
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Mechanically robust cationic cellulose nanofibril 3D scaffolds with tuneable biomimetic porosity for cell culture†
James C.
Courtenay
 ab,
Jefferson G.
Filgueiras
c,
Eduardo Ribeiro
deAzevedo
c,
Yun
Jin‡
b,
Karen J.
Edler
ab,
Ram I.
Sharma
ad and
Janet L.
Scott
*ab
ab,
Jefferson G.
Filgueiras
c,
Eduardo Ribeiro
deAzevedo
c,
Yun
Jin‡
b,
Karen J.
Edler
ab,
Ram I.
Sharma
ad and
Janet L.
Scott
*ab
aCentre for Sustainable Chemical Technologies, University of Bath, Bath, BA2 7AY, UK. E-mail: j.l.scott@bath.ac.uk
bDepartment of Chemistry, University of Bath, Bath, BA2 7AY, UK
cInstituto de Física de São Carlos, Universidade de São Paulo, CP 369, 13560-970, São Carlos, SP, Brazil
dDepartment of Chemical Engineering, University of Bath, Bath, BA2 7AY, UK
First published on 23rd November 2018
Abstract
3D foam scaffolds were produced in a “bottom-up” approach from lyophilised cationic cellulose nanofibril (CCNF) dispersions and emulsions (CCNF degree of substitution 23.0 ± 0.9%), using a directional freezing/lyophilisation approach, producing internal architectures ranging from aligned smooth walled micro channels, mimicking vascularised tissue, to pumice-like wall textures, reminiscent of porous bone. The open, highly porous architecture of these biomimetic scaffolds included mesopores within the walls of the channels. A combination of SEM and NMR cryoporometry and relaxometry was used to determine the porosity at different length scales: CCNF foams with aligned channels had an average macropore (channel) size of 35 ± 9 μm and a mesopore (wall) diameter of 26 ± 2 nm, while CCNF foams produced from directional freezing and lyophilisation of Pickering emulsions had mesoporous walls (5 ± 3 μm) in addition to channels (54 ± 20 μm). Glyoxal crosslinking both enhanced robustness and stiffness, giving Young's moduli of 0.45 to 50.75 MPa for CCNF foams with degrees of crosslinking from 0 to 3.04 mol%. Porosity and channels are critical scaffold design elements for transport of nutrients and waste products, as well as O2/CO2 exchange. The viability of MG-63 cells was enhanced on crosslinked, mechanically stiff scaffolds, indicating that these exquisitely structured, yet robust, foams could provide biomaterial scaffolds suitable for industrial applications requiring 3D cell culturing.
Introduction
Current demand for donor organs and tissues for transplantation vastly surpasses availability. For example, more than 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 US patients wait on the organ donor list in 2018 and, on average, 22 will die per day.1 The goal of tissue engineering is to develop new cell constructs that can be implanted into a patient to restore function to the damaged organs.2 This process requires a biocompatible scaffold to support the cells, often by mimicking the extracellular matrix (ECM), as these proliferate and grow into tissues.3 Traditionally, adherent cells are cultured on a two-dimensional (2D) scaffold in vitro. Cells growing on 2D scaffolds tend to only be attached to the substrate at their periphery, which forces the cells into a monolayer culture, rather than promoting layering as would be found in vivo, as well as limiting the size of the cell population produced.4 Once the cell layer has reached 100% confluence (all available surface is covered), cell viability can decrease, and cell death can occur as mass transfer of nutrients and oxygen diffusion is limited to depths of 100–200 μm.5 To retain cell viability beyond this depth, a vascularised network is required.6–8
000 US patients wait on the organ donor list in 2018 and, on average, 22 will die per day.1 The goal of tissue engineering is to develop new cell constructs that can be implanted into a patient to restore function to the damaged organs.2 This process requires a biocompatible scaffold to support the cells, often by mimicking the extracellular matrix (ECM), as these proliferate and grow into tissues.3 Traditionally, adherent cells are cultured on a two-dimensional (2D) scaffold in vitro. Cells growing on 2D scaffolds tend to only be attached to the substrate at their periphery, which forces the cells into a monolayer culture, rather than promoting layering as would be found in vivo, as well as limiting the size of the cell population produced.4 Once the cell layer has reached 100% confluence (all available surface is covered), cell viability can decrease, and cell death can occur as mass transfer of nutrients and oxygen diffusion is limited to depths of 100–200 μm.5 To retain cell viability beyond this depth, a vascularised network is required.6–8
Furthermore, if primary keratinocytes are grown to 100% confluence, the phenomenon of “terminal differentiation” can arise: cells receiving insufficient nutrients die and those that remain alive become senescent, limiting the size of the cell construct that can be grown on a 2D scaffold.
On the other hand, 3D scaffolds require internal porosity to mimic the vascular morphology of native tissue, facilitating transfer of nutrients and gases and allowing larger tissue fragments to be grown. Ideally, the scaffolds should be resorbed and replaced, over time, by the newly regenerated tissue at the site of implant.9 A 3D arrangement of cells enables more complex cell–cell and cell–ECM interactions as cells are surrounded by ECM or scaffold, due to the greater surface area available for adhesion. Thus, cells cultured in a 3D scaffold more accurately mimic the response and behaviour of cells in vivo, beneficial for applications in both cell culturing for tissue engineering and as model tissues for drug development.10
Interconnecting porous networks can be used to promote cell growth, migration and mass transfer of nutrients.11 Generally, a larger pore size is considered to be beneficial for improving both cell migration and nutrient flow, however, this decreases the specific surface area of the scaffold, in turn reducing the matrix ligand density available for cell binding. Instead of migrating into the bulk of the material, cells tend to cluster around the edges of the scaffold.12 O’Brien et al., demonstrated that increasing pore size from 96 to 151 μm increased nutrient permeability through scaffolds,12 however, a pore size of between 20 and 120 μm was required for optimal balance between nutrient flow and ligand binding density.11 Therefore, a compromise must be reached between the two factors, pore size and specific surface area, of a 3D scaffold.13
Decellularised tissue has been investigated as a “top-down” method of obtaining the complex tissue structure and ECM composition needed for tissue regeneration, whilst retaining the microvasculature (<10 μm in diameter) that cannot readily be fabricated by current techniques such as 3D printing.14 Human donor or animal tissues can be used, but need to be sterilised and decellularised by enzymatic or detergent methods to remove the native cells and proteins. The remaining material is the ECM, which can vary in composition and structural properties, depending on the source.8 It is important that the decellularising process does not disrupt the ECM or adversely affect the biological activity or mechanical integrity of the remaining structure.15 The performance of the tissue can degrade with age and human tissue in particular, is in short supply.16 Decellularised scaffolds based on plant tissue have been reported,17 but, despite much of the vascularised structure being in place, scaffold shapes are limited to the shape of the plant tissue from which it was derived and further shape modulation is challenging.
Complementing the “top-down” approaches to the creation of porous 3D scaffolds, there are also a range of “bottom-up” methodologies to produce complex structures for cell culture, including: electrospinning polymer solutions to form 3D nanofiber meshes,18–20 3D printing of “bioinks” to fabricate complex scaffold architectures21 and the lyophilisation of dispersions or solutions to produce open porous aerogels or foams.22 Additive manufacturing approaches can reportedly give access to structures with minimum feature lengths of <100 μm,21 while reverse templating can be used to achieve porosities of 50–85% and interconnecting pores of 50 μm in size.23,24
There are several other “bottom-up” approaches to fabricate scaffolds suitable for applications in tissue engineering. For example, foam templating by generation of bubbles in solutions of polysaccharides containing surfactants,25 followed by freeze drying and crosslinking with a carbodiimide cross-linker yields a porous structure with pore diameters of 206–250 μm.26 Foam templating routes have also been applied to poly(vinyl alcohol) solutions27 and dextran–methacrylate solutions.28 Scaffolds so produced present highly ordered pore arrays with uniform and tuneable pore sizes.29 As pores are interconnected, mass transfer of nutrients to cells within such scaffolds is enhanced.
Freeze drying, or lyophilisation, is a popular method to introduce porosity into the scaffold due to its low cost, simplicity and versatility.24 In this process, ice crystals act as a porogen, leaving a porous structure post sublimation under vacuum.30 The rate of cooling during the freezing step can affect the size and distribution of ice crystals formed and thus the size and form of the pores within in the resulting foam.31 An advantage of using this technique is that water is commonly the solvent; hence impurities, such as surfactants, are not added during the lyophilisation stage, making it especially beneficial for biological applications. The introduction of emulsions, nanoparticles and dilute polymer solutions into the material can further modulate macro- and mesopore formation.30 Ice crystals grown in a unidirectional fashion leave behind channels of aligned pores, mimicking the vascularity of native tissue32 and can be applied to a range of scaffold materials, including: hydroxyapatite,33 silk fibroin,34 gelatin,35 dextran,36 and, more recently, chitosan–alginate blends,37 cellulose–chitosan blends,38 and cellulose solutions.39 The resultant scaffolds have the potential to expand the use of these materials from 2D films to 3D porous scaffolds and many authors suggest that the channels can promote vascularisation in vivo, although corroborating cell studies are only reported for some materials.
Scaffolds fabricated from cellulose, using both “top-down” and “bottom-up” approaches, have been tested in a range of cell culture applications.40–43 Cellulose offers many beneficial attributes to tissue engineering, such as biocompatibility, versatile chemical and physical properties, and ease of processing. It is also a cost effective and sustainable material, which makes it suitable for industrial applications.44–48 However, previous reports of directionally frozen/freeze dried cellulose hydrogels for scaffolds have largely focussed on the materials characterisation aspects – few reports of the growth of cells on these 3D scaffolds have appeared.38,39
Previously, we have demonstrated that cellulose surfaces grafted with cationic moieties allow for attachment of MG-63 cells in the absence of matrix ligands.49 Here we use similarly modified nanofibrillar materials to develop novel porous 3D scaffolds that we suggest offer many advantages over “top-down” scaffolds such as those derived from decellularised plant tissue. In particular the use of a scalable method offering ease of manufacture, tuneable porosity and mechanical properties, to prepare materials that can be stored without special requirements, yet which mimic vascularised tissue, may provide supports beneficial for various cell culture applications.
Materials and methods
Cellulose powder (C8002), sodium hydroxide pellets (≥98%), glycidyl trimethylammonium chloride (GTMAC, ≥90%), DMSO (≥99%), 1-ethyl-3-methylimidazolium acetate (97%), cyclohexane (99.5%), absolute ethanol (≥99%) and N,N-dimethylacetamide (DMAc) were purchased from Sigma-Aldrich and used as received (except DMAc, which was dried over 3 Å molecular sieves prior to use). For crosslinking modifications, glyoxal 40 w/w% aqueous solution was purchased from Alfa Aesar and made up to required concentrations with deionised (DI) water. Mobile phases for HPLC were prepared from H2SO4 (99.99%) and sodium phosphate monobasic monohydrate, sodium phosphate dibasic and sodium azide powders purchased from Sigma Aldrich.For cell studies, Dulbecco's Modified Eagle Medium (DMEM, GlutaMAX™), non-essential amino acids, sodium pyruvate, trypsin (0.05%) and trypan blue (0.4%) were purchased from Gibco and stored at 4 °C. Foetal bovine serum (FBS, non-USA origin), MG-63 human osteosarcoma cells, and methylthiazolyldiphenyltetrazolium bromide (MTT) powder were purchased from Sigma-Aldrich. Phosphate buffer solution (PBS, 0.1 μm sterile filtered) was purchased from HyClone and penicillin streptomycin from Life Technologies. For cell fixation, a glutaraldehyde solution (25 wt% in H2O), hexamethyl-disilazane (HMDS, >99%), dry acetone and 1 wt% osmium tetroxide solution were purchased from Sigma Aldrich.
Surface modification by derivatisation
3D scaffold formation
500 rpm for 15 min. Dispersion to form hydrogels was completed by sonication using a Sonic Dismembrator Ultrasonic Processor (Fisher Scientific) with a 3.2 mm tip at a power output of 45 W cm−2 as 1 s on/off pulses for 2 min. Dispersions were stored at 4 °C until required. To form the crosslinked hydrogels, stock glyoxal solutions were prepared at 5, 10 and 20 wt% in DI water. These were added to appropriate quantities of a 2 wt% CCNF dispersion to yield 1 wt% CCNF aqueous dispersions containing 2.5, 5 or 10 wt% glyoxal. To introduce macropores into the structure of the hydrogel, oil-in-water emulsions were prepared using oil: aqueous phase volume ratio of 30:70. Typically, 0.3 mL of cyclohexane was mixed with 0.7 mL of CCNF aqueous dispersion (1 wt% prepared as described above) and sonicated (45 W cm−2 as 1 s on/off pulses for 30 s) to effect emulsion formation.
Cast films were flash frozen in liquid nitrogen prior to lyophilisation. CCNF hydrogels and emulsions were subjected to directional freezing: 250 μL of the sample was placed into the well of a 48 tissue culture well plate, which was set atop a metal block partially submerged in a bath of liquid nitrogen, resulting in directional growth of ice-crystals from the bottom of the sample. Once fully frozen, samples were lyophilised to remove the ice crystals. More complex shaped foams were produced by freezing the CCNF hydrogels in silicon moulds (Fig. S7, ESI†). The lyophilised foams containing glyoxal were heated in an oven for 1 h at 125 °C to allow the crosslinking reaction to proceed.
:70 w/w, EMImOAc:DMSO, and the mixture stirred for 1 h at room temperature, resulting in a 4 wt% CC solution. For comparison, a solution of unmodified cellulose (UC) was prepared using the same procedure.
CC and UC films were formed by casting the solutions described above onto a clean glass sheet using an Elcometer 3700 reservoir and an Elcometer 4340 Automatic film applicator, with a gap of 0.8 mm between the blade and glass surface. The films were regenerated by immersion in an ethanol anti-solvent bath for 24 h. Residual EMImOAc and DMSO were removed by Soxhlet extraction with ethanol overnight. The films were washed twice with copious amounts of DI water to remove excess EtOH before being stored in 20 wt% MeOH solution to inhibit bacterial growth.
Scaffold characterisation
The melting point depression, ΔT, is related to the pore radius, r, via bulk properties of the probe liquid, P, as described by the Gibbs–Thomson equation:
| ΔT ∝ P/(r − s) | (1) |
000 echoes were acquired with echo time of 70 μs and recycle delay of 15 s. The CPMG decay curves were processed to obtain the T2 distribution using a non-negative least square procedure known as a numerical Inverse Laplace Transform, ILT.58,59 The obtained T2 distributions were deconvoluted using log-gaussian functions to provide the contribution of each component in the pore structure. Fluid bound in a pore interacts with the pore surface, restricting the molecular mobility of the fluid. Such restriction is reflected by a decrease of the transverse relaxation time, T2, and can be quantified, in the fast diffusion regime, by the relation:60,61| 1/T2 = ρ(S/V) = 2ρ/r, | (2) |
To assess the pore structure of the cellulose foams, DMAc (HPLC grade) was used as a molecular probe, due to its weak interaction with the cellulose structure. DMAc does not significantly modify the cellulose pore structure, while water, often used as molecular probe for relaxometry experiments, alters the pore distributions in biomass samples.62 Lyophilised modified cellulose foams and films were dried under reduced pressure (640 mmHg) for 24 h at 80 °C. Samples were soaked in DMAc and kept in a desiccator under a 600 mmHg vacuum for 36 h, following which excess DMAc was removed by centrifuge filtration at 600 g for 1 min (Corning Costar Spin-X, 0.45 μm, nylon membrane filter). All measurements were carried out in duplicate and the CPMG decay of the mean used in the ILT procedure (Fig. S14–S16, ESI†).
000×.
Mechanical properties
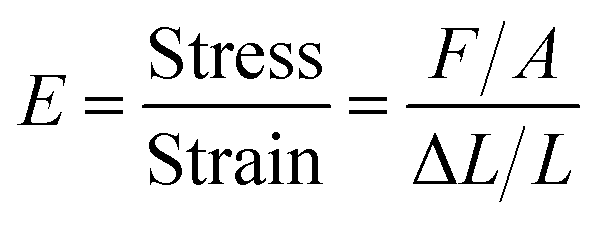 | (3) |
Cell studies
000 cells cm−2 (corresponding to a total cell number of 9500 cells per sample) in growth media and placed in an incubator at 37 °C in 5% CO2 to proliferate for a given time. Tissue culture plastic was used as a control “scaffold” for the MTT assay and material controls were performed for each sample. N = 8 replicates per sample were measured.
A MTT stock solution of 50 mg mL−1 in PBS was prepared. After appropriate incubation time (1, 4 and 7 days), the growth medium was replaced with 250 μL of 5 mg mL−1 MTT in growth medium and incubated for a further 4 h. The culture medium was gently removed from the well plate and 200 μL of DMSO added to dissolve the formazan crystals generated in this assay. Well plates were placed on a plate shaker and gently agitated for 10 min. The DMSO-formazan crystal suspension was transferred to a 96 well plate and the absorption intensity recorded at 570 nm and 690 nm, using a Biotek® Synergy HT plate reader. To determine the activity the following equation was used:
| MTT activity = (S570nm) − (MC570nm) | (4) |
The culture medium was very slowly removed from the scaffold keeping the pipette at the edge of the scaffold away from the cells. The samples were gently washed in 2 × 1 mL fresh medium (normal strength culture medium without serum) again keeping the pipette at edge of the scaffold. The wash medium was removed as above, replaced with the fixative solution and left for 2 h. The samples were rinsed three times with 1 mL wash medium. The cells were post-fixed in aqueous 1 wt% osmium tetroxide for 1 h in a fume hood at ambient temperature, then washed in 1 mL DI H2O three times at 15 min intervals.
The cells were dehydrated by sequential washing with solutions of acetone in H2O gradually increasing in acetone content to 100% dry acetone. Specifically solutions of 50, 70, 90, 95% and 100% acetone in H2O were applied to the cells (on scaffold) for 15 min, repeated three times for each concentration. Acetone was replaced with 1:1 dry acetone:HMDS solution for 15 min, followed by three sequential treatments with 100% HMDS for 30 min each. Excess HMDS was removed and the samples allowed to air dry in a fume hood for 2 h prior to mounting onto SEM stubs.
Results and discussion
Application of directional freezing and lyophilisation to aqueous dispersions of CCNF and CCNF stabilised oil-in-water Pickering emulsions, led to preparation of 3D cationic cellulose scaffolds in the form of foams with exquisite tuneable structure. Unidirectional ice crystal formation resulted in channels in the tens of micron range, mimicking vascular structures, and preservation of spherical pores arising from emulsions, yielded pumice-like wall textures, reminiscent of porous bone (Fig. 1).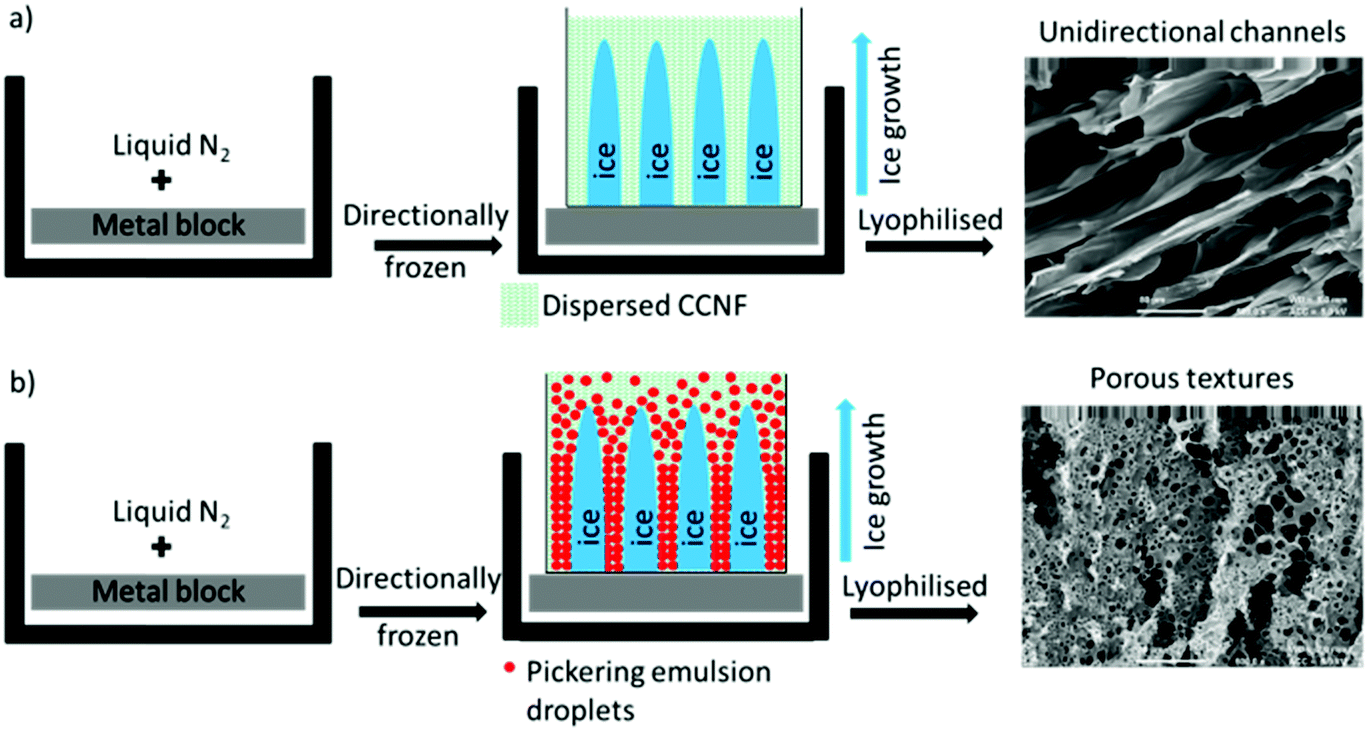 | ||
| Fig. 1 Directional freezing followed by lyophilisation process: (a) CCNF dispersions (b) CCNF stabilised oil-in-water Pickering emulsions. | ||
Crosslinking, previously shown to enhance cell spreading63 (indicative of cell viability and scaffold compatibility), was used to render the delicate structures robust enough for handling and repeated immersion in cell growth media, as would be required for use in tissue engineering applications.
To illustrate their utility as tissue culture scaffolds, the attachment and viability of MG-63 cells was assessed using MTT assays and SEM analysis to discern how well cells became integrated into the bulk of the putative scaffold material and whether or not these cells began to proliferate.
“Bottom-up” fabrication of vascularised 3D scaffolds
3D scaffolds, with variable form, internal architecture and robustness, were constructed using a “bottom-up” approach from “cationic cellulose”, CC, bearing quaternary ammonium groups derived from glycidyl trimethylammonium chloride (GTMAC) grafting, either as CC nanofibrils, or dissolved and regenerated CC films.To induce directional freezing, low weight% CCNF dispersions (weak hydrogels) were placed, in containers, on metal blocks immersed in liquid nitrogen. This resulted in exclusion of the CCNF into “walls” surrounding oriented ice crystals, which, upon lyophilisation, yielded aligned channels, reflecting the size of the ice crystals formed. Cyclohexane in water CCNF stabilised Pickering emulsions treated similarly resulted in both channel-like porosity and walls bearing much smaller pores, 3–5 μm in diameter, due to oil droplet templation of the hydrogel. These materials are designated “CCNF PE” henceforth.
Although the foams contained both aligned micro channels reflecting the longitudinal ice crystal growth, and pores, reflecting the emulsion structure, these were very delicate and easily compressed, or even dispersed. Even gentle agitation in cell culture medium caused the scaffolds to break apart, rendering these useless as scaffolds in cell culture applications. Hence, the foams were crosslinked to enhance robustness,64 especially when hydrated, and to retain the complex internal architecture.
To reinforce these delicate porous materials, glyoxal, a crosslinker, was added to the CCNF dispersions and later reacted by heating, in order to fix the complex internal structure, through in situ glyoxalation – these materials are designated as “CCNF XL”. In common with most reactive cross-linkers, unreacted glyoxal is cytotoxic, but once reacted to form acetal and hemiacetal linkages with cellulose, does not inhibit cell viability.63
This modification served to support the delicate structures, maintaining the integrity of the vascularised or porous internal architecture, but it has also been shown previously that crosslinking further increases the spreading of attached MG-63 cells on cationised cellulose scaffolds.63 Furthermore, modification of the mechanical properties of the scaffold can act as a stimulus to up-regulate physiological processes and signalling pathways within the cell cycle, thus promoting cell growth.65,66 Thus, these materials could serve as very flexible scaffolds, allowing external shape/size, internal architecture, rigidity and robustness to be varied while also offering opportunities to modulate cell response, as described later.
Scaffold porosity
To determine the effect of lyophilisation and glyoxalisation on macroporosity, the CCNF, CCNF XL, and CCNF PE scaffolds were characterised using SEM and compared to regenerated lyophilised films (Fig. 2 and Fig. S17–S19, ESI†). Structures accessible ranged from unidirectional, vascularised materials with smooth walls (Fig. 2a), foamed walls (Fig. 2c) to more open pore structure materials (Fig. 2b), compared with regenerated cationic cellulose films (Fig. 2d). (For comparison, an SEM image and pore size distribution for unmodified cellulose films is included in the ESI† (Fig. S19).) To determine the average pore size diameter SEM images were analysed using ImageJ software yielding the following average pore sizes: CCNF = 35 ± 9 μm, CCNF XL = 60 ± 20 μm, CCNF PE = 54 ± 20 μm and 5 ± 3 μm, and CC film = 20 ± 10 μm. The presence of 5 wt% glyoxal in the CCNF XL hydrogel appeared to reduce the alignment of ice-crystals formed during the freezing process, while emulsion templating introduced smaller pores, ∼5 μm in diameter, into the walls of the lyophilised hydrogel structure. Film structures, while porous, appear to show less connectivity between pores and a dense “skin” layer.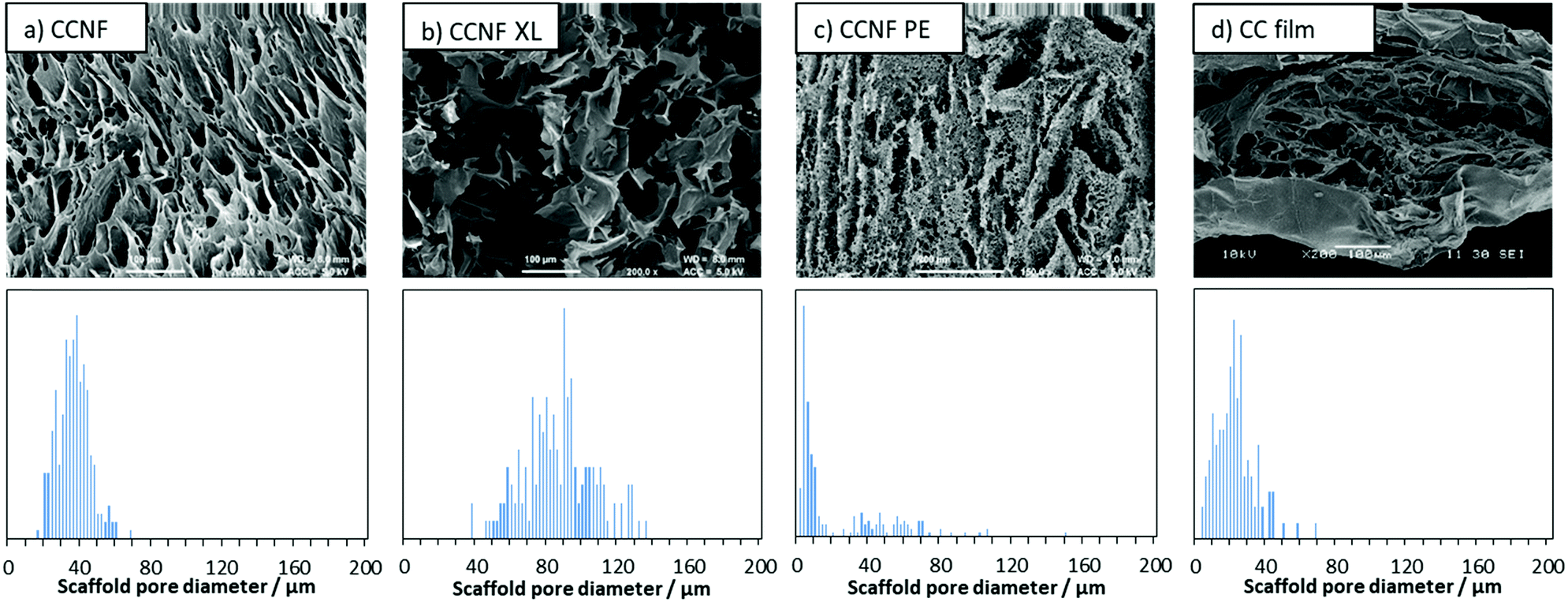 | ||
| Fig. 2 SEM images and pore size distributions of lyophilised foams (a) 2 wt% CCNF hydrogel, CCNF; (b) 2 wt% CCNF hydrogel + 5 wt% glyoxal, CCNF XL; (c) 1 wt% CCNF Pickering emulsion templated hydrogel, CCNF PE; and (d) regenerated 4 wt% cationic cellulose film, CC. | ||
Along with the macroporous network, visible in SEM images, mesopores occur within the walls of the freeze dried foams. NMR cryoporometry was used to probe the permeability of the foam walls to PBS (Fig. 3 and Table 1). PBS was used to reflect typical cell culture media. Importantly, this technique allows the pore structure of hydrogels to be analysed in the hydrated materials, thus more accurately mimicking the microenvironment cells would experience in scaffolds. The smallest mesopore diameter, 20 ± 2 nm, was observed in CCNF XL foams, where the pore size was found to be very similar to that in the control regenerated CC films, 18 ± 2 nm. The wall pore modal diameter determined for CCNF foams was 25 ± 2 nm and was not significantly affected by the concentration of initial CCNF dispersion used. Crosslinking appeared to lead to a slight reduction in wall pore diameter to 20 ± 2 nm. In contrast the CCNF PE foam does not allow for penetration of PBS solution into the walls, as reflected by a modal diameter matching that determined for the bulk PBS solution control. It is likely that this reflects the densification of CCNF at the oil/water interface in the Pickering emulsion,67 yielding structures permeated by a plethora of pores arising from oil droplets, but with dense close-packed CCNF walls. These 3D scaffolds, formed from CCNF dispersions and emulsions, thus have porosity at three different scales: large pores or channels resulting from ice-crystal templation; smaller pores arising from templation by oil droplets in Pickering emulsions; and nanoscale pores reflecting packing of the CCNF within the walls of the foamed structures. As this differing porosity could be beneficial for both penetration of cells into the scaffold and mass transfer of nutrients, gases and waste products to and from cells during cell growth, we sought to unify the porosity measurements to allow comparison of potential scaffold materials and thus to inform future selection of the form of templates.
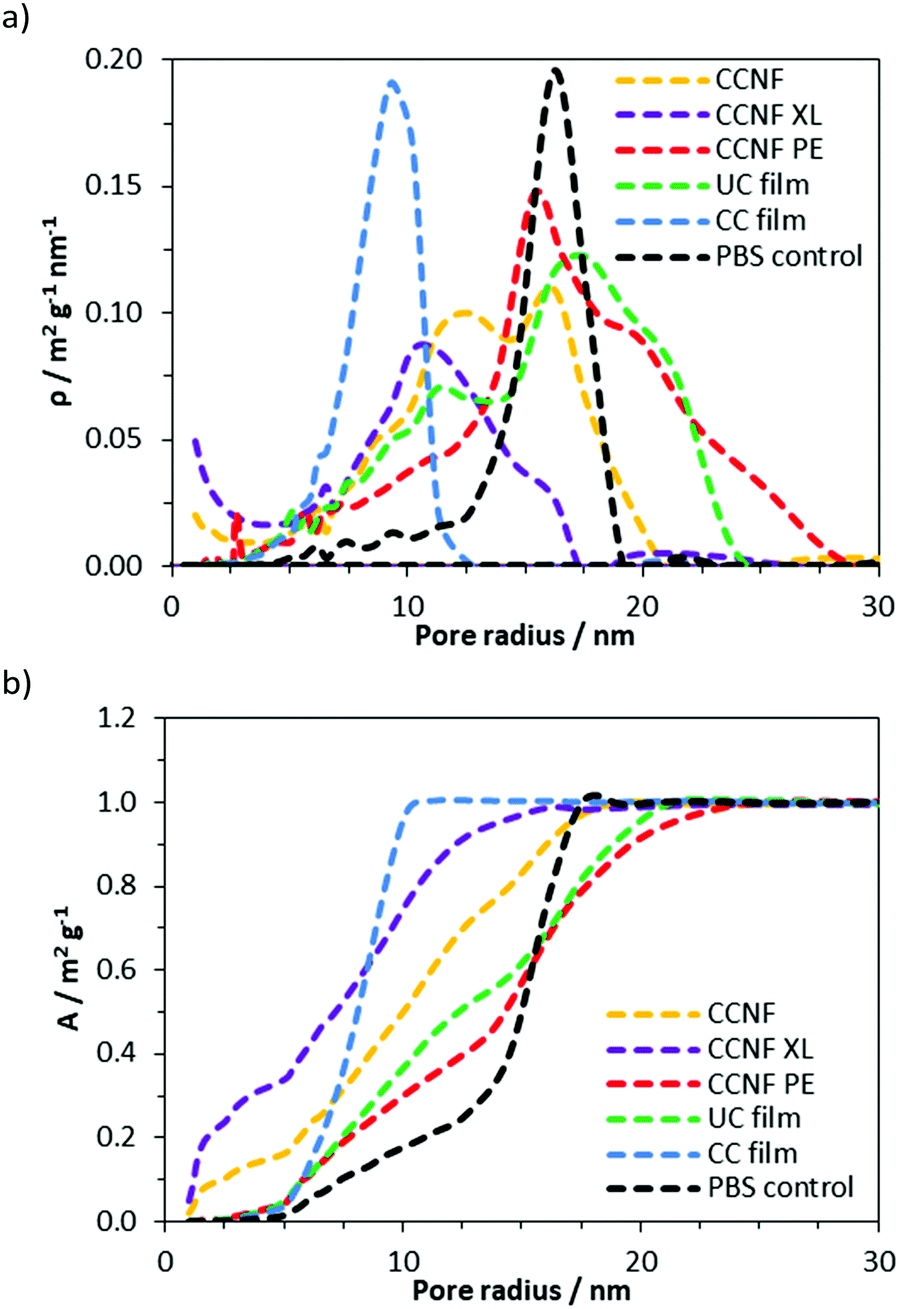 | ||
| Fig. 3 Size distribution of mesopores in hydrated CCNF foams from NMR cryoporometry measurements: (a) specific pore surface area density, ρ, and (b) specific cumulative pore surface area. The walls of the CCNF freeze dried foams are permeable to the PBS solution, with 1 wt% foams exhibiting the largest pores ascribed to the more open network formed by dispersed nanofibrils. Crosslinked foams have the smallest pores, at 20 nm compared to 25 nm for uncrosslinked foams. The CCNF PE material appears to have dense (albeit very thin – SEM) walls that do not allow for penetration of PBS as the modal diameter calculated is the same as that arising from a bulk PBS solution. This reflects dense packing of nanofibrillar walls formed from fibrils adsorbed at the oil/water interface in the emulsion. | ||
| Scaffold type | Sample | Mesopore diameter/nm | Macropore diameter/μm |
|---|---|---|---|
| a Directionally frozen dispersions. b Directionally frozen Pickering emulsions. c Cast regenerated films. d Crosslinker concentration in solution = 5 wt%. e Macropore diameter – emulsion droplets. f Macropore diameter – channels. | |||
| CCNF 1 wt% | 25 ± 2 | 35 ± 9 | |
| CCNF 2 wt% | 26 ± 2 | ||
| CCNF 4 wt% | 27 ± 2 | ||
| CCNF XL 2 wt%d | 20 ± 2 | 60 ± 20 | |
| CCNF PE 1 wt% | 30 ± 2 | 5 ± 3,e 54 ± 20f | |
| CC 4 wt% | 18 ± 2 | 20 ± 10 | |
| UC 4 wt% | 35 ± 2 | 0.6 ± 0.3 | |
| Control | PBS | 32 ± 2 | n/a |
NMR T2 relaxometry experiments were used to probe the accessibility of DMAc to three different scales on the materials, given by each component in the T2 distribution. Such scales are related to pores of a few nanometers, for T2 values of about 10−3 s, mesopores of dozens of nanometers, for T2 values ranging from 10−2 to 10−1 s and large pores of hundreds of nanometers up to one micrometer, for T2 values above 10−1 s. The T2 distributions for all scaffolds are shown in the ESI† (Fig. S14–S16, ESI†).
The proportion of DMAc accessing pores on each length scale can be estimated from the respective relative areas of the log-gaussian obtained from deconvolution (Fig. 4). The greater the accessibility, the more pores are filled with the fluid at a particular length scale. Thus, CCNF XL has a greater accessibility of pores on the smallest scale in comparison to CCNF and CCNF PE, suggesting that crosslinking increases the number of accessible nanopores in the cell wall. This reflects the observation that crosslinked cellulose materials are less prone to hornification than uncrosslinked celluloses.
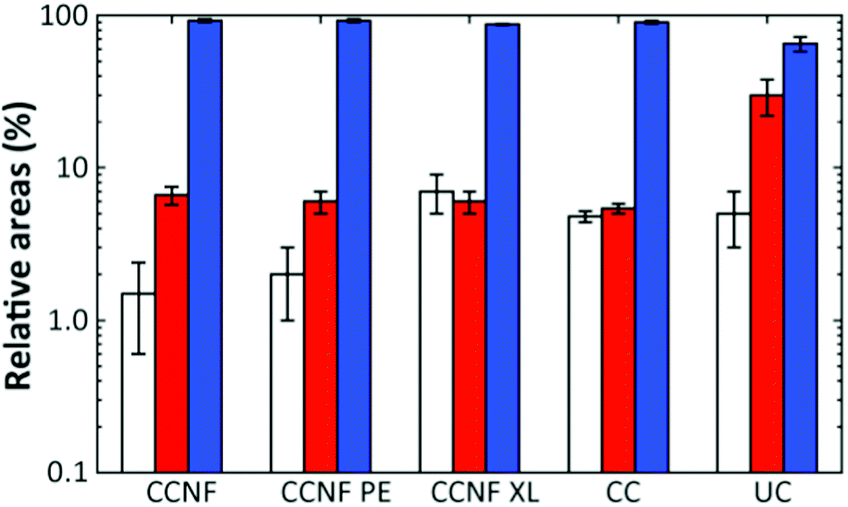 | ||
| Fig. 4 Relative areas of the log-gaussian obtained from deconvolution for CCNF, CCNF PE, and CCNF XL foams, compared to CC and unmodified cellulose (UC) films as controls. The left (white) bars indicate the nanopores, the centre (red) bars the mesopores and the right (blue) bars the large pores. | ||
While the T2 distributions do not give precise pore sizes due to the unknown surface relaxivity for each sample, if the (reasonable) assumption is made that the surface relaxivity is similar in the different pore categories, the linear relationship between the pore size and T2 (eqn (2)) allows estimation of the ratio between the mean of the components seen in the T2 distribution. There is a reasonable correlation between the cryoporometry data and the central T2 values for the mesopore components (Fig. S16, ESI†). Thus, based on the average pore sizes determined by NMR cryoporometry results and the T2 values for the mesopore components, it is possible to estimate the pore sizes in the hundreds of nm length scale, which is neither accessible by NMR cryoporometry nor by SEM. The pore sizes thus calculated are shown in Table 2.
| Scaffold type | Sample | T 2 (ms) | Large pore diameter (nm) |
|---|---|---|---|
| a Directionally frozen dispersions. b Directionally frozen Pickering emulsions. c Cast regenerated films. d Crosslinker concentration in solution = 5 wt%. | |||
| CCNF 2 wt% | 382 ± 2 | 300 ± 30 | |
| CCNF XL 2 wt%d | 353 ± 2 | 350 ± 40 | |
| CCNF PE 1 wt% | 209 ± 1 | 320 ± 20 | |
| CC 4 wt% | 135 ± 1 | 180 ± 20 | |
| UC 4 wt% | 117 ± 5 | 70 ± 7 | |
Mechanical properties and robustness
As mentioned previously, uncrosslinked CCNF foams were very delicate, becoming broken up and dispersing readily in DI H2O or buffer upon even very gentle agitation. Foams formed in moulds to create more complex 3D shapes, as may be required in the production of suitable tissue samples in ex vivo cultures, completely dispersed in cell culture medium (Fig. 5). Thus, in their native state, the foams were not compatible with the handling requirements for cell culture techniques.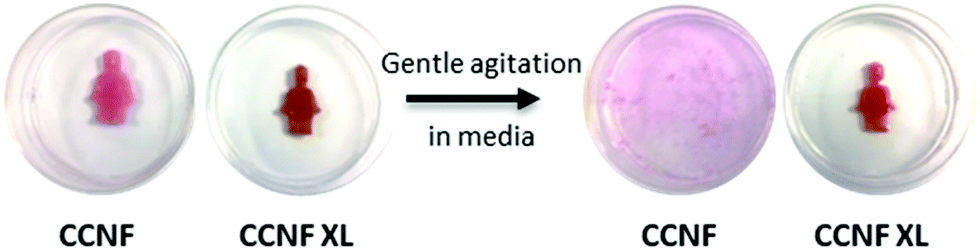 | ||
| Fig. 5 CCNF and CCNF XL foams soaked in DMEM cell culture medium. The CCNF foams swelled by 10% upon hydration in H2O, whereas the CCNF XL foams retained their dry dimensions. After shaking in a centrifuge vial for 10 s CCNF foams broke up and dispersed in H2O, whereas CCNF XL foams remained intact. | ||
To avoid compromising the exquisite structures formed by sequential soaking in glyoxal solution and heating (as used previously63), glyoxalation was effected in situ: glyoxal solutions were added to the hydrogels prior to freezing and the curing step took place post lyophilisation on the now dehydrated foam. This simple procedure is attractive for manufacturing as it reduces the number of processing steps required in foam scaffold fabrication, making scaffold manufacture readily scalable. The dry foams thus produced had degrees of crosslinking from 1.18 to 3.04 mol%, as determined by HPLC analysis (Fig. S6, ESI†), comparable with glyoxalation conducted on cellulose films.63
As the concentration of glyoxal crosslinker used increased from 2.5 to 10 wt%, the crosslinked scaffolds became more robust with mechanical properties, such as Young's modulus and compressive strength, increasing accordingly (Fig. 6a and b). Compressive strength, in particular, was enhanced, with the Young's modulus, E, increasing from 0.1 MPa for uncrosslinked CCNF foams to 50.8 ± 8 MPa at 10 wt% glyoxal (Fig. 6a). It was apparent from the compressive load versus compressive extension profiles that compression was occurring in two stages; initial compression at lower loading was assigned to the collapse of the large porous network and a second stage, at much higher loads, reflected the bulk of the material under compression (Fig. 6b). E was determined from the stress/strain at 30% deformation. Although the highest E value occurred in the most crosslinked foams, these tended to fracture more quickly, reflecting a more brittle structure; hence foams up to 5 wt% were used in cell culture studies (Fig. S20–S27, ESI†).
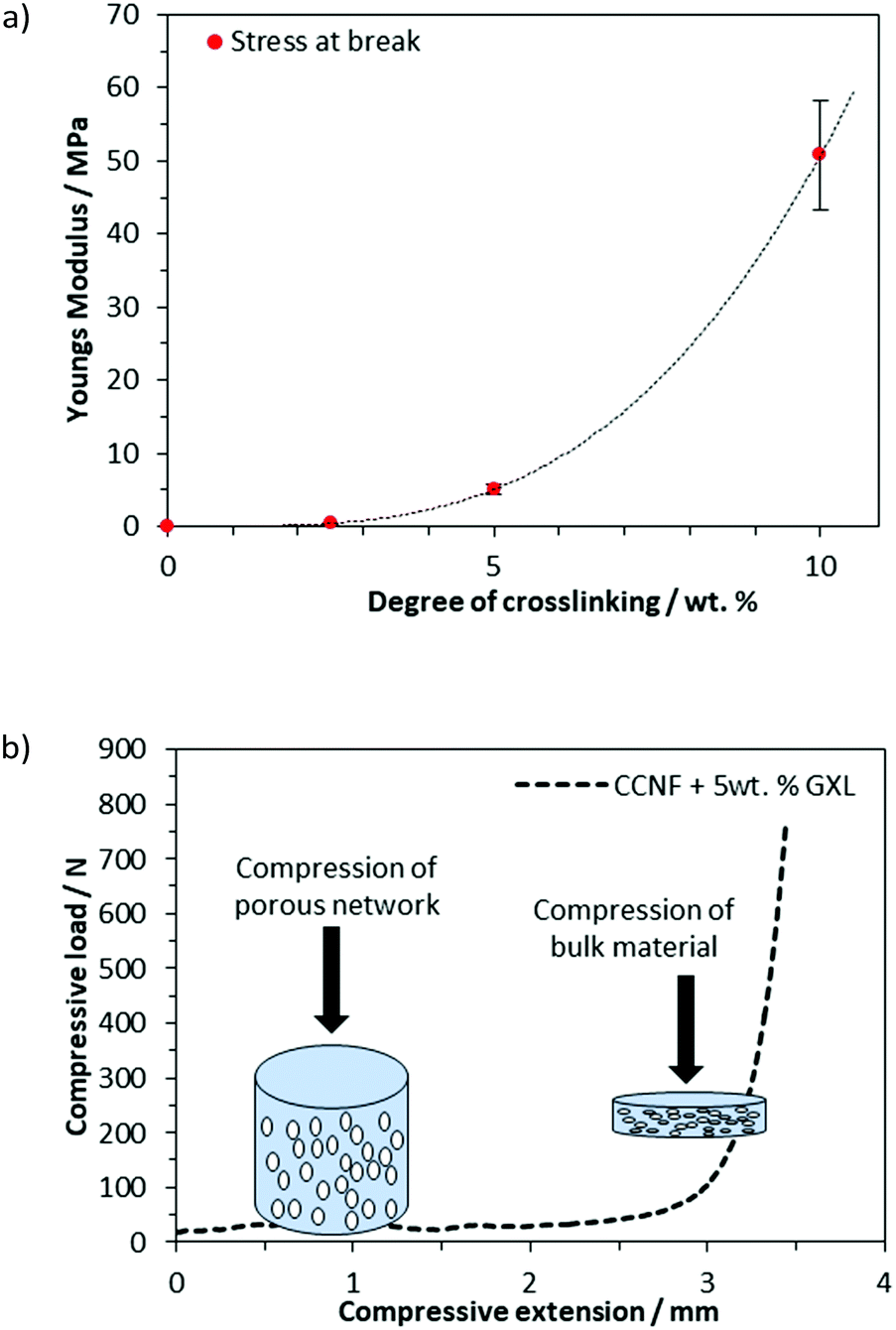 | ||
| Fig. 6 (a) The Young's modulus, determined from the stress/strain at 30% deformation, showed that crosslinking the CCNF foams had a significant influence on the robustness of the materials. (b) Compressive load versus compressive extension graph showing the two phases of compression: first, compression of the porous network, followed by the greater load required to compress the bulk material (illustrated for CCNF XL). | ||
Such foams represented a “sweet spot”: materials were robust enough to handle and survive the manipulations required for cell studies (e.g., aspiration of media), yet were not rendered too brittle to be of utility: CCNF foams with a moderate level of crosslinking (exposed to 2.5 and 5 wt% glyoxal, yielding DXL of 1.18 and 3.04 mol%) remained intact in PBS and only disintegrated if subjected to very vigorous agitation. This enabled the foams to be handled without breaking during cell viability studies.
Cell response to 3D scaffolds
We have previously investigated the attachment of cells on the surface of CC films,68 but here the presence of a “skin” on the surface of the regenerated CC films (Fig. S17, ESI†) rendered the porous internal structure beneath inaccessible to cells, so that cell studies were confined to the foamed scaffolds produced by directional freezing, lyophilisation and crosslinking.The cell viability was determined by assessing cell metabolism using an MTT assay, which showed that MG-63 cells were viable on all 3D CCNF scaffolds after 7 days incubation at 37 °C in 5% CO2 (Fig. 7a).
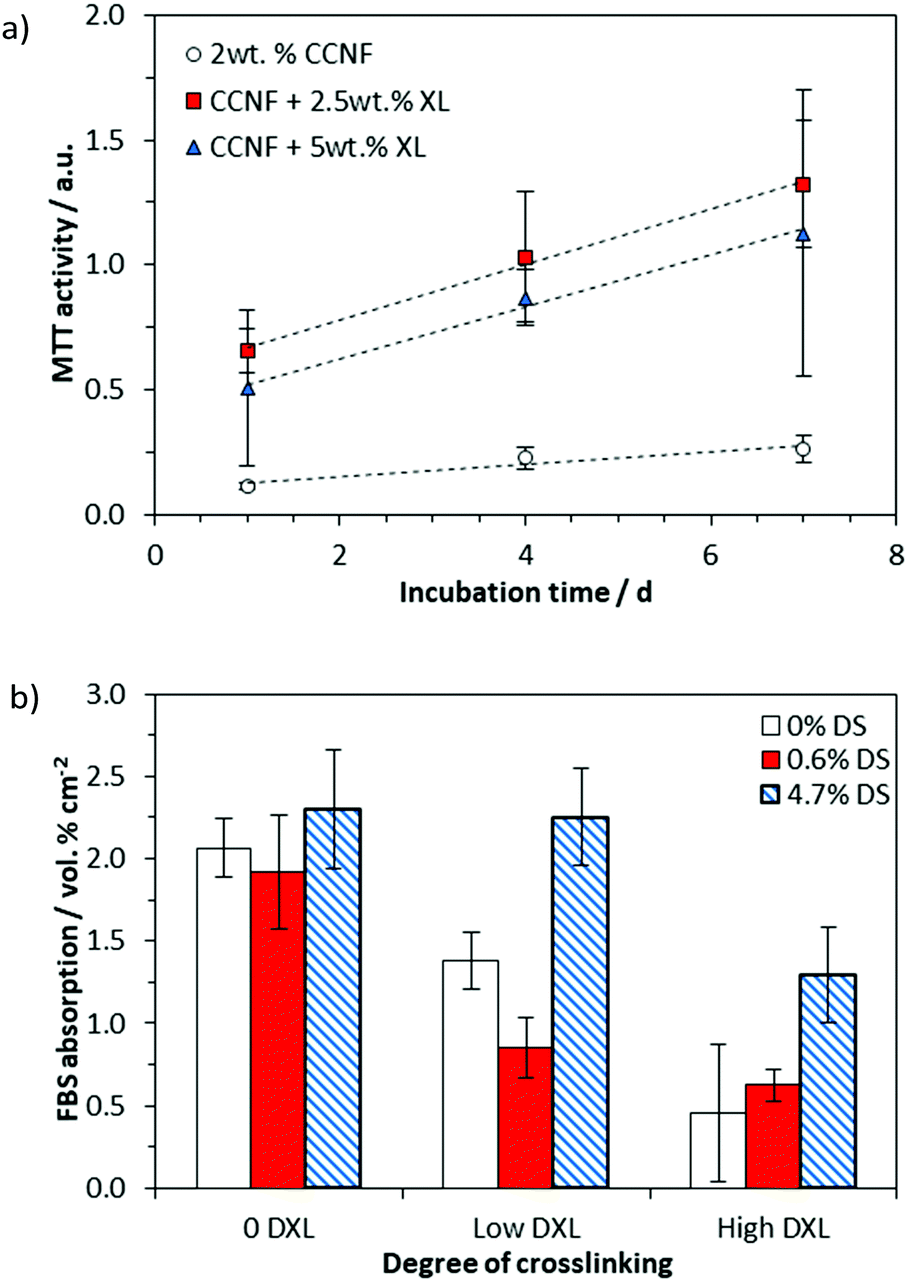 | ||
| Fig. 7 (a) Cell viability on 2 wt% CCNF 3D scaffolds after 1, 4 and 7 days incubation at 37 °C in 5% CO2. Tissue culture plastic was used as the control and the sample values have had the material control subtracted. It is evident that glyoxalation provides the foams with the structural support to use as 3D scaffolds as well as enhancing cell viability. (b) HPLC analysis of the quantity of FBS, specifically bovine serum albumin (BSA), adsorbed onto the scaffold surface after 24 h incubation at 37 °C in 5% CO2. By comparison TCP bore 0.95 vol% cm−2 BSA absorbed onto the surface. | ||
We have reported previously that cell elongation can be promoted on crosslinked 2D CC scaffolds, an effect that was ascribed to increased elastic and shear moduli.63 Thus, the enhanced level of cell viability on CCNF XL materials was expected. Nonetheless, to test whether or not the enhancement could be ascribed to enhanced sorption of proteins (present in FBS) to the cationic surface, protein adsorption studies were conducted. The quantity of protein (specifically bovine serum albumin, BSA) adsorbed to the scaffolds after incubation with cell culture medium for 24 h at 37 °C in 5% CO2 was quantified by HPLC analysis (Fig. 7b and Fig. S28–S30, ESI†). While increased cationisation (increased DS) enhances protein absorption, there is no such correlation with crosslinking, suggesting that the enhanced cell viability on CCNF XL resulted from the structural properties of the scaffold (SEM images of growing cells on the 3D scaffolds are provided as Fig. S28, ESI†). Not surprisingly, measured mechanical strength decreased on hydration of scaffolds, but there was no evidence of further degradation after 7 days incubation (Fig. S24–S27, ESI†).
It has been reported previously that the mechanical properties of a cell scaffold affect the mechanosensitive cell response, although this can be dependent on the cell type.69 For example fibroblasts, smooth muscle cells and epithelial cells spread more and develop larger focal adhesions and actin stress fibres on stiffer scaffolds than on compliant ones.70 Cells receive mechanical feedback from the substrate to which they adhere, even in the absence of externally applied forces,71 as they attempt to deform the substrate. As cells elongate on stiff scaffolds they experience a higher stress than on softer more compliant scaffolds, which promotes the assembly of the cytoskeleton into actin stress fibres and focal adhesions, and subsequently triggers signalling cascades that ultimately promote cell expansion.72
Thus, we suggest that the cells adhered to the CCNF XL scaffolds due to the cationic surface charge, which promotes the initial phase of cell attachment, facilitating cell binding through electrostatic interactions and possibly aided by enhanced FBS sorption, particularly in the second phase of cell adhesion (Fig. 8). Once attached, the stiffer CCNF XL scaffolds provided the cells with a larger feedback force, promoting mechanotransduction signalling within by activating the Rho GTPase family to promote cell spreading and migration in the third phase of cell adhesion.73
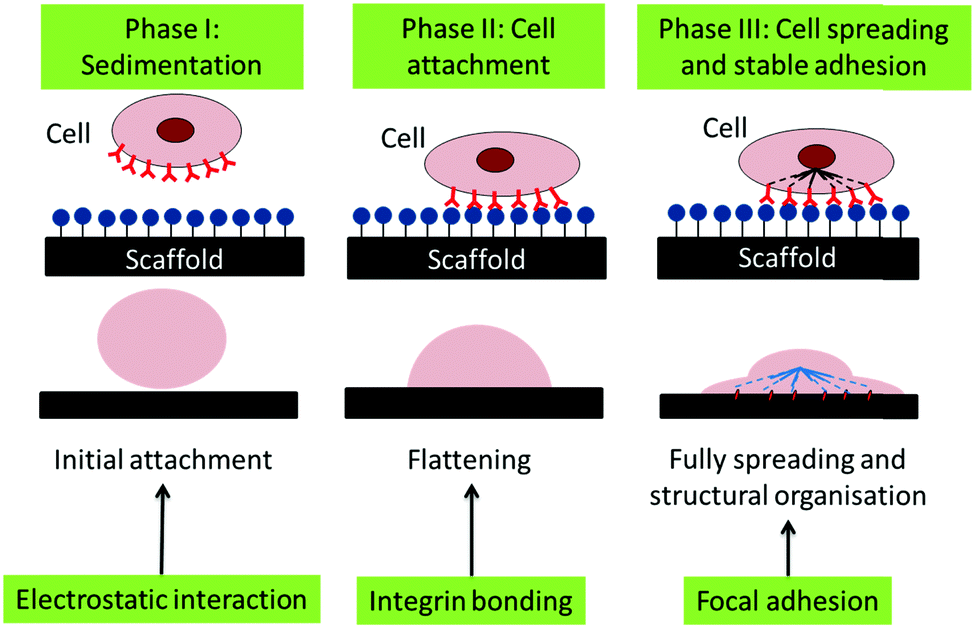 | ||
| Fig. 8 Schematic of the phases of passive in vitro cell adhesion. Phase I: sedimentation of cells can be enhanced through electrostatic interactions, Phase II: cell attachment is facilitated through the formation of integrin binding sites between the cell and scaffold and Phase III: cell spreading occurs through focal adhesions the interactions with the force experienced by the cell actin cytoskeleton via mechanotransduction.73 | ||
These “bottom-up” scaffolds complement the current “top-down” decellularised plant tissue scaffolds available and offer a wide range of potential cell culture applications, potentially promoting the differentiation of tissue on the variable stiffness CCNF foams. Furthermore, the porosity and vascular structure of the foams facilitates mass transfer of O2 and nutrients to the cells and the removal of waste products, required to maintain viable cultures, e.g. in and on larger 3D scaffolds shaped to mimic organs or to match specific damaged areas of tissue.
Conclusion
Robust 3D modified cellulose scaffolds with exquisite tuneable structure in the form of foams, with meso and macro scale pores were prepared by a “bottom-up” approach. Directionally freezing CCNF dispersions and emulsions, followed by lyophilisation, was used to produce a range of scaffolds with a variety of internal architectures from aligned micro channels with smooth walls, mimicking vascularised tissue, and pumice-like wall textures, reminiscent of porous bone. Directional freezing is a facile and low cost method to introduce porosity into tissue engineering scaffold materials and this process would be attractive for (relatively) large scale manufacturing as it has fewer steps than previous methodology.To overcome the poor structural integrity of the CCNF foams, crosslinking by glyoxalisation was used. This also enhanced the Young's modulus and yielded scaffold materials suitable for cell culture as demonstrated by the improved viability of MG-63 grown on the scaffolds. It is postulated that the more porous structures arising, allowed for enhanced mass transfer of nutrients and gases into the scaffolds and removal of cell waste products, beneficial for cell growth. Porosity was characterised at all length scales by a combination of SEM image analysis, NMR cryoporometry and T2 relaxation studies.
These scaffolds can complement the library of 3D porous materials derived from decellularised plant tissue, yet have the advantage over the “top-down” scaffolds, which usually require several treatments prior to application and can be limited in structure and shape, whereas, a variety of bulk 3D shapes can be prepared through process moulding the frozen hydrogel prior to lyophilisation and crosslinking.
These “bottom-up” scaffolds derived from dispersed CCNF require minimal modification, use known and scalable chemistries, and can be easily processed through the use of moulding techniques to create the desired 3D constructs, with modulated vascularity and wall porosity.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge funding from the EPSRC Centre for Doctoral Training in Sustainable Chemical Technology (EP/L016354/1), in the form of a PhD studentship for JCC. SEM images were obtained at the Microscopy and Analysis Suite in Bath University with the assistance of Ursula Potter and the authors thank John Lowe for his assistance with NMR spectroscopy at the University of Bath.References
- J. Colaneri, Am. J. Transplant., 2016, 16, 8–10 CrossRef.
- T. Dvir, B. P. Timko, D. S. Kohane and R. Langer, Nat. Nanotechnol., 2011, 6, 13–22 CrossRef CAS.
- J. K. Kular, S. Basu and R. I. Sharma, J. Tissue Eng., 2014, 5, 1–17 Search PubMed.
- D. Anton, H. Burckel, E. Josset and G. Noel, Int. J. Mol. Sci., 2015, 16, 5517–5527 CrossRef.
- W. Sekine, Y. Haraguchi, T. Shimizu, A. Umezawa and T. Okano, J. Biochips Tissue Chips, 2011, S1(007), 1–9 Search PubMed.
- P. Carmeliet and R. K. Jain, Nature, 2000, 407, 249–257 CrossRef CAS.
- J. Rouwkema, B. F. J. M. Koopman, C. A. V. Blitterswijk, W. J. A. Dhert and J. Malda, Biotechnol. Genet. Eng. Rev., 2009, 26, 163–178 CrossRef.
- J. R. Gershlak, S. Hernandez, G. Fontana, L. R. Perreault, K. J. Hansen, S. A. Larson, B. Y. K. Binder, D. M. Dolivo, T. Yang, T. Dominko, M. W. Rolle, P. J. Weathers, F. Medina-Bolivar, C. L. Cramer, W. L. Murphy and G. R. Gaudette, Biomaterials, 2017, 125, 13–22 CrossRef CAS.
- J. Mitra, G. Tripathi, A. Sharma and B. Basu, RSC Adv., 2013, 3, 11073–11094 RSC.
- F. A. Saleh and P. G. Genever, Cytotherapy, 2011, 13, 903–912 CrossRef.
- F. J. O’Brien, B. A. Harley, M. A. Waller, I. V. Yannas, L. J. Gibson and P. J. Prendergast, Technol. Heal. Care, 2007, 15, 3–17 Search PubMed.
- F. J. O’Brien, B. A. Harley, I. V. Yannas and L. J. Gibson, Biomaterials, 2005, 26, 433–441 CrossRef.
- Z. Ma, M. Kotaki, R. Inai and S. Ramakrishna, Tissue Eng., 2005, 11, 101–109 CrossRef CAS.
- P. M. Crapo, T. W. Gilbert and S. F. Badylak, Biomaterials, 2011, 32, 3233–3243 CrossRef CAS.
- T. W. Gilbert, T. L. Sellaro and S. F. Badylak, Biomaterials, 2006, 27, 3675–3683 CAS.
- J. J. Song and H. C. Ott, Trends Mol. Med., 2011, 17, 424–432 CrossRef CAS PubMed.
- D. J. Modulevsky, C. Lefebvre, K. Haase, Z. Al-Rekabi and A. E. Pelling, PLoS One, 2014, 9, e97835 CrossRef.
- J. Doshi and D. H. Reneker, J. Electrost., 1995, 35, 151–160 CrossRef CAS.
- H. S. Yoo, T. G. Kim and T. G. Park, Adv. Drug Delivery Rev., 2009, 61, 1033–1042 CrossRef CAS.
- H. W. Kim, H. S. Yu and H. H. Lee, J. Biomed. Mater. Res., Part A, 2008, 87, 25–32 CrossRef.
- B. Derby, Science, 2012, 338, 921–926 CrossRef CAS.
- K. R. Hixon, T. Lu and S. A. Sell, Acta Biomater., 2017, 62, 29–41 CrossRef CAS PubMed.
- C. R. Almeida, T. Serra, M. I. Oliveira, J. A. Planell, M. A. Barbosa and M. Navarro, Acta Biomater., 2014, 10, 613–622 CrossRef CAS.
- J. G. Torres-Rendon, T. Femmer, L. De Laporte, T. Tigges, K. Rahimi, F. Gremse, S. Zafarnia, W. Lederle, S. Ifuku, M. Wessling, J. G. Hardy and A. Walther, Adv. Mater., 2015, 27, 2989–2995 CrossRef CAS.
- M. Costantini, C. Colosi, P. Mozetic, J. Jaroszewicz, A. Tosato, A. Rainer, M. Trombetta, W. Święszkowski, M. Dentini and A. Barbetta, Mater. Sci. Eng., C, 2016, 62, 668–677 CrossRef CAS.
- A. Barbetta, A. Carrino, M. Costantini and M. Dentini, Soft Matter, 2010, 6, 5213–5224 RSC.
- C. Colosi, M. Costantini, A. Barbetta, R. Pecci, R. Bedini and M. Dentini, Langmuir, 2013, 29, 82–91 CrossRef CAS.
- M. Costantini, C. Colosi, J. Guzowski, A. Barbetta, J. Jaroszewicz, W. Święszkowski, M. Dentini and P. Garstecki, J. Mater. Chem. B, 2014, 2, 2290–2300 RSC.
- M. Costantini, C. Colosi, J. Jaroszewicz, A. Tosato, W. Święszkowski, M. Dentini, P. Garstecki and A. Barbetta, ACS Appl. Mater. Interfaces, 2015, 7, 23660–23671 CrossRef CAS.
- L. Qian and H. Zhang, J. Chem. Technol. Biotechnol., 2011, 86, 172–184 CrossRef CAS.
- C. M. Murphy and F. J. O’Brien, Cell Adhes. Migr., 2010, 4, 377–381 CrossRef.
- H. Zhang and A. I. Cooper, Adv. Mater., 2007, 19, 1529–1533 CrossRef CAS.
- S. Deville, E. Saiz and A. P. Tomsia, Biomaterials, 2006, 27, 5480–5489 CrossRef CAS.
- B. B. Mandal and S. C. Kundu, Biomaterials, 2009, 30, 2956–2965 CrossRef CAS PubMed.
- X. Wu, Y. Liu, X. Li, P. Wen, Y. Zhang, Y. Long, X. Wang, Y. Guo, F. Xing and J. Gao, Acta Biomater., 2010, 6, 1167–1177 CrossRef CAS PubMed.
- A. Autissier, C. Le Visage, C. Pouzet, F. Chaubet and D. Letourneur, Acta Biomater., 2010, 6, 3640–3648 CrossRef CAS PubMed.
- S. Reed, G. Lau, B. Delattre, D. D. Lopez, A. P. Tomsia and B. M. Wu, Biofabrication, 2016, 8, 015003 CrossRef PubMed.
- G. Li, A. G. Nandgaonkar, Y. Habibi, W. E. Krause, Q. Wei and L. A. Lucia, RSC Adv., 2017, 7, 13678–13688 RSC.
- S. Flauder, T. Heinze and F. A. Müller, Cellulose, 2014, 21, 97–103 CrossRef CAS.
- M. Bhattacharya, M. M. Malinen, P. Lauren, Y.-R. Lou, S. W. Kuisma, L. Kanninen, M. Lille, A. Corlu, C. GuGuen-Guillouzo, O. Ikkala, A. Laukkanen, A. Urtti and M. Yliperttula, J. Controlled Release, 2012, 164, 291–298 CrossRef CAS.
- B. Wang, X. Lv, S. Chen, Z. Li, J. Yao, X. Peng, C. Feng, Y. Xu and H. Wang, Carbohydr. Polym., 2018, 181, 948–956 CrossRef CAS.
- E. Entcheva, H. Bien, L. Yin, C.-Y. Chung, M. Farrell and Y. Kostov, Biomaterials, 2004, 25, 5753–5762 CrossRef CAS.
- M. Zaborowska, A. Bodin, H. Bäckdahl, J. Popp, A. Goldstein and P. Gatenholm, Acta Biomater., 2010, 6, 2540–2547 CrossRef CAS.
- D. Klemm, B. Heublein, H.-P. P. Fink and A. Bohn, Angew. Chem., Int. Ed., 2005, 44, 3358–3393 CrossRef CAS PubMed.
- D. J. Modulevsky, C. M. Cuerrier and A. E. Pelling, PLoS One, 2016, 11, 1–19 CrossRef PubMed.
- A. Sannino, C. Demitri and M. Madaghiele, Materials, 2009, 2, 353–373 CrossRef CAS.
- A. Svensson, E. Nicklasson, T. Harrah, B. Panilaitis, D. L. Kaplan, M. Brittberg and P. Gatenholm, Biomaterials, 2005, 26, 419–431 CrossRef CAS PubMed.
- F. G. Torres, S. Commeaux and O. P. Troncoso, J. Funct. Biomater., 2012, 3, 864–878 CrossRef CAS PubMed.
- J. C. Courtenay, M. A. Johns, F. Galembeck, C. Deneke, E. M. Lanzoni, C. A. Costa, J. L. Scott and R. I. Sharma, Cellulose, 2017, 24, 253–267 CrossRef CAS.
- J. C. Courtenay, S. M. Ramalhete, W. J. Skuze, R. Soni, Y. Z. Khimyak, K. J. Edler and J. L. Scott, Soft Matter, 2018, 14, 255–263 RSC.
- C. Schramm and B. Rinderer, Anal. Chem., 2000, 72, 5829–5833 CrossRef CAS PubMed.
- M. A. Johns, A. Bernardes, E. R. De Azevêdo, F. E. G. Guimarães, J. P. Lowe, E. M. Gale, I. Polikarpov, J. L. Scott and R. I. Sharma, J. Mater. Chem. B, 2017, 5, 3879–3887 RSC.
- S. Berger and S. Braun, 200 and more NMR experiments: a practical course, Wiley-VCH Weinheim, Weinheim, 2004 Search PubMed.
- H. Y. Carr and E. M. Purcell, Phys. Rev., 1954, 94, 630–638 CrossRef CAS.
- S. Meiboom and D. Gill, Rev. Sci. Instrum., 1958, 29, 688–691 CrossRef CAS.
- Å. Östlund, A. Idström, C. Olsson, P. T. Larsson and L. Nordstierna, Cellulose, 2013, 20, 1657–1667 CrossRef.
- C. Boissier, F. Feidt and L. Nordstierna, J. Pharm. Sci., 2012, 101, 2512–2522 CrossRef CAS PubMed.
- G. C. Borgia, R. J. S. Brown and P. Fantazzini, J. Magn. Reson., 2000, 147, 273–285 CrossRef CAS PubMed.
- S. W. Provencher, Comput. Phys. Commun., 1982, 27, 213–227 CrossRef.
- K. R. Brownstein and C. E. Tarr, Phys. Rev. A: At., Mol., Opt. Phys., 1979, 19, 2446–2453 CrossRef.
- D. Capitani, V. Di Tullio and N. Proietti, Prog. Nucl. Magn. Reson. Spectrosc., 2012, 64, 29–69 CrossRef CAS PubMed.
- C. Zhang, P. Li, Y. Zhang, F. Lu, W. Li, H. Kang, J.-F. Xiang, Y. Huang and R. Liu, Polymer, 2016, 98, 237–243 CrossRef CAS.
- J. C. Courtenay, C. Deneke, E. M. Lanzoni, C. A. Costa, Y. Bae, J. L. Scott and R. I. Sharma, Cellulose, 2018, 25, 925–940 CrossRef CAS.
- F. Quero, M. Nogi, K. Y. Lee, G. Vanden Poel, A. Bismarck, A. Mantalaris, H. Yano and S. J. Eichhorn, ACS Appl. Mater. Interfaces, 2011, 3, 490–499 CrossRef CAS PubMed.
- R. G. Wells, Hepatology, 2008, 47, 1394–1400 CrossRef CAS PubMed.
- E. A. Klein, L. Yin, D. Kothapalli, P. Castagnino, F. J. Byfield, T. Xu, I. Levental, E. Hawthorne, P. A. Janmey and R. K. Assoian, Curr. Biol., 2009, 19, 1511–1518 CrossRef CAS PubMed.
- J. C. Courtenay, Y. Jin, J. Schmitt, K. J. Edler and J. L. Scott, personal communication.
- M. A. Johns, Y. Bae, F. E. G. Guimarães, E. M. Lanzoni, C. A. R. Costa, P. M. Murray, C. Deneke, F. Galembeck, J. L. Scott and R. I. Sharma, ACS Omega, 2018, 3, 937–945 CrossRef PubMed.
- P. C. Georges and P. A. Janmey, J. Appl. Physiol., 2005, 98, 1547–1553 CrossRef PubMed.
- J. D. Humphrey, E. R. Dufresne and M. A. Schwartz, Nat. Rev. Mol. Cell Biol., 2014, 15, 802–812 CrossRef CAS PubMed.
- R. G. M. Breuls, T. U. Jiya and T. H. Smit, Open Orthop. J., 2008, 2, 103–109 CrossRef PubMed.
- M. Tamada, M. P. Sheetz and Y. Sawada, Dev. Cell, 2004, 7, 709–718 CrossRef CAS PubMed.
- A. A. Khalili and M. R. Ahmad, Int. J. Mol. Sci., 2015, 16, 18149–18184 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Further experimental details for material preparation, modification and characterisation. See DOI: 10.1039/c8tb02482k |
| ‡ Current address: FiberLean® Technologies Ltd, Par Moor Centre, Par Moor Rd, Par PL24 2SQ, UK. |
| This journal is © The Royal Society of Chemistry 2019 |