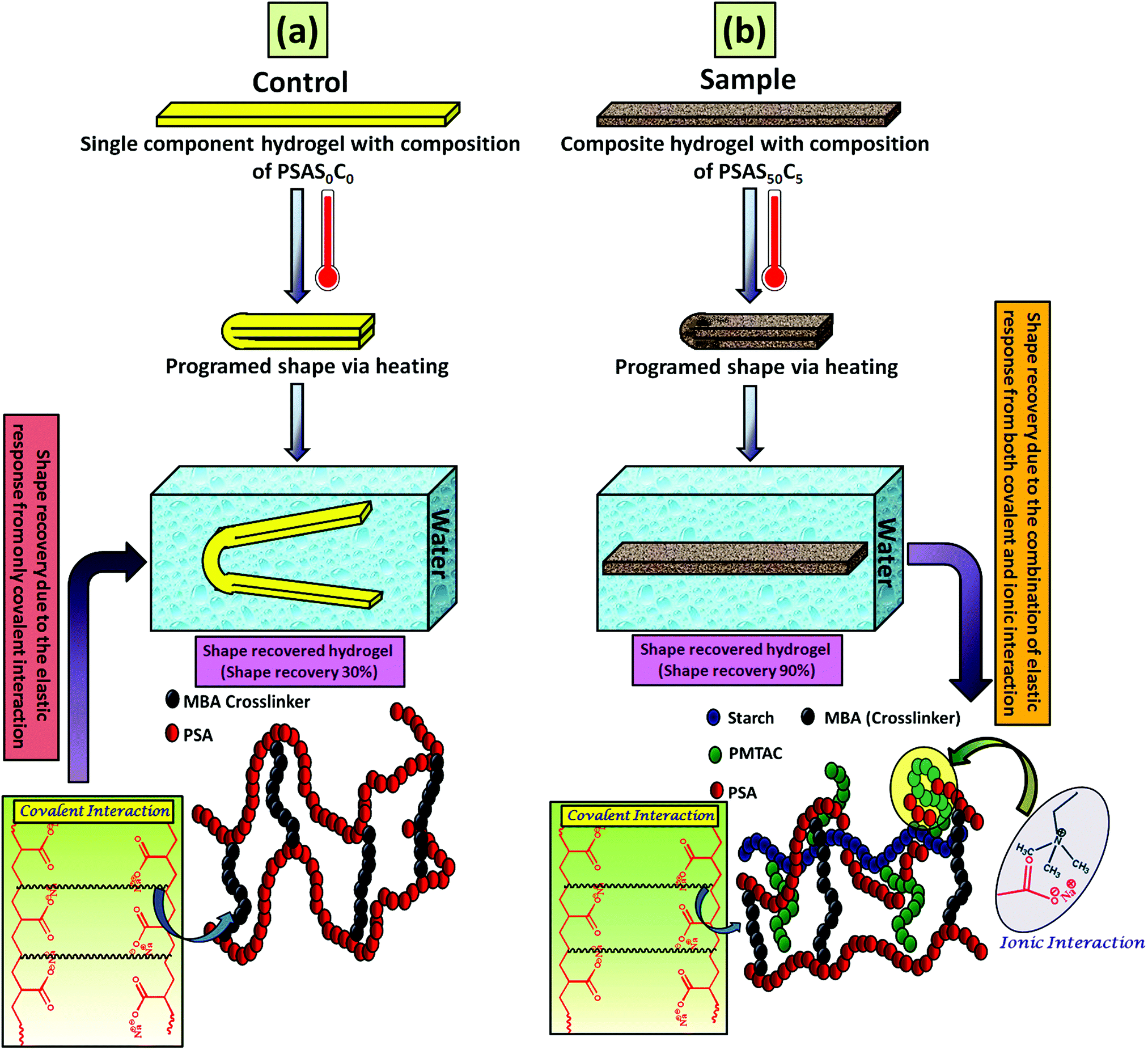 | ||
| Scheme 2 Schematic representation of the shape memory effect shown by the hydrogel having a composition of (a) PSAS0C0 (control) and (b) PSAS50C5 (sample) after subsequent treatment with heat followed by water and the respective mechanisms acting behind the shape memory effect. | ||
In the case of the control sample, the shape recovery of the deformed hydrogel sample is governed by the residual elastic force of the covalent bond that regenerates in the presence of water (Scheme 2a). Whereas, in the case of the composite hydrogel, due to the presence of covalent bonds as well as the ionic interaction, a synergistic effect accelerates the process of shape recovery and a 90% shape recovery was observed after deformation at the programmed shape and subsequent dipping into water (Scheme 2b). Along with the shape-recovery ratio (Rr), the shape fixity ratio (Rf) also plays a vital role in the shape memory property. It was observed that in every composition, more than 95% shape fixity was observed. The restriction of the polymer chain movement upon drying of the water is the main reason behind the significant shape-fixity of the hydrogels.
4.5. Actuation property of the hydrogel in the presence of salt, acid and electrical field
In recent days, several reports on the design of actuators by using the piezoelectric effect,63 magnetic64 and electrical field excitation,65 fluid flow kinetics66etc have been available. Among them, hydrogel-based stimuli-responsive actuators are of great importance due to their soft and elastic nature which allows their use in the integrated regions. Importantly their movement can be controlled by altering the stimuli. Here we have designed a polyelectrolyte based hydrogel system that consists of both soft acid moieties [poly(sodium acrylate), anionic segment] and cationic moieties [Starch-g-PMTAC].The actuation of the hydrogel system was studied in the presence of salt solution (2 M NaCl solution), acidic medium (pH 1.2) and electric impulse.
It is reported that the actuation property of the polyelectrolyte based hydrogel system depends on the (a) elasticity of the system, (b) polymer–liquid interaction parameters (Flory–Rehner Theory), (c) flow of the electrolyte through the pore of the hydrogel and finally (d) electrophoretic interactions.67
To examine the pore-density of the prepared hydrogel, we carried out the Micro CT analysis of the composite hydrogel system [PSAS50C5] consisting of cationically modified clay (Fig. 8a) and without clay [PSAS50C0] (Fig. 8b). From the figure, it was observed that a drastic drop in the number of voids and the diameter of the pore had occurred. The reduction in the void percentage clearly defines the presence of strong interaction between all the ionic components present in the hydrogel system. In the absence of the cationic clay, ionic interactions present between the poly(sodium acrylate) segment and the cationically modified starch segment determine the void density. The presence of cationic clay imparts additional ionic interaction with the anionic segments and the presence of a silicate layer reduces the size of pores. As the sizes of the pores were reduced in the composite hydrogel system, the holding time of the solvent was higher inside the hydrogel resulting in significant interaction with the polymer chains.68
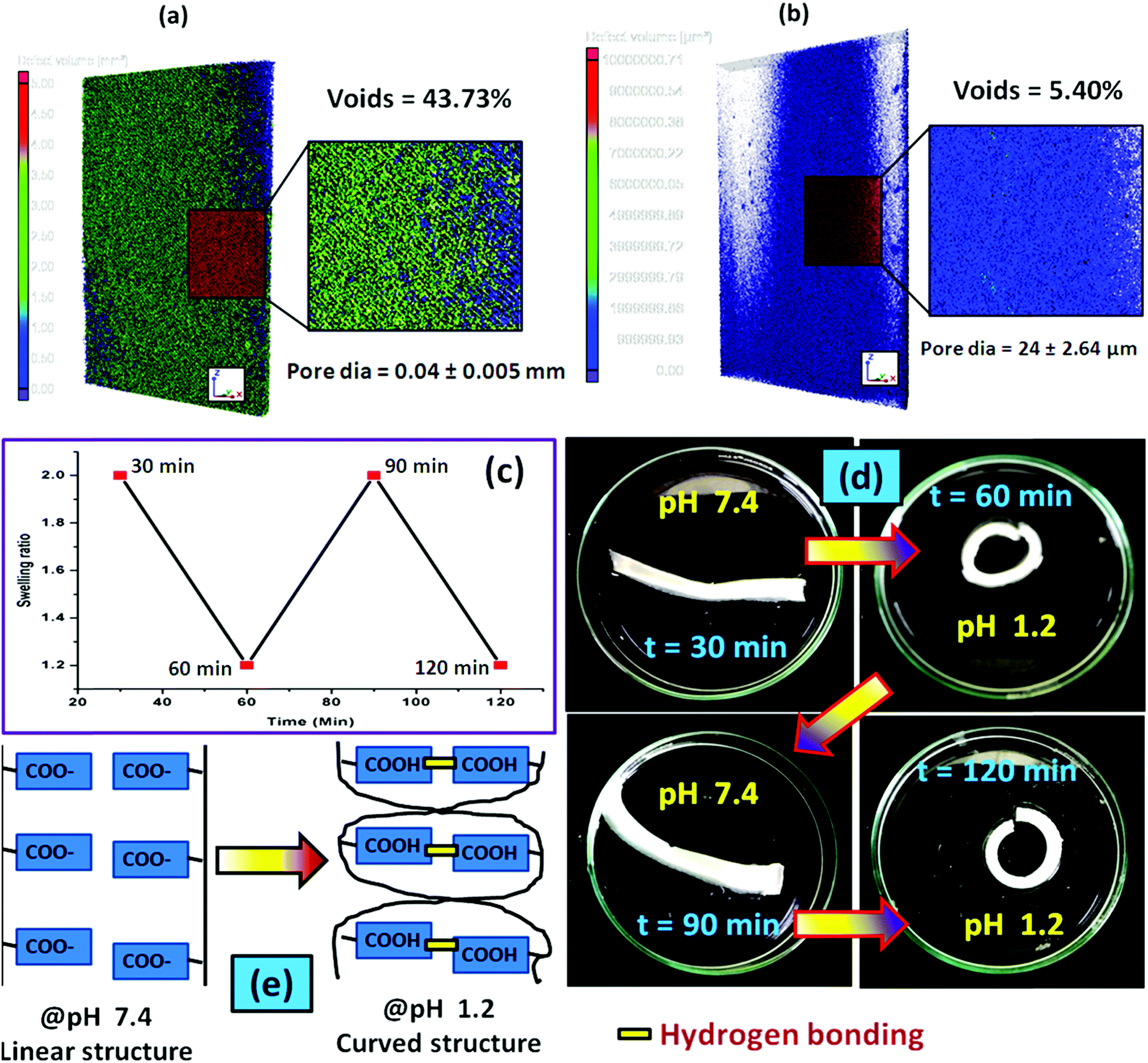 | ||
| Fig. 8 Micro CT analysis of the hydrogel having a composition of (a) PSAS50C0 and (b) PSAS50C5; (c) actuation cycle of the composite hydrogel (PSAS50C5) in the presence of acid (pH 1.2); (d) acid induced actuation in the composite hydrogel system (PSAS50C5) and (e) probable mechanism lying behind the actuation. | ||
During the acid based actuation study, when we switched the surrounding environment of the hydrogel from pH 7.4 to pH 1.2, a cyclic expansion and contraction was observed as shown in Fig. 8c. As a result, the hydrogel strip changed its shape from linear to coil as observed from Fig. 8d. We programmed each cycle for 30 minutes, and after a predetermined time interval, the surrounding environment of the hydrogel was changed. The cyclic change in the hydrogel shape from linear to coil can be explained by considering the protonation of the carboxylate moiety (–COO−) in the presence of an acidic medium which accelerates the formation of the hydrogen bonding between the carboxylic acid (–COOH) moieties. Formation of the hydrogen bonding generates a nanoscale force gradient inside the hydrogel system as shown in Fig. 8e resulting in the coiling of the hydrogel. After a certain interval when it was again placed in the buffer solution having a pH 7.4, –COOH was converted to –COO−, providing the drives to return to its original position (linear shape).69
Like under the acidic conditions, the presence of a high concentration of the Na+ ions also affects the shape of the hydrogel strip. The change in the shape of the polyelectrolyte hydrogel strip in the presence of 2 M NaCl aqueous solution (a typical concentration) has been shown in Fig. 9a. The nature of the cyclic swelling property of the hydrogel in the sequential immersion of the hydrogel at pH 7.4 buffer solution and the salt solution has been imaged in Fig. 9b. The probable reason behind the reversible coiling–uncoiling behaviour of the hydrogel has been shown in Fig. 9c. It is reported that in the presence of a high salt concentration muscles contract as a result of the Donnan osmotic pressure difference that balances the Na+/K+ concentration between two phases separated by a semi-permeable membrane. At a very high concentration of the salt in the body, Donnan osmotic pressure became lower resulting in the contraction of the muscle system.70 Similar to this process, when the Na+ concentration is very high, the existing –COO− ions are masked. As a result, the repulsion force between the carboxylate ions no longer exists resulting in the bending of the hydrogel like in the muscle system. Upon application of the buffer solution (pH 7.4) due to the dilution of the Na+ ions, the hydrogel strip again reverts to its original shape.71
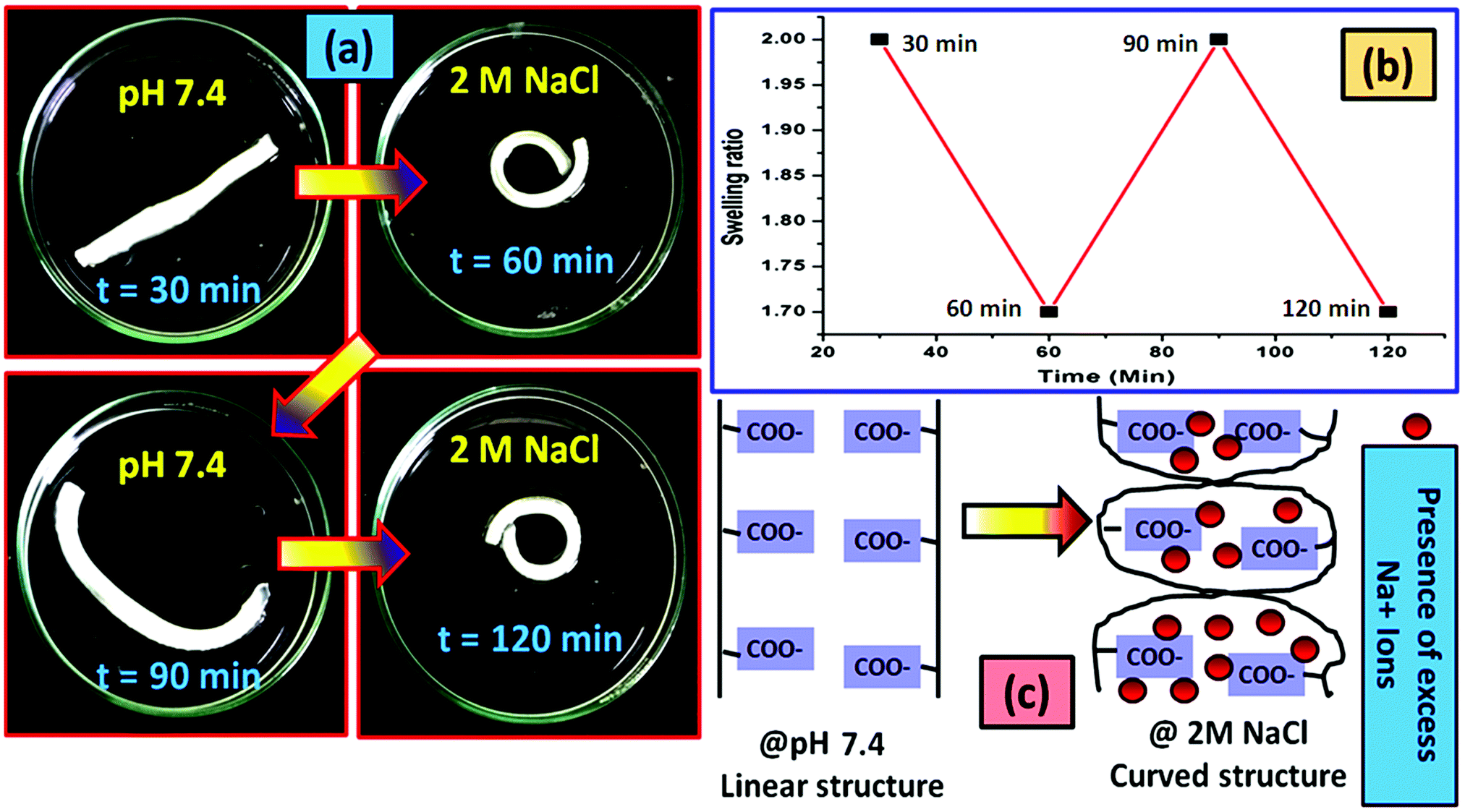 | ||
| Fig. 9 (a) Salt induced actuation in the composite hydrogel system (PSAS50C5), (b) actuation cycle of the composite hydrogel (PSAS50C5) in the presence of NaCl solution (2 M, a typical concentration) and (c) probable mechanism lying behind the actuation. | ||
For the control study, the pristine hydrogel underwent the same experimentation as in the case of the composite one. In this case we have changed the cycle time to 45 min so that the hydrogel can properly achieve its deformed state and get back to its actual shape after deformation. It was observed that in both salt (Fig. S5, ESI†) and acidic (Fig. S6, ESI†) solutions the hydrogel bend to a certain shape after providing the external stimuli (acid or salt) and re-achieve its original shape when it gets back to the neutral environment (pH 7.4).
The actuation property of the hydrogel strip was analyzed in the presence of an electric field. For this study, a polyelectrolyte composite hydrogel strip having a typical dimension of 40 mm × 10 mm × 1 mm was dipped into the electrolyte solution (0.1 M NaCl solution). A biased current of 18 V was passed through the solution using carbon electrodes placed 50 mm apart from each other. Fig. 10a shows the arrangement of the electrodes and the hydrogel strip between them. When the electrical impulse was applied for 5 min in the electrolyte solution the hydrogel strip starts twisting with time as shown in Fig. 10b. Upon withdrawal of the electrical impulse, the sample reverted to its initial shape (rectangular shape) after 40 min of rest. The presence of the opposite directional force gradient exerted by the ionic components (cationic and anionic segments) inside the hydrogel strip was the governing factor that controls the twisting of the hydrogel. Upon application of the electric field, the poly(sodium acrylate) segments moved towards the cathode whereas the cationic segment (starch-g-PMTAC) tried to move towards the anode as shown in the image (Fig. 10b). Due to the generation of the nanoscale force gradient, a macroscopic change in the shape of the hydrogel strip was observed. The images of the custom-built voltameter and orientation of the polyelectrolyte hydrogel strip upon application of the electric field are shown in Fig. S5 (ESI†). The macroscopic movement of the hydrogel strips in the presence of the nanoscale inhomogeneity inside the hydrogel strip induced by an electric field makes it a potential simulant material for the muscle.72,73 The behavior of the pristine anionic hydrogel (PSAS0C0, control sample) was also monitored to compare it with that of the composite hydrogel under an electrical impulse. It was observed that in the presence of an electrical impulse (18 V) the anionic hydrogel leaned towards the anode (Fig. S8, ESI†). This is due to the attraction of the anionic carboxylate group (–COO−) towards the anode that accelerates its deformation towards the anode.
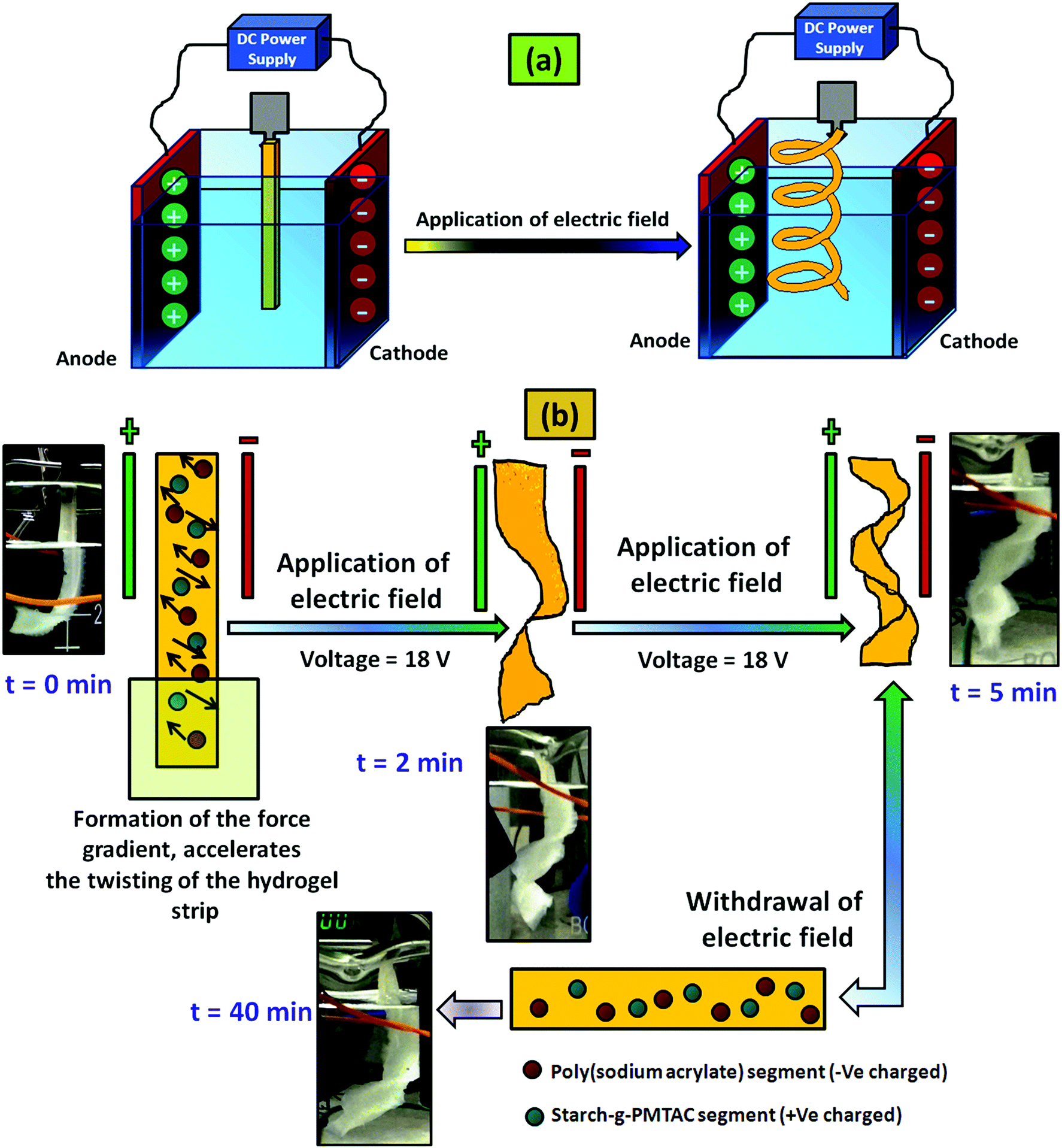 | ||
| Fig. 10 (a) Schematic representation of the actuation in the polyelectrolyte composite hydrogel (PSAS50C5) system in the presence of an electric field and (b) behaviour of the composite hydrogel (PSAS50C5) system with the time of electric field application. | ||
4.6. In vitro cell cytotoxicity assay
To study the in vitro cell cytotoxicity of the synthesized hydrogel, a cell proliferation assay was carried out against NIH 3T3 fibroblast cells. From the MTT assay results, it was observed that cell proliferation was dependent on the quantity of the OMMT clay. Here, a single component hydrogel was taken as a control sample (PSAS50C0) (during the experiment, the amount of cationic starch remained constant (50 wt%)) (Fig. 11a). With the increase in the OMMT content from 5 wt% (Fig. 11b) to 7 wt% (Fig. 11c) cell viability reduced (Fig. 11d). The cell viability value was taken based on the control result. This observation is very much similar to the experimental statement reported by Arundhati et al.,74 where they have prepared a chitosan/zirconium oxide/OMMT clay based nanocomposite film and employed it in bone tissue engineering. Pernodet et al. have also observed that higher loading of the natural cloisite clay has a detrimental effect on the growth of the dermal fibroblast cells.75 It has been reported by Angele et al. that in the presence of higher loading of CTAB modified OMMT clay, a significant toxic effect over the Caco-2 (human colon cell line) cell line has been observed. This is due to the generation of the reactive oxygen species (ROS) in the presence of the CTAB modified MMT that can damage the cell membrane resulting in cell death.76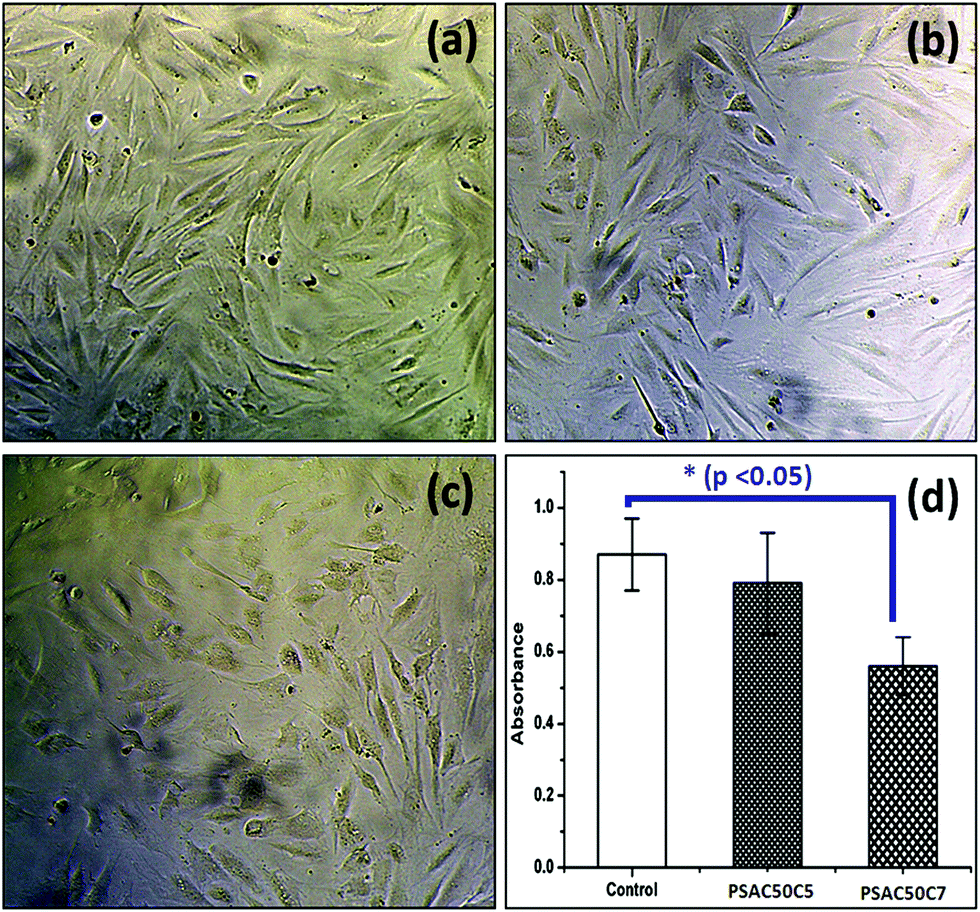 | ||
| Fig. 11 In vitro cell cytotoxicity images against the (a) control sample, (b) PSAS50C5 hydrogel (p > 0.05), (c) PSAS50C7 hydrogel (p < 0.05) and (d) respective absorbance values. | ||
5. Conclusions
In conclusion, we have successfully prepared a muscle mimetic self-healable nanocomposite polyelectrolyte hydrogel. The hydrogel was synthesized utilizing conventional free radical polymerization reactions to generate a brush like cationic starch and anionic poly(sodium acrylate) moieties along with CTAB modified MMT clay. The formation of cationic brush-like macromolecules is confirmed by 1H NMR and DOSY analyses. This hydrogel can self-heal in a wide range of pH (1.2 to 7.4 tested) by altering the self-healing mechanism to allow both hydrogen bonding in acidic media and the formation of ionic interaction at higher pH. This synthesized composite hydrogel is capable of water-based self-recovery which is another important property of muscle tissues. Along with this, the polyelectrolyte hydrogel can show actuation property in the presence of salt solution, acidic medium and electrical impulse. Importantly the hydrogel is non-toxic against fibroblast cells and even after self-healing can achieve a comparable ultimate tensile strength value (125 ± 21 kPa at pH 7.4) to extensor digitorum longus (EDL) muscle of a New Zealand white rabbit (163 ± 75.7 kPa). This smart and easy to synthesize polyelectrolyte hydrogel can have several biomedical applications.Conflicts of interest
There are no conflicts to declare.Acknowledgements
SLB acknowledges IIT Kharagpur for providing the Institute fellowship, and IIT KGP central research facility (CRF) for providing characterization support. SLB would also like to acknowledge the University of Bradford (UoB) and UoB CRF for providing the DOSY NMR analysis facility. MRC (MR/N501888/2) is acknowledged for the financial support to SLB for his visit to UoB and providing a post-doctoral fellowship for R. Hoskins.References
- E. M. Ahmed, J. Adv. Res., 2015, 6, 105–121 CrossRef CAS PubMed.
- I. M. El-Sherbiny and M. H. Yacoub, Global Cardiol. Sci. Pract., 2013, 38 CrossRef PubMed.
- J. Zhu and R. E. Marchant, Expert Rev. Med. Devices, 2011, 8, 607–626 CrossRef CAS PubMed.
- H. Tan and K. G. Marra, Materials, 2010, 3, 1746–1767 CrossRef CAS.
- T. V. Chirila, H. H. Lee, M. Oddon, M. M. L. Nieuwenhuizen, I. Blakey and T. M. Nicholson, J. Appl. Polym. Sci., 2014, 131, 39932 CrossRef.
- S. L. Banerjee, M. Khamrai, P. Kundu and N. K. Singha, RSC Adv., 2016, 6, 81654–81665 RSC.
- H. Liang, Z. Zhang, Q. Yuan and J. Liu, Chem. Commun., 2015, 51, 15196–15199 RSC.
- O. Okay, Supramolecular Polymer Networks and Gels, Springer, 2015, pp. 101–142 Search PubMed.
- J. Yu, W. Ha, J.-n. Sun and Y.-p. Shi, ACS Appl. Mater. Interfaces, 2014, 6, 19544–19551 CrossRef CAS PubMed.
- S. Biswas, D. B. Rasale and A. K. Das, RSC Adv., 2016, 6, 54793–54800 RSC.
- S. L. Banerjee, R. Hoskins, T. Swift, S. Rimmer and N. K. Singha, Polym. Chem., 2018, 9, 1190–1205 RSC.
- S. L. Banerjee and N. K. Singha, Soft Matter, 2017, 13, 9024–9035 RSC.
- M. Nadgorny, Z. Xiao and L. A. Connal, Mol. Syst. Des. Eng., 2017, 2, 283–292 RSC.
- S. Mukherjee, M. R. Hill and B. S. Sumerlin, Soft Matter, 2015, 11, 6152–6161 RSC.
- V. X. Truong, M. P. Ablett, S. M. Richardson, J. A. Hoyland and A. P. Dove, J. Am. Chem. Soc., 2015, 137, 1618–1622 CrossRef CAS PubMed.
- E. Zhang, T. Wang, L. Zhao, W. Sun, X. Liu and Z. Tong, ACS Appl. Mater. Interfaces, 2014, 6, 22855–22861 CrossRef CAS PubMed.
- N. A. Sirajuddin, M. S. M. Jamil and A. M. Lazim, AIP Conf. Proc., 2014, 1614, 469–475 CrossRef CAS.
- J. Wang, L. Lin, Q. Cheng and L. Jiang, Angew. Chem., 2012, 124, 4754–4758 CrossRef.
- J. P. Gong, Y. Katsuyama, T. Kurokawa and Y. Osada, Adv. Mater., 2003, 15, 1155–1158 CrossRef CAS.
- J. Collins, M. Nadgorny, Z. Xiao and L. A. Connal, Macromol. Rapid Commun., 2017, 38, 1600760 CrossRef PubMed.
- T. Corrales, I. Larraza, F. Catalina, T. Portolés, C. Ramírez-Santillán, M. Matesanz and C. Abrusci, Biomacromolecules, 2012, 13, 4247–4256 CrossRef CAS PubMed.
- M. Fu, Z. Zhang, L. Wu, G. Zhuang, S. Zhang, J. Yuan and L. Liao, Appl. Clay Sci., 2016, 132, 694–701 CrossRef.
- N. Ogata, S. Kawakage and T. Ogihara, J. Appl. Polym. Sci., 1997, 66, 573–581 CrossRef CAS.
- L. Wang and A. Wang, J. Hazard. Mater., 2008, 160, 173–180 CrossRef CAS PubMed.
- P. J. Flory, Principles of polymer chemistry, Cornell University Press, 1953 Search PubMed.
- T. Hussain, M. Ansari, N. M. Ranjha, I. U. Khan and Y. Shahzad, Sci. World J., 2013, 2013, 340737 Search PubMed.
- J. Crank, The mathematics of diffusion, Oxford university press, 1979 Search PubMed.
- N. Peppas, Y. Huang, M. Torres-Lugo, J. Ward and J. Zhang, Annu. Rev. Biomed. Eng., 2000, 2, 9–29 CrossRef CAS PubMed.
- Z. Ding, J. Aklonis and R. Salovey, J. Polym. Sci., Part B: Polym. Phys., 1991, 29, 1035–1038 CrossRef CAS.
- L. Gras, S. Laporte, D. Mitton, N. Crevier-Denoix and P. Viot, Proceedings International Research Council on Biomechanical Injuries, Dublin, Ireland (IRC-12-61: 515–523), 2012.
- D. Sgouras and R. Duncan, J. Mater. Sci.: Mater. Med., 1990, 1, 61–68 CrossRef CAS.
- T. Mosmann, J. Immunol. Methods, 1983, 65, 55–63 CrossRef CAS PubMed.
- G. Fotakis and J. A. Timbrell, Toxicol. Lett., 2006, 160, 171–177 CrossRef CAS PubMed.
- A. Pourjavadi, P. E. Jahromi, F. Seidi and H. Salimi, Carbohydr. Polym., 2010, 79, 933–940 CrossRef CAS.
- N. J. Penfold, Y. Ning, P. Verstraete, J. Smets and S. P. Armes, Chem. Sci., 2016, 7, 6894–6904 RSC.
- G. Moreira, E. Fedeli, F. Ziarelli, D. Capitani, L. Mannina, L. Charles, S. Viel, D. Gigmes and C. Lefay, Polym. Chem., 2015, 6, 5244–5253 RSC.
- T. Swift, L. Swanson, M. Geoghegan and S. Rimmer, Soft Matter, 2016, 12, 2542–2549 RSC.
- Y.-M. Lim, S. Yao, S. L. Gras, C. McSweeney, T. Lockett, M. A. Augustin and P. R. Gooley, Food Hydrocolloids, 2014, 40, 16–21 CrossRef CAS.
- S. Shen, Z. Zhu and F. Liu, Carbohydr. Polym., 2016, 138, 280–289 CrossRef CAS PubMed.
- J.-P. Wang, S.-J. Yuan, Y. Wang and H.-Q. Yu, Water Res., 2013, 47, 2643–2648 CrossRef CAS PubMed.
- R. Das, R. Kumar, S. L. Banerjee and P. Kundu, RSC Adv., 2014, 4, 59265–59274 RSC.
- S. Kim, J. Huang, Y. Lee, S. Dutta, H. Y. Yoo, Y. M. Jung, Y. Jho, H. Zeng and D. S. Hwang, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, E847–E853 CrossRef CAS PubMed.
- M. C. Stuparu, A. Khan and C. J. Hawker, Polym. Chem., 2012, 3, 3033–3044 RSC.
- M. Zhao, D. Yi, G. Camino, A. Frache and R. Yang, RSC Adv., 2017, 7, 861–869 RSC.
- N. Peng, D. Hu, J. Zeng, Y. Li, L. Liang and C. Chang, ACS Sustainable Chem. Eng., 2016, 4, 7217–7224 CrossRef CAS.
- M. M. Abeer, M. Amin, M. C. Iqbal and C. Martin, J. Pharm. Pharmacol., 2014, 66, 1047–1061 CAS.
- K. Jlassi, M. M. Chehimi and S. Thomas, Clay-Polymer Nanocomposites, Elsevier, 2017 Search PubMed.
- C.-C. Lin and A. T. Metters, Adv. Drug Delivery Rev., 2006, 58, 1379–1408 CrossRef CAS PubMed.
- A. Pourjavadi and S. Barzegar, Starch-Stärke, 2009, 61, 173–187 CrossRef CAS.
- R. Hamm, Advances in food research, Elsevier, 1961, vol. 10, pp. 355–463 Search PubMed.
- G. Offer and J. Trinick, Meat Sci., 1983, 8, 245–281 CrossRef CAS PubMed.
- A. Bajpai and A. Giri, Carbohydr. Polym., 2003, 53, 271–279 CrossRef CAS.
- C. Chang, M. He, J. Zhou and L. Zhang, Macromolecules, 2011, 44, 1642–1648 CrossRef CAS.
- F. Hofmeister, Arch. Exp. Pathol. Pharmakol., 1888, 25, 1–30 Search PubMed.
- M. Sadeghi and H. Hosseinzadeh, Turk. J. Chem., 2010, 34, 739–752 CAS.
- Y. M. Mohan, P. K. Murthy and K. M. Raju, React. Funct. Polym., 2005, 63, 11–26 CrossRef CAS.
- M. J. Legato and G. A. Langer, J. Cell Biol., 1969, 41, 401–423 CrossRef CAS PubMed.
- M. Alexandre and P. Dubois, Mater. Sci. Eng., R, 2000, 28, 1–63 CrossRef.
- L. Sun, R. F. Gibson, F. Gordaninejad and J. Suhr, Compos. Sci. Technol., 2009, 69, 2392–2409 CrossRef CAS.
- B.-H. Kim, J.-H. Jung, S.-H. Hong, J. Joo, A. J. Epstein, K. Mizoguchi, J. W. Kim and H. J. Choi, Macromolecules, 2002, 35, 1419–1423 CrossRef CAS.
- W. Li, R. Feng, R. Wang, D. Li, W. Jiang, H. Liu, Z. Guo, M. J. Serpe and L. Hu, J. Mater. Chem. B, 2018, 6, 4799–4807 RSC.
- D. A. Morrow, T. L. H. Donahue, G. M. Odegard and K. R. Kaufman, J. Mech. Behav. Biomed. Mater., 2010, 3, 124–129 CrossRef PubMed.
- A. A. Bent, N. W. Hagood and J. P. Rodgers, J. Intell. Mater. Syst. Struct., 1995, 6, 338–349 CrossRef.
- P. R. Buckley, G. H. McKinley, T. S. Wilson, W. Small, W. J. Benett, J. P. Bearinger, M. W. McElfresh and D. J. Maitland, IEEE Trans. Biomed. Eng., 2006, 53, 2075–2083 Search PubMed.
- Y. Bar-Cohen, Electroactive polymer (EAP) actuators as artificial muscles: reality, potential, and challenges, SPIE press, Bellingham, WA, 2004 Search PubMed.
- M. E. Harmon, M. Tang and C. W. Frank, Polymer, 2003, 44, 4547–4556 CrossRef CAS.
- W. T. Huck, Mater. Today, 2008, 11, 24–32 CrossRef CAS.
- M. Drupitha, B. Das, R. Parameswaran, S. Dhara, G. B. Nando and K. Naskar, Mater. Today Commun., 2018, 16, 264–273 CrossRef.
- Y. Li, Y. Sun, Y. Xiao, G. Gao, S. Liu, J. Zhang and J. Fu, ACS Appl. Mater. Interfaces, 2016, 8, 26326–26331 CrossRef CAS PubMed.
- M. E. Holman, J. Physiol., 1957, 136, 569–584 CrossRef CAS.
- L. Ionov, Adv. Funct. Mater., 2013, 23, 4555–4570 CrossRef CAS.
- L. Ionov, Mater. Today, 2014, 17, 494–503 CrossRef.
- S. Liu, G. Gao, Y. Xiao and J. Fu, J. Mater. Chem. B, 2016, 4, 3239–3246 RSC.
- A. Bhowmick, P. Jana, N. Pramanik, T. Mitra, S. L. Banerjee, A. Gnanamani, M. Das and P. P. Kundu, Carbohydr. Polym., 2016, 151, 879–888 CrossRef CAS PubMed.
- H. M. Lewkowitz-Shpuntoff, M. C. Wen, A. Singh, N. Brenner, R. Gambino, N. Pernodet, R. Isseroff, M. Rafailovich and J. Sokolov, Biomaterials, 2009, 30, 8–18 CrossRef CAS PubMed.
- S. Maisanaba, D. Gutiérrez-Praena, S. Pichardo, F. J. Moreno, M. Jordá, A. M. Cameán, S. Aucejo and Á. Jos, J. Appl. Toxicol., 2014, 34, 714–725 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8tb02852d |
| This journal is © The Royal Society of Chemistry 2019 |