
Semicrystalline physical hydrogels with shape-memory and self-healing properties

Abstract
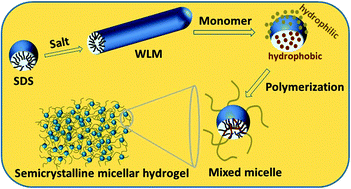
Synthetic hydrogels are generally amorphous in nature without any order at the molecular level. This is in contrast to biological gels containing ordered aggregates contributing significantly to their mechanical performance. Semicrystalline hydrogels, first developed in 1994, are moderately water-swollen hydrogels containing crystalline domains. Recent work shows that physically cross-linked semicrystalline hydrogels belong to one of the groups of mechanically strong and highly stretchable hydrogels exhibiting melt-processability, self-healing and shape-memory functions. They can undergo an abrupt and reversible change from a solid-like to a liquid-like state at the melting temperature, opening up several applications such as shape-memory hydrogels, injectable gels, chemical motors, and smart inks for 3D or 4D printing. In this review article, recent advances in the field of semicrystalline physical hydrogels prepared from hydrophilic and hydrophobic vinyl monomers via a free-radical mechanism are summarized. Synthesis–molecular structure–property relations of semicrystalline hydrogels, current challenges and future directions are also discussed.
- This article is part of the themed collection: Hydrogel properties and applications
 Please wait while we load your content...
Please wait while we load your content...

