
A pH-responsive self-healing hydrogel based on multivalent coordination of Ni2+ with polyhistidine-terminated PEG and IDA-modified oligochitosan†
Quan
Tang
,
Dinglei
Zhao
,
Haiyang
Yang
 *,
Lijun
Wang
and
Xingyuan
Zhang
*
*,
Lijun
Wang
and
Xingyuan
Zhang
*
CAS Key Laboratory of Soft Matter Chemistry, School of Chemistry and Materials Science, University of Science and Technology of China, Hefei, 230026, P. R. China. E-mail: yhy@ustc.edu.cn; zxym@ustc.edu.cn
First published on 23rd October 2018
Abstract
Metal coordination hydrogels have drawn intensive attention in controlled drug release due to their facile incorporation of stimuli and dynamic self-healing properties. However, many metal coordination hydrogels which are responsive to mild physiologically relevant stimuli are restricted by their poor stability in non-triggering environments. Here, we reported a new strategy for enhancing the neutral stability of a pH-responsive self-healing hydrogel based on multivalent metal coordination without loss of its responsiveness. The hydrogel was formed by multivalent coordination of Ni2+ with polyhistidine (PHis) and multiple iminodiacetic acid (IDA) ligands which were modified on the terminal groups of polyethylene glycol (PEG) and the side groups of oligochitosan (OChi), respectively. By incorporating multivalent coordination, the hydrogel maintains its self-healing character and weak acid responsiveness; it can also be used as an injectable hydrogel by injecting it into a neutral environment. Through varying the PHis![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :IDA ratio, the mechanical strength and dynamic relaxation of the hydrogel can be regulated conveniently. More importantly, the hydrogels with multivalent coordination can remain stable and integral in neutral buffer (pH 7.4); meanwhile, they dissolve quickly in a weak acid environment (pH 5.5), which is similar to some tissue disease conditions. In contrast, the control hydrogel without multivalent coordination disintegrated within 4 hours in the neutral non-triggering buffer. Because the hydrogel consists of large amounts of coordinative Ni2+ ions, it naturally possesses relatively high affinity to molecules containing polyhistidine motifs. Thus, pH-tuned controlled release of a model molecule, rhodamine-modified polyhistidine, was accomplished, with limited neutral leakage and quick release in weak acid. These results indicate potential applications for this multivalent coordination hydrogel in the controlled release field.
:IDA ratio, the mechanical strength and dynamic relaxation of the hydrogel can be regulated conveniently. More importantly, the hydrogels with multivalent coordination can remain stable and integral in neutral buffer (pH 7.4); meanwhile, they dissolve quickly in a weak acid environment (pH 5.5), which is similar to some tissue disease conditions. In contrast, the control hydrogel without multivalent coordination disintegrated within 4 hours in the neutral non-triggering buffer. Because the hydrogel consists of large amounts of coordinative Ni2+ ions, it naturally possesses relatively high affinity to molecules containing polyhistidine motifs. Thus, pH-tuned controlled release of a model molecule, rhodamine-modified polyhistidine, was accomplished, with limited neutral leakage and quick release in weak acid. These results indicate potential applications for this multivalent coordination hydrogel in the controlled release field.
Introduction
Hydrogels, which have characteristics of porous structures and biocompatibility, have been widely applied in both sustained and controlled drug release systems for decades.1–3 Compared to stable covalent crosslinking hydrogels, physical hydrogels crosslinked by non-covalent interactions, including hydrogen bonds,4–6 hydrophobic interactions,7,8 supramolecular interactions9 and metal coordination interactions,10,11 have drawn special attention due to their dynamic properties.12 These dynamic properties, including self-healing, prolong the lifetime of the hydrogel inside the body and decrease the damage to the human body if the hydrogel is subjected to a strong external force.13–15 Also, some physical hydrogels can form instantly when they are transferred to physiological conditions, such as body temperature and pH.16,17 This property can be termed “injectable hydrogels”, which can decrease injury to patients by a minimal invasion without surgery. For controlled release systems, physical hydrogels with weak physical interactions can be easily endowed with responsiveness.18,19 Some responsiveness can be related to abnormal physiological environments, including the pH conditions of inflammation sites (pH 5.4, compared to 7.4 in normal tissues),20 which can trigger disease-controlled drug release and increase the therapeutic effects.21–23 Upon triggering by environmental stimuli, the vanishing of the responsive physical interaction in physical hydrogels will induce a gel–sol transition to decompose the hydrogel to solution.24 The gel–sol transition ensures not only convenient payload encapsulation in the hydrogel, but also full clearance of the hydrogel frameworks after release.25 Although physiological environment-responsive physical hydrogels show great benefits, the limited stability of the physical hydrogels is further weakened by the incorporation of these physiological environmental responses.26,27 The narrow gap between the environment of triggering disease tissues and non-triggering normal tissues complicates the regulation of the stability of these hydrogels in normal conditions and their responsiveness in abnormal conditions.28 Generally, a method to enhance the stability of physiologically relevant environmentally responsive hydrogels may be helpful to solve this problem.As a promising weak acid environment-responsive moiety, polyhistidine (PHis), which has a pKa of approximately 6.5, has been applied in many fields related to biomedicine due to the biocompatibility of this polypeptide.29–31 Also, PHis shows metal ion (Ni2+, Cu2+, Co2+, etc.) coordination ability to itself or to multidentate ligands, such as IDA (iminodiacetic acid) and NTA (nitrilotriacetic acid).32 This chemistry was widely verified by incorporating PHis segments into recombinant proteins for purification,33 labelling,34 encapsulation35 and immobilization.36 The metal coordination structures endow the hydrogels with desirable properties, such as enhanced mechanical properties, self-healing and injectability, by the exchangeable and reversible nature of the relatively strong coordination bonds.37 Because the pH responsiveness and metal coordination ability of PHis arise from the same nitrogen atoms on the imidazole rings of PHis, a competitive relationship between the two abilities can be developed to pH-tune the metal coordination and pH responsiveness to the hydrogel.38 Thus, this type of hydrogel meets all the requirements described above. However, due to dynamic connections inside the hydrogel network, some responsive metal coordination hydrogels show low stability in water and disintegrate within several hours in the absence of stimuli, which limits their applications in drug release.39
To enhance the stability of metal coordination hydrogels in neutral water without changing the pH responsiveness, some modifications of the metal coordination structures should be performed. After numerous studies on the physical properties of metal coordination gels, it was found that the swelling and stability of the gel tend to be determined by the dissociation kinetics of the coordination structures, not the bond energy or thermodynamic equilibrium.40–42 For coordination hydrogels formed by bipyridyl-branched polyoxazoline and different metal ions, the stability of the gels exactly follows the reverse order of the dissociation rate; this order is Ru2+ > Fe2+ > Ni2+ for stability.41 Moreover, multivalent interactions, which comprise connection structures through multiple ligands and multiple binding sites or receptors, appear to be an effective method to decrease the dissociation rates of interactions, which plays an important role in biological events.43 The chemical linkage of the multiple ligands restricts the movement of the dissociated ligand, increases its effective concentration and, thus, decreases the total dissociation rate by enhancing its rebinding to receptors, when some of the ligands in the multivalent interactions partially dissociate.44 Therefore, a promising method to enhance the stability of metal coordination hydrogels by multivalent metal coordination is revealed. Some previous studies on metal coordination interactions between the PHis segment and mono- to multiple chemically linked NTA structures have proven the effects of multivalent coordination on decreasing the dissociation rate of the coordinated complexes.45,46 Also, because this method enhances stability without changing the individual structure around each Ni2+, the final weak acid responsiveness may not be seriously influenced, which is an exceptional advantage for the multivalent strategy45 and is different from some other methods.47
Here, we report a pH-responsive hydrogel with enhanced neutral stability based on multivalent Ni2+ coordination between its ligand PHis and IDAs. As shown in Scheme 1a, an IDA-modified oligochitosan (OChi-IDA) serves as the crosslinker and a PHis-b-PEG-b-PHis triblock copolymer (PEG-PHis) serves as the soft linker; these form a coordination hydrogel when mixed in a proper ratio with Ni2+ ions and by tuning the pH to neutral. Due to the multivalent coordination between multiple IDA groups and a PHis segment, the hydrogels showed prolonged relaxation times, which is related to decreased dissociation rates and results in enhanced stability.38 Moreover, the multivalence of the coordination in this hydrogel can be simply tuned by the PHis:IDA molar ratio and the length of PHis, which further leads to regulation of its mechanical properties and stability in the neutral environment. For the multivalent coordination hydrogels, the pH responsiveness and self-healing properties are maintained. The necessity of multivalence was also verified by an interchange of the two polymers as a control, in which PHis-grafted oligochitosan (OChi-g-PHis) and IDA-modified PEG (PEG-IDA) were used to prepare coordination hydrogels (Scheme 1b). These hydrogels could not undergo multivalent metal coordination and showed short relaxation times and very poor neutral stability; they decomposed rapidly even in non-triggering neutral buffer. Compared to our previous work, in which we exploited multivalent coordination between PHis segments to construct a metal coordination hydrogel, this work involving a donor–acceptor structure between the PHis segment and multiple IDAs can regulate the mechanical properties and stability of the hydrogel in a more facile and controllable manner, which facilitates adjustments to balance the pH responsiveness and neutral stability.37 Due to its pH responsiveness and enhanced neutral stability, we believe this multivalent coordination hydrogel is a promising candidate for application in stimuli-responsive drug release hydrogels.
 | ||
| Scheme 1 Schematic of the metal coordination hydrogel based on PHis-(Ni2+-IDA)n complexes. (a) The composition of the OChi-IDA/PEG-PHis series coordination hydrogel and its proposed hydrogel structure by multivalent coordination. (b) The composition of the OChi-g-PHis/PEG-IDA control coordination hydrogel and its proposed hydrogel structure by monovalent coordination. | ||
Experimental
Materials
Oligochitosan, obtained by enzymatic hydrolysis of chitosan, was purchased from Zhejiang Golden-Shell Pharmaceutical Co., Ltd; the oligochitosan had a molecular weight of less than 1500 Da and a degree of deacetylation of 90%. Polyethylene glycol (PEG) with a molecular weight of 4000 Da was purchased from Sinopharm Chemical Reagent Co. Ltd. Triethylamine (TEA) and dimethyl sulfoxide (DMSO) were purchased from Sinopharm Reagents and dried with calcium hydride (CaH2) before usage. All other chemicals were purchased from commercial sources and were used as received without further purification unless otherwise specified.Characterization
All 1H NMR spectra were recorded on a Bruker AV300 NMR 300 MHz spectrometer using CDCl3 or D2O as the solvent. The molecular weights and distributions (Mw/Mn) were determined by a gel permeation chromatograph (GPC) equipped with a G1310B Iso. pump, a G1316A PLgel column, and a G1362A differential refractive index detector. The eluent was DMF with 1 g L−1 LiBr at a flow rate of 1.0 mL min−1. The rheological properties of all the prepared hydrogels were studied on a rheometer (TA Instruments, AR-G2) with a stainless-steel cone-plate upper geometry (40 mm in diameter, 1° angle). Frequency-sweep measurements were performed in constant-strain (1%) mode over the frequency range of 0.1 to 100 rad s−1 at 25 °C. UV-Vis spectra were collected on a Shimadzu UV-2401PC instrument. The morphologies of the hydrogels before and after swelling were observed using field emission SEM (Zeiss Supra 40 and Zeiss Merlin compact) at an acceleration voltage of 5 kV. The hydrogels were freeze-dried in advance, and the samples were sputter-coated with gold before observation.Synthesis of 2-(iminodiacetic acid)-ethanethiol (IDA-SH)
The synthesis of IDA-SH included three steps. The procedures and characterization are shown in Fig. S1 (ESI†). 2-(Triphenylmethylether)ethylamine was synthesized according to a previously reported literature procedure.48Synthesis of IDA modified oligochitosan (OChi-IDA)
The modification of the oligochitosan involved two steps, including modification of allyl groups onto the amino groups of oligochitosan and a thiol–ene reaction between IDA-SH and the allyl groups on oligochitosan. The former step was reported previously.49 Also, the modification efficiency was approximately 75% as calculated from the 1H NMR spectra.Synthesis of PHis-b-PEG-b-PHis triblock copolymer (PEG-PHis)
The triblock copolymer was synthesized by polymerizing histidine monomer (PhO-His(Trt)-OH) using amino-terminated PEG4k (PEG4k-NH2) as the initiator. The polymerization method was previously reported by us37 and the PEG4k-NH2 macro-initiator was synthesized according to the literature.50Synthesis of IDA modified PEG (PEG-IDA)
The synthesis of PEG-IDA was based on a thiol–ene click reaction between allyl-modified PEG4k and IDA-SH. The allyl-modified PEG was prepared according to the general method reported in the literature.51 The thiol–ene reaction was similar to that used in the synthesis of OChi-IDA.Synthesis of PHis-grafted oligochitosan (OChi-g-PHis)
OChi-g-PHis was synthesized by direct polymerization of PhO-His(Trt)-OH on the pendant amino groups of oligochitosan. The oligochitosan was soluble in DMSO;52 therefore, the polymerization was conducted in dry DMSO. Specifically, the initiator oligochitosan and monomer PhO-His(Trt)-OH (5% excess to the targeted degree of polymerization) were placed in a round-bottom ampere bottle with toluene, which was evaporated in vacuum to azeotropically dehydrate the initiator and monomer. After that, dry DMSO was injected into the reaction bottle, followed by sealing and stirring at 60 °C for 36 h. The reaction mixture was poured into ethyl ether, and the precipitate was subjected to the former process again after being dissolved in THF/DMF = 1/1. The dried product was sequentially deprotected in trifluoroacetic acid with 5 vol% triethylsilane and 2.5 vol% water. The reaction mixture was precipitated in excess diethyl ether and the precipitate was subjected to the former process again after being dissolved in THF/DMF = 1/1. After centrifugation, the precipitate was further dialysed in deionized water for 2 days. The final product (78%) was obtained by lyophilization.Synthesis of rhodamine 6G-labelled PHis (Rh6G-PHis)
The amino-functioned rhodamine 6G (Rh6G-NH2) was synthesized according to the literature.53 Using Rh6G-NH2 as the initiator to polymerize the histidine monomer, Rh6G-PHis was obtained. The detailed procedure is similar to that of the previous PHis synthesis.Determination of the molecular weight of oligochitosan
The molecular weight of the purchased oligochitosan was measured by end group analysis. This method was reported in the literature.54 Here, we followed this method with a slight modification. To put it simply, this method determines the end group content of a pre-weighed oligochitosan solution by fitting the UV absorbance of the color-developed sample to a standard curve. The color-developing reagent was obtained by dissolving 0.5 g potassium ferricyanide in 1 L of 0.5 mol L−1 sodium carbonate solution. The standard solution was composed of 0.1 g D-glucosamine in 100 mL deionized water. For color development, several different volumes of standard solution and 3.0 mL of color-developing reagent were mixed at room temperature, and the final volume was adjusted to 5 mL by adding deionized water. This mixture was sealed and placed in a boiling water bath for 15 min, followed by instant cooling to room temperature. The color of the final mixture was determined by a UV-Vis spectrometer, and the absorbance at 420 nm was used to obtain a standard curve. To determine the end group content, pre-weighed dry oligochitosan was also dissolved in 3.0 mL color-developing reagent, and the final volume was adjusted to 5.0 mL. After the same heating process, the UV-Vis absorbance at 420 nm of the sample was used to calculate the molar contents of the end groups, and the corresponding molecular weight of the oligochitosan was then calculated.Hydrogel preparation
The hydrogel was prepared according to the literature55 using HEPES (100 mM, pH 7.4) as the buffer solution and 1 M NaOH with 100 mM HEPES as the alkaline solution. After tuning to the target pH, the hydrogel was crushed for uniformity and centrifuged for bubble removal. The hydrogel was placed in a refrigerator (4 °C) for at least 12 h for stabilization and uniformity.Self-healing test
Before studying the self-healing properties of the hydrogel, strain-sweep measurements were performed at a constant frequency of 6.283 rad s−1 over the strain range of 0.1% to 300% at 25 °C. Then, a strain step cycled between 1% and 300% was performed at 25 °C and 6.283 rad s−1. The recovery rate of the hydrogel was determined by the quotient of the average storage modulus of the last and first 1% strain steps. The self-healing test of the bulk hydrogel was conducted in the original mould with slight pressing to ensure complete contact.Sol–gel transition and injectability
To test the pH-induced gel–sol transition, a certain amount of conc. HCl solution (according to the amount of added NaOH used to tune the mixture from pH 5.0 to 7.4 in preparation) was added to the hydrogel, and it was shaken for one minute at room temperature. To test the injectability, a 15 wt% mixture solution comprising OChi-IDA, PEG-PHis6 and Ni2+ ions was adjusted to pH 5.3 and injected into pH 7.4 phosphate buffer (100 mM) using a 50 μL HPLC injector.Swelling behavior and stability of the hydrogel
A certain weight of hydrogel was placed in a small alumina crucible (weighed previously) and immersed in excess pH 7.4 phosphate saline buffer (1×, containing 10 mM phosphate salt and diluted with commercial 10× PBS) or pH 5.5/4.5 sodium acetate–acetic acid buffer (10 mM, plus the same NaCl and KCl concentration in the 1× PBS). Also, the weight was measured at predetermined time intervals. The swelling ratio was calculated as:where Wt is the weight at time t and W0 is the weight before swelling.
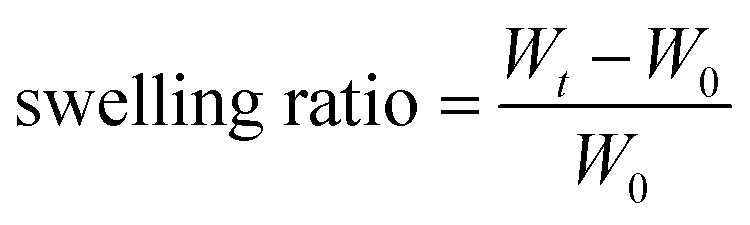
In vitro cytotoxicity evaluation
HeLa cells were seeded onto 96-well plates at a density of 1 × 104 cells per well in 100 μL DMEM with 10% FBS at 37 °C under a 5% CO2 humidified atmosphere. After 24 h of incubation, the DMEM medium was replaced with fresh culture medium; then, solutions of 15 wt% OChi-IDA, OChi-PHis6, PEG-IDA and PEG-PHis6 polymer, OChi-IDA/PEG-PHis6 (PHis:IDA = 1:2), OChi-PHis6/PEG-IDA (PHis:IDA = 1:1) hydrogel and NiCl2 solution were added to the cell culture. Due to the dilution, the final concentrations of the polymer and gel were about 1.5 wt%. Then, the cells were incubated for 24 h. MTT solution (20 μL, 5 mg mL−1 in PBS buffer) was added to each well and incubated for 4 h to allow the reaction to proceed. The medium in each well was then removed and 200 μL of DMSO was added to dissolve the internalized purple formazan crystals. The plate was subjected to gentle agitation for 30 min until all the crystals were dissolved. The absorbance at the wavelength of 480 nm was recorded using a micro-plate reader (Thermo Fisher).
Model molecule release
To study the release of Rh6G-PHis6, the hydrogel was prepared with 7.4 HEPES buffer with 4 mg mL−1 Rh6G-PHis6 to load the model drug. Then, the hydrogel (around 40 mg) was placed in a wire mesh and placed at the bottom of a glass bottle. Then, the hydrogel was dialyzed against 9 mL buffer (pH 7.4, pH 5.5 or pH 4.5) for certain time intervals. The released Rh6G-PHis6 concentrations in the buffer were quantified by measuring the absorption intensity at 529 nm against the corresponding standard calibration curves for Rh6G-PHis6. The total loaded Rh6G-PHis6 was determined by direct addition of an excess amount of HCl into a methylene blue-loaded hydrogel (weighed previously) and the solution was diluted to 9 mL using phosphate buffer (pH 7.4).Results and discussion
Preparation of the polymers and hydrogels
To construct a metal coordination hydrogel based on the PHis-Ni2+-IDAs multivalent coordination structure, it is necessary to conjugate PHis and IDA ligands to different hydrophilic polymer backbones. Also, the incorporation of multivalence relies on chemical linkages to restrict the movement of the multiple ligands; this condition is naturally satisfied for polymer ligand PHis. However, for mono-metal-ion IDA ligands, extended conjugation to the pendant of a polymer backbone is needed. Here, we chose oligochitosan with multiple modifiable pendant amino groups as the crosslinking polymer backbone and PEG with two modifiable ends as the soft linear backbone.As shown in Fig. 1a, allyl groups were modified on the amino groups of the oligochitosan. Also, a special IDA molecule with a thiol group (IDA-SH) was synthesized; the IDA group was attached to the oligochitosan backbone via thiol–ene click chemistry with nearly complete conversion. After all these processes, the OChi-IDA molecules were prepared. By evaluating the molecular weight, deacetylation degree and modification efficiency through end group analysis or 1H NMR spectroscopy, it was found that the final OChi-IDA molecules possess approximately 5.8 IDA moieties on a single oligochitosan backbone (see the ESI†). For the PHis segment, we previously reported a novel convenient polymerization method to obtain PHis from an activated urethane derivative of histidine which uses a primary amino group as the initiator.8,37 Through this method, the PHis segments were incorporated by the two amino ends of the PEG-NH2 backbone; a series of PHis-b-PEG-b-PHis triblock copolymers (PEG-PHis) were synthesized by tuning the ratio of the initiator and monomer. Three different degrees of polymerization (DP) of the PHis block were used here to verify the influence of multivalent coordination; these are 3, 6, or 9 per block and are represented as PEG-PHisn (Fig. 1(b) and (c)). According to the GPC and 1H NMR characterization, all the block polymers show narrow dispersities around 1.1 and good monomer conversion, which elucidates the good control of the polymerization of PHis by this method. Moreover, we synthesized a pair of PHis and IDAs polymers with a reverse backbone combination as a control to clarify the mechanism of multivalence. The PHis-grafted oligochitosan (OChi-g-PHis) and IDAs-modified PEG (PEG-IDA) were synthesized by similar chemistry, including amino-initiated polymerization in dry DMSO solvent and the thiol–ene reaction. The DP of PHis per branch for OChi-g-PHis is approximately 6, and the efficiency of the IDA modification is around 100%; these data were calculated from 1H NMR spectra.
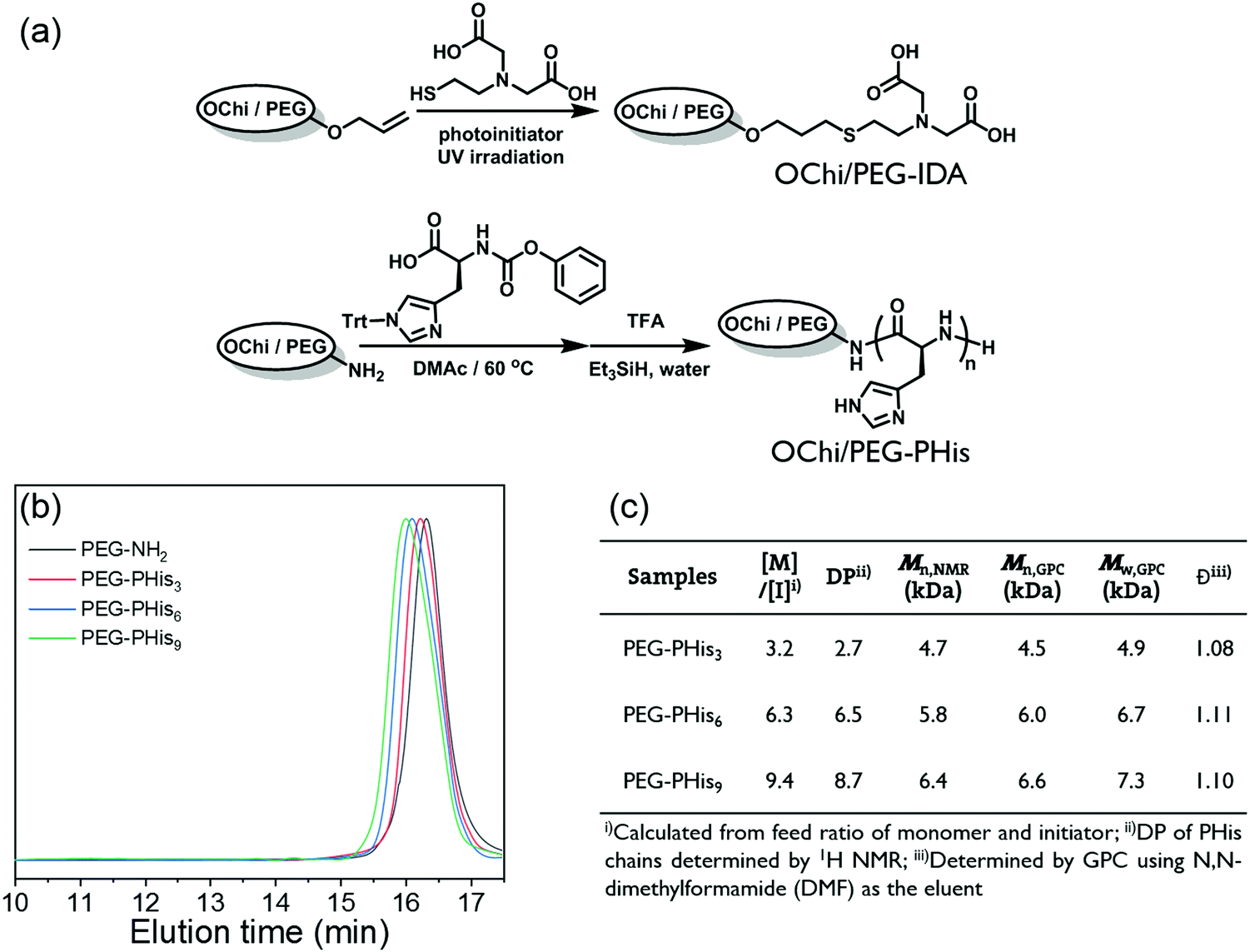 | ||
| Fig. 1 (a) Synthetic routes of OChi/PEG-IDA and PEG/OChi-PHis. (b) GPC traces for polymer PEG-NH2 and PEG-PHis3,6,9. (c) Table summarizing the structure parameters of the synthetic polymers. | ||
The metal coordination hydrogel was formed simply by mixing the PHis and IDAs-containing polymers with Ni2+ ions. To stabilize the pH of the hydrogel, 100 mM HEPES buffer solution (pH 7.4) was applied in the system. The HEPES buffer shows a negligible coordinating ability to metal ions and thus has minimal influence on the coordination hydrogel.56 Due to the acidity of the Ni2+ solution, the final pH was tuned by the addition of NaOH solution to about pH 7.4, which recovered the coordination ability of the pH-responsive PHis blocks and induced the formation of the hydrogel. The solid concentration (total weight percentage of the polymers) of this hydrogel was fixed at 15 wt%, and the molar ratio between PHis blocks and IDA groups was set at a predetermined value. Also, the molar concentration of the Ni2+ ions is the same as that of the IDA groups.
Mechanical and dynamic properties tuned by multivalent coordination
The rheological properties of the formed hydrogel were further investigated for deep observation and understanding of the effects of its special multivalent coordination. Because the IDA groups show insensitivity to mildly acidic environments,57 the Ni2+ ions are initially coordinated to the IDA groups, forming the Ni2+-IDA coordinating structure. When the pH is tuned to around neutral, the PHis block will coordinate with the Ni2+-IDA structures in donor–acceptor type linkages, as widely reported in studies about polyhistidine-tag chemistry.33,45 Due to the coordinating ability of PHis to multiple Ni2+-IDA structures, the multivalence of the final coordination in the hydrogel can be tuned by altering the molar ratio of the PHis blocks to the IDA groups. Here, we named this multivalent coordination structure PHis-(Ni2+-IDA)n, where the “n” in the subscript represents the coordinated Ni2+-IDA structures connected by chemical linkages. Thus, through changing the PHis:IDA ratio, this multivalence of this special PHis-(Ni2+-IDA)n complex can be regulated.
As shown in Fig. 2b, the frequency-sweeping rheology of the OChi-IDA/PEG-PHis6 hydrogel system was measured with different PHis:IDA ratios from 1:1 to 1:3. An increase in the plateau storage modulus and a decrease in the crossover frequency of G′ and G′′ was found when the PHis:IDA values were changed from 1:1 to 1:3. This change is mainly a result of strengthening of the hydrogel by multivalent coordination, especially for the crossover frequency. For a dynamic hydrogel that fits well with the Maxwell liquid model, the inverse of the crossover frequencies of the G′ and G′′ spectra in frequency-sweeping rheology equals its relaxation time. The larger the crossover frequency, the longer the relaxation time and the more dynamic the hydrogel. Based on previous studies, the gel relaxation dynamics are correlated with the bond dissociation rate for a hydrogel crosslinked by dynamic interactions.38 Hence, the relaxation time calculated from the crossover frequency is proportional to the bond dissociation rate. According to the former description in the Introduction, the incorporation of multivalent coordination will decrease the bond dissociation rate.43 There is a final conclusion that the incorporation of multivalence will increase the relaxation time of the coordination hydrogel and decrease the crossover frequency correspondingly. Therefore, as illustrated in Fig. 2a, the OChi-IDA/PEG-PHis6 hydrogel tends to form a monovalent PHis-(Ni2+-IDA)1 coordination structure at PHis:IDA = 1:1, and the weak linkage leads to a low plateau storage modulus and high crossover frequency. After the PHis:IDA was changed to 1:2, the coordination structure trend moved to the PHis-(Ni2+-IDA)2 coordination structure. The strengthened coordination linkage enhances the plateau storage modulus and decreases the crossover frequency, which leads to a prolonged relaxation time. When the PHis:IDA ratio was further changed to 1:3 for the OChi-IDA/PEG-PHis6 hydrogel system, the limited PHis block length restricted further strengthening of the multivalence, and the frequency-sweeping spectrum was similar to that of PHis:IDA = 1:2.
 | ||
| Fig. 2 (a) The proposed coordination structures obtained by varying the PHis:IDA ratio for the OChi-IDA/PEG-PHis hydrogels. Frequency-sweep measurements of (b) the OChi-IDA/PEG-PHis6 hydrogels with different PHis:IDA ratios, (c) the OChi-IDA/PEG-PHis hydrogels (PHis:IDA = 1:1) with different PHis block lengths, (d) the OChi-IDA/PEG-PHis9 hydrogels with different PHis:IDA ratios. | ||
The length restriction for the above OChi-IDA/PEG-PHis6 hydrogel indicates a correlation between the mechanical properties and the PHis block length. A series of PEG-PHis polymers with different PHis block lengths were used to form the OChi-IDA/PEG-PHis hydrogel at a fixed PHis:IDA ratio of 1:1; the frequency-sweeping rheology spectra of these gels are shown in Fig. 2c. The gradual increase of the plateau storage modulus and the decrease of the crossover frequency from PHis3 to PHis9 indicates enhancement of the coordination. This enhancement may result from the increase in both the coordination ability and the tendency of multivalent coordination due to the elongation of the PHis segment. Additional experiments were performed on the PHis9 series with different PHis:IDA ratios for the OChi-IDA/PEG-PHis hydrogel. As shown in Fig. 2d, the OChi-IDA/PEG-PHis9 hydrogels display a monotonic increase in storage modulus as the PHis:IDA ratio changes from 1:1 to 1:3, similar to the frequency-sweeping rheology spectra of the gels with PHis:IDA ratios of 1:2 and 1:3 for PHis6. This monotonic increase may be a result of the sufficient length of the PHis9 block, which can guarantee PHis-(Ni2+-IDA)3 coordination, unlike the restricted length of PHis6. All the results above demonstrate that the mechanical properties of this OChi-IDA/PEG-PHis hydrogel can be regulated by the molar ratio of PHis:IDA and the length of the PHis block, both of which can change the multivalence of the linkage coordination structure.
Control hydrogels were fabricated to clarify the necessity of multivalence by removing the multivalent coordination ability without changing the selection of functional groups. An interchange between PHis/IDA and the polymer backbone was designed to realize this concept. The control hydrogels were composed of oligochitosan-graft-PHis6 (OChi-g-PHis6), IDAs-modified PEG (PEG-IDA) and Ni2+ ions; these hydrogels were named OChi-g-PHis6/PEG-IDA. Although the PHis block has the potential to coordinate with multiple Ni2+-IDA structures, the multivalent coordination structure could not form due to the lack of efficient chemical linkages between multiple coordinated IDA groups. As shown in Fig. 3b, the control hydrogel with PHis:IDA = 1:1 showed a 13 kPa plateau storage modulus and a similar crossover frequency to the OChi-IDA/PEG-PHis6 hydrogel (PHis:IDA = 1:1). By varying the PHis:IDA ratio from 1:1 to 1:3, an opposite trend was observed for the control hydrogels, which displayed a decreasing plateau storage modulus (from 13 kPa to 1.4 kPa) and an increasing crossover frequency (1.6 rad s−1 to 5.0 rad s−1) (Fig. 3c and d). Unlike the OChi-IDA/PEG-PHis6 hydrogels, which can undergo multivalent coordination as the PHis:IDA ratio changes and increase both their plateau storage moduli and relaxation times, these control hydrogels can only undergo weak monovalent coordination; this coordination is so weak that the gels cannot withstand the increasing steric hindrance as the number of Ni2+-IDA structures becomes relatively enriched around the OChi-g-PHis6 by changing the PHis:IDA ratio from 1:1 to 1:3 (Fig. 3a). The increasing steric hindrance induced an increase in the number of uncoordinated Ni2+-IDA structures, which decreased the crosslinking degree and accelerated the dissociation rate of the coordinated PHis-Ni2+-IDA complexes. Thus, an opposite trend appeared, although the control hydrogels possessed the same functional groups as the multivalent hydrogels.
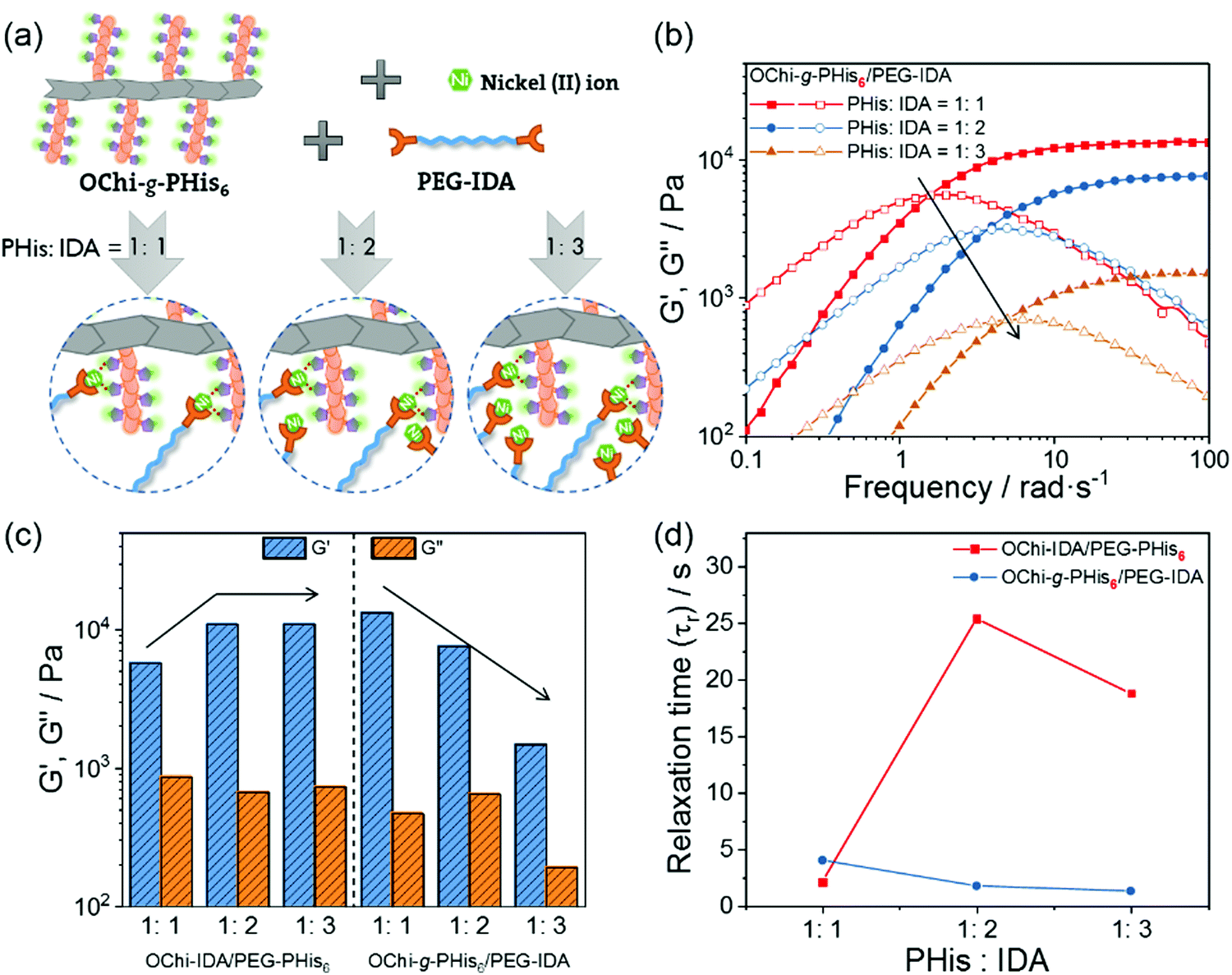 | ||
| Fig. 3 (a) The proposed coordination structures by varying the PHis:IDA ratio of the OChi-g-PHis/PEG-IDA hydrogel. (b) Frequency-sweep measurements of the OChi-g-PHis6/PEG-IDA hydrogels with different PHis:IDA ratios; (c) G′ and G′′ (d) relaxation times versus the PHis:IDA ratios of the OChi-IDA/PEG-PHis6 and OChi-g-PHis6/PEG-IDA hydrogels. | ||
By decreasing the dissociation rate, multivalent coordination decreases the dynamics of the hydrogels and endows them with stiff and freestanding shapes. As shown in Fig. S8 (ESI†), a series of hydrogels were allowed to stand at room temperature for 12 hours after centrifugation to generate a slope. The slopes of some of the hydrogels (e.g. hydrogels i, iv, and v) fell to near horizontal planes with relatively short relaxation times, indicating reconfiguration under gravity. In contrast, hydrogel ii (OChi-IDA/PEG-PHis6, PHis:IDA = 1:2) showed little change in the angle of its slope with a relatively long relaxation time as a consequence of the decrease of the dynamics. Upon further prolonging the relaxation time by using PHis9, the roughness on the surface of the slope before and after a 12 hour standing period appeared to be stiffer and less dynamic for hydrogel iii (OChi-IDA/PEG-PHis9, PHis:IDA = 1:3).
On the other side of the loss of dynamics, multivalent coordination may induce setbacks in some important dynamic properties, including self-healing. Gratifyingly, the stiff OChi-IDA/PEG-PHis6 hydrogel (PHis:IDA = 1:2) maintained remarkable self-healing properties; therefore, we could evaluate the related properties by a direct hydrogel healing process and rheological characterization. As shown in Fig. 4a, two OChi-IDA/PEG-PHis6 hydrogels (PHis:IDA = 1:2) were dyed and cut into two pieces. After placing the two different pieces in full contact and applying a limited press, the two pieces could self-heal to a whole piece within 1 hour at room temperature. The minimal shape change in the hanging test (the right most photo in Fig. 4a) was consistent with the free-standing tests and demonstrated the stiff mechanical properties and the relatively long relaxation time of the gels. For the rheological measurement, Fig. 4b presents that G′ and G′′ did not change under a strain of less than 40%, indicating resistance to deformation of the gel. The sharp decreases of G′ and G′′ at a strain of around 100% and the crossover at approximately 170% suggested disruption of the gel and transformation into a sol state. Therefore, the dynamic strain steps were set at 1% and 300% to test the recovery of the gel after disruption (Fig. 4c). The nearly 90% recovery after 3 disruption-recovery cycles indicates the self-healing properties of this relatively stiff coordination hydrogel.
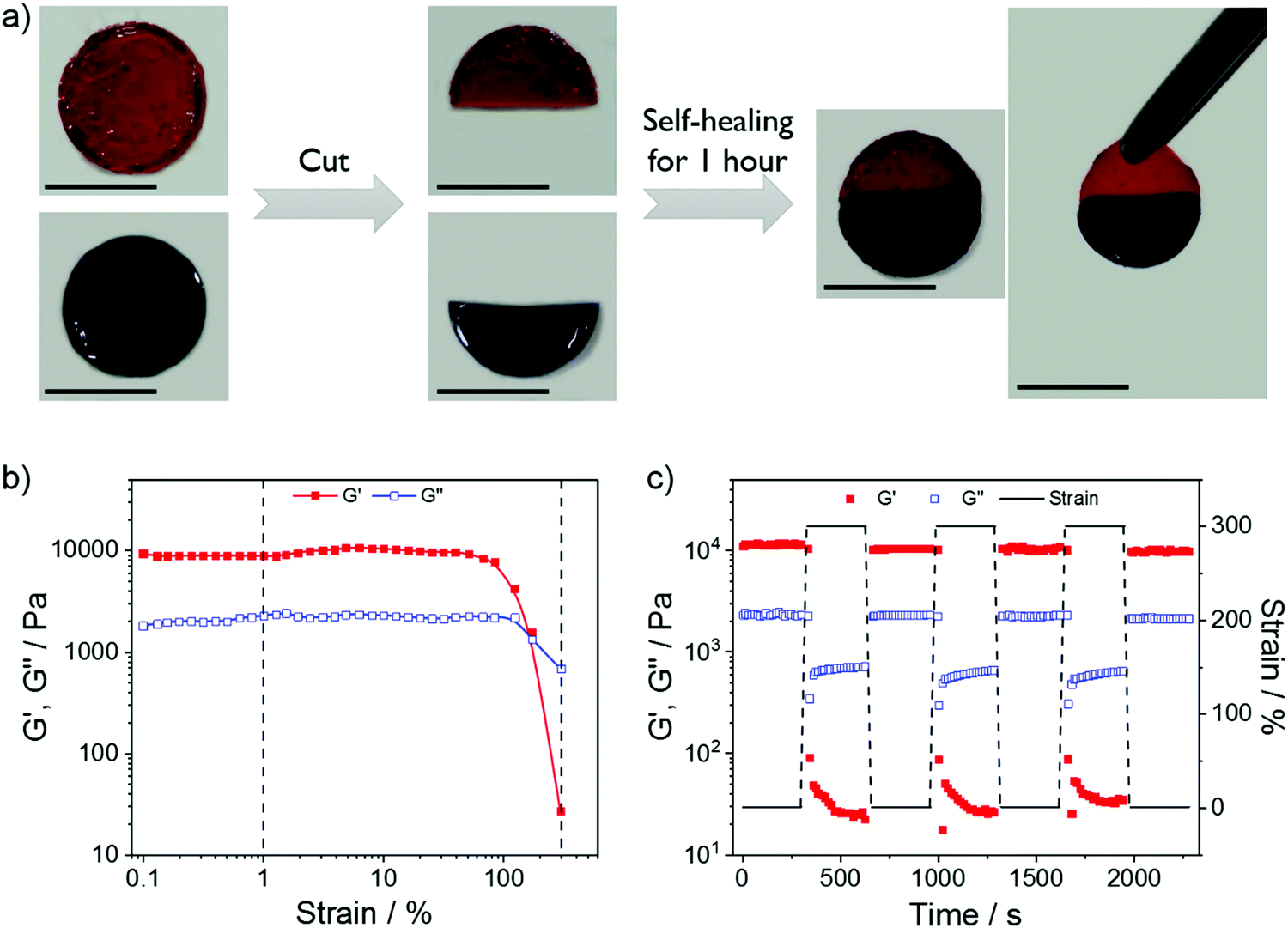 | ||
| Fig. 4 (a) Macroscopic self-healing photos for the OChi-IDA/PEG-PHis6 hydrogel (PHis:IDA = 1:2) stained with rhodamine 6G (red) and methylene blue (blue) (scale bars: 10 mm). (b) Strain-sweep measurements and (c) repeated dynamic strain step tests (strain = 1% or 300%) for the OChi-IDA/PEG-PHis6 hydrogel (PHis:IDA = 1:2). | ||
The OChi-IDA/PEG-PHis6 hydrogel (PHis:IDA = 1:2) appears to show balanced mechanical and dynamic properties. The multivalent coordination enhances its mechanical properties so that the hydrogel will be relatively stiff and maintain a specific shape on a large time scale, while the dynamic properties such as self-healing are retained to repair damage from external forces. These balanced properties manifest great potential for this multivalent coordination method in hydrogel performance regulation.
pH responsiveness and stability
After confirming the regulation of the mechanical properties of the hydrogels due to multivalent coordination, we proceeded to study their pH responsiveness. The PHis block naturally possesses mild acid responsiveness and should be protonated on the nitrogen atoms of its imidazole rings. Because the coordination ability of the PHis block originates from the same nitrogen atoms, this pH responsiveness can serve as a switch of the coordination ability. When the hydrogel pH is changed to weakly acidic, the formed PHis-Ni2+-IDA complexes will be disrupted and the hydrogel will undergo a pH-induced sol–gel transition (Fig. 5a). For the OChi-IDA/PEG-PHis6 hydrogel (PHis:IDA = 1:2), the hydrogel was formed at neutral pH; then, several drops of 1 M HCl solution were added to decrease the pH of the mixture to approximately 5.0. After shaking, the relatively stiff hydrogel collapsed to a non-viscous solution with protonated PHis blocks (Fig. 5c). As a result of the reversible nature of the protonation of the PHis block, an inverse sol-to-gel transition was realized by addition of equivalent NaOH solution. This sol-to-gel transition can also be processed by injecting the weak acid mixture solution into a neutral phosphate buffer, which will immediately neutralize the mixture and induce gel formation (Fig. 5b). Thus, this hydrogel showed neutral pH-induced injectability, which is promising due to the physiological gel-forming conditions. Moreover, the relatively mild pH response decreases the proton concentration of the initial sol mixture and quickens the neutralization process, which contributes to the instant gel formation at the tip of the injector. For the OChi-IDA/PEG-PHis9 hydrogel (PHis:IDA = 1:3), the pH responsiveness was delayed to more acidic conditions. Although initial dissolution was observed, the hydrogel did not completely dissolve at pH 5.0 even after 1 hour of shaking. Upon addition of more HCl, the mixture finally collapsed to a complete solution at pH 4.3. This stronger resistance to acidic conditions may be a result of the longer PHis blocks, which induce stronger coordination.
 | ||
| Fig. 5 (a) Graphic illustration of the proposed mechanism of pH-induced gel–sol transition. (b) pH-induced injectability by injecting near-gelation sol into neutral phosphate buffer (100 mM). The hydrogel was stained with methylene blue. (c) Photos of the pH-induced gel–sol transition of the OChi-IDA/PEG-PHis6 hydrogel (PHis:IDA = 1:2) and its reversibility. (d) The OChi-IDA/PEG-PHis9 hydrogel (PHis:IDA = 1:3) dissolved under more acidic conditions (all scale bars: 1 cm). | ||
The previous experiments on the hydrogels concentrated only on their responsiveness to the pH conditions of their own systems. To actually use these hydrogels as controlled drug release carriers and for other functions, the hydrogels must make contact with external environments, including various stimuli; thus, additional properties, such as swelling behavior, stability, etc., should be taken into consideration. Here, we further evaluated the swelling and stability properties of different OChi-IDA/PEG-PHis hydrogels and the control OChi-g-PHis6/PEG-IDA hydrogel in buffer with different pH conditions using the weight-loss method. As shown in Fig. 6a, the OChi-IDA/PEG-PHis6 hydrogel (PHis:IDA = 1:1) and the control OChi-g-PHis6/PEG-IDA hydrogel (PHis:IDA = 1:1) with comparatively short relaxation times showed a 3-hour survival time in pH 7.4 PBS solution (1×) for both hydrogels. The OChi-IDA/PEG-PHis6 hydrogel (PHis:IDA = 1:1) with the shortest relaxation time immediately lost weight after being immersed in the buffer; meanwhile, the control gel with a relatively longer relaxation time initially swelled slightly, followed by complete dissolution. It should be noted that the plateau storage modulus of the control gel is approximately 13 kPa, which is larger than that of most of the multivalent coordination hydrogels used here. This supplies powerful evidence to clarify that the modulus of the hydrogel is not the main factor in the stability of a hydrogel, especially a dynamic hydrogel. As for the mainly used OChi-IDA/PEG-PHis6 hydrogel (PHis:IDA = 1:2), this multivalent coordination hydrogel with a relaxation time as long as 25 s survived after 130 hours of immersion in pH 7.4 PBS solution. The hydrogel swelled in the initial 6 hours and then lost weight afterwards; this downtrend slowed after 80 hours. By increasing the relaxation time from approximately 2 s to 25 s, a dramatic enhancement of the stability of the coordination hydrogels was observed. This enhancement of the stability in excess water elucidates the effectiveness of the multivalence. The multivalence of the ligands strengthens the coordination linkage and decreases its dissociation rate, which further improves the stability in excess water. To examine the pH-responsive properties, an acetic acid/sodium acetate (HOAc/NaOAc) buffer at pH 5.5 was used with a similar composition to the PBS solution (1×). The results indicate the possibility that a physiological environment will trigger degradation, which may be effective in some disease tissues. For the stronger coordination hydrogel OChi-IDA/PEG-PHis9 (PHis:IDA = 1:3), a limited weight change at pH 7.4 was observed, and the hydrogel did not dissolve in pH 5.5 buffer after 130 hours of immersion; this is in accordance with the hydrogel pH response results above. Upon further decreasing the pH of the HOAc/NaOAc buffer to pH 4.5, the hydrogel showed rapid dissolution in approximately 2 hours.
 | ||
| Fig. 6 (a) Stabilities of the OChi-g-PHis6/PEG-IDA (PHis:IDA = 1:1) and OChi-IDA/PEG-PHis6 (PHis:IDA = 1:1) hydrogels in pH 7.4 PBS solution (1×). Stabilities and pH responsiveness of (b) the OChi-IDA/PEG-PHis6 (PHis:IDA = 1:2) and (c) the OChi-IDA/PEG-PHis9 (PHis:IDA = 1:3) hydrogels in different pH buffers. Each error bar represents mean ± SD, n = 3. | ||
Due to the multivalent coordination, the hydrogels with relatively long relaxation times showed enhanced stability in a neutral environment. The PBS solution (1×) used here had similar osmolality and ion concentrations to the human body, which implies stability inside the human body. Moreover, this enhancing method did not decrease the pH responsiveness of the hydrogel, and the responsiveness around pH 5.5 demonstrates a potential physiological relation to some disease tissues, including some inflammation tissues.
pH-regulated model molecule release
The main components of this hydrogel, the PHis block and IDA chelating ligand, are all widely used in polyhistidine-tag related recombinant peptides or protein purification.32 These recombinant peptides or proteins contain a polyhistidine motif that consists of at least six histidine residues, often at the N- or C-terminus. By their coordination ability, the peptides or proteins are immobilized at the surface of resin beads modified by Ni2+-IDA or Ni2+-NTA structures. Inspired by the abovementioned immobilization, the coordination hydrogels composed of similar PHis blocks and IDA chelating ligands should have great potential to immobilize recombinant peptides or proteins containing the polyhistidine tag and to release their payloads in response to weak acid stimuli. On the basis of the above discussion, we designed a rhodamine 6G-labelled model molecule containing a polyhistidine-tag (Rh6G-PHis6) to probe the release of molecules with a polyhistidine-tag. As showed in Fig. 7a, the Rh6G-PHis6 molecule was synthesized by polymerizing histidine monomers using an amino-functioned Rh6G as the initiator (Rh6G-NH2). The model molecule Rh6G-PHis6 was mixed with OChi-IDA, PEG-PHis6 and Ni2+ ions to form an Rh6G-loaded hydrogel. Benefitting from the coordination bonds between Rh6G-PHis6 and OChi-IDA, the payload was strongly embedded in the hydrogel framework and not simply physically entangled with the framework (Fig. 7b). By immersing this Rh6G-loaded hydrogel in PBS solution (pH 7.4), a negligible background release of Rh6G-PHis6 was observed in the whole release process (Fig. 7c). When the environment outside of the hydrogel changed to weak acid, the protonated PHis blocks of the PEG-PHis6 and Rh6G-PHis6 dissociated from the OChi-IDA and the hydrogel collapsed to a sol, resulting in a fast Rh6G-PHis6 payload release. For this Rh6G-loaded multivalent coordination hydrogel, the pH 5.5 HOAc/NaOAc buffer triggered a gradual degradation of the hydrogel, and the payload was nearly completely released to the weak acid environment within approximately 7 hours. When we used a longer PHis block as the hydrogel framework, the OChi-IDA/PEG-PHis9 hydrogel released the model Rh6G-PHis6 payload in a more acidic environment. As shown in Fig. S11 (ESI†), the pH 5.5 external environment could only trigger slow release, with around 10% release after 120 hours of incubation; the model molecule-loaded hydrogel remained intact. Upon more acidic stimuli, pH 4.5 buffer resulted in nearly complete release in approximately 5 hours. | ||
| Fig. 7 (a) Synthetic route of the Rh6G-PHis6 model molecule. (b) The proposed mechanism of the pH-regulated release of the model molecule from the OChi-IDA/PEG-PHis6 (PHis:IDA = 1:2) hydrogel. (c) The cumulative release of the Rh6G-PHis6 model molecule versus incubation time in pH 7.4 and 5.5 buffers. Each error bar represents mean ± SD, n = 3. | ||
Conclusions
In summary, we developed a new strategy for enhancing the neutral stability of pH-responsive metal coordination hydrogels without decreasing their responsiveness. In this strategy, we replaced the traditional monovalent metal coordination structure with a multivalent one, which can decrease the dissociation rate of the coordination bonds and further strengthen the swelling/stability properties of the hydrogels. To realize this multivalent coordination, we used IDA-modified oligochitosan (OChi-IDA) and a PHis-terminated PEG copolymer (PEG-PHis), and multivalent coordination with multiple Ni2+-IDA complexes was formed by PHis. This multivalent coordination strengthens not only the plateau storage modulus but also the relaxation time of the hydrogel. The mechanical and dynamic properties of the hydrogel can be regulated by tuning the ratio of PHis to IDA groups, which can vary the coordination preference of the multivalence. Meanwhile, the multivalent coordination hydrogel with weakened dynamic relaxation maintains self-healing properties and can repair itself after cracking. Also, a control experiment hydrogel without efficient linkages between its IDA groups proved the necessity of multivalence. For pH responsiveness and stability, the multivalent coordination hydrogel can quickly dissolve in response to a pH 5.5 external mildly acidic environment while remaining integral and intact in a pH 7.4 neutral environment; this is a great improvement compared to the low neutral stability of the monovalent control hydrogel with a short relaxation time. Due to this pH responsiveness and neutral stability after regulation by multivalent coordination, these coordination hydrogels are promising candidates in controlled drug release systems, which was preliminarily confirmed by the pH-tuned release of a model molecule.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 51273189), the National Science and Technology Major Project of the Ministry of Science and Technology of China (No. 2016ZX05016), and the National Science and Technology Major Project of the Ministry of Science and Technology of China (No. 2016ZX05046).Notes and references
- C.-C. Lin and A. T. Metters, Adv. Drug Delivery Rev., 2006, 58, 1379–1408 CrossRef CAS.
- P. Gupta, K. Vermani and S. Garg, Drug Discovery Today, 2002, 7, 569–579 CrossRef CAS.
- X. Qi, W. Wei, J. Li, G. Zuo, X. Pan, T. Su, J. Zhang and W. Dong, Mol. Pharmaceutics, 2017, 14, 431–440 CrossRef CAS.
- M. Guo, L. M. Pitet, H. M. Wyss, M. Vos, P. Y. W. Dankers and E. W. Meijer, J. Am. Chem. Soc., 2014, 136, 6969–6977 CrossRef CAS.
- J. Cui and A. d. Campo, Chem. Commun., 2012, 48, 9302–9304 RSC.
- X. Qi, X. Hu, W. Wei, H. Yu, J. Li, J. Zhang and W. Dong, Carbohydr. Polym., 2015, 118, 60–69 CrossRef CAS.
- I. Molina, S. Li, M. B. Martinez and M. Vert, Biomaterials, 2001, 22, 363–369 CrossRef CAS.
- D. Zhao, Q. Tang, Q. Zhou, K. Peng, H. Yang and X. Zhang, Soft Matter, 2018, 14, 7420–7428 RSC.
- Z. Deng, Y. Guo, X. Zhao, P. X. Ma and B. Guo, Chem. Mater., 2018, 30, 1729–1742 CrossRef CAS.
- N. Holten-Andersen, M. J. Harrington, H. Birkedal, B. P. Lee, P. B. Messersmith, K. Y. C. Lee and J. H. Waite, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 2651–2655 CrossRef CAS.
- S. Y. Zheng, H. Ding, J. Qian, J. Yin, Z. L. Wu, Y. Song and Q. Zheng, Macromolecules, 2016, 49, 9637–9646 CrossRef CAS.
- W. E. Hennink and C. F. van Nostrum, Adv. Drug Delivery Rev., 2012, 64, 223–236 CrossRef.
- M. Black, A. Trent, Y. Kostenko, J. S. Lee, C. Olive and M. Tirrell, Adv. Mater., 2012, 24, 3845–3849 CrossRef CAS.
- D. L. Taylor and M. in het Panhuis, Adv. Mater., 2016, 28, 9060–9093 CrossRef CAS.
- X. Zhao, H. Wu, B. Guo, R. Dong, Y. Qiu and P. X. Ma, Biomaterials, 2017, 122, 34–47 CrossRef CAS.
- L. Yu and J. Ding, Chem. Soc. Rev., 2008, 37, 1473–1481 RSC.
- J. Qu, X. Zhao, Y. Liang, T. Zhang, P. X. Ma and B. Guo, Biomaterials, 2018, 183, 185–199 CrossRef CAS.
- Y. Tabata, Tissue Eng., 2003, 9, 5–15 CrossRef.
- S.-k. Ahn, R. M. Kasi, S.-C. Kim, N. Sharma and Y. Zhou, Soft Matter, 2008, 4, 1151–1157 RSC.
- D. Schmaljohann, Adv. Drug Delivery Rev., 2006, 58, 1655–1670 CrossRef CAS.
- Y. Qiu and K. Park, Adv. Drug Delivery Rev., 2001, 53, 321–339 CrossRef CAS.
- X. Qi, W. Wei, J. Li, Y. Liu, X. Hu, J. Zhang, L. Bi and W. Dong, ACS Biomater. Sci. Eng., 2015, 1, 1287–1299 CrossRef CAS.
- J. Qu, X. Zhao, P. X. Ma and B. Guo, Acta Biomater., 2017, 58, 168–180 CrossRef CAS.
- B. Jeong, S. W. Kim and Y. H. Bae, Adv. Drug Delivery Rev., 2012, 64, 154–162 CrossRef.
- B. Jeong, Y. H. Bae, D. S. Lee and S. W. Kim, Nature, 1997, 388, 860 CrossRef CAS PubMed.
- S. J. Buwalda, K. W. M. Boere, P. J. Dijkstra, J. Feijen, T. Vermonden and W. E. Hennink, J. Controlled Release, 2014, 190, 254–273 CrossRef CAS PubMed.
- Joseph V. Accardo and J. A. Kalow, Chem. Sci., 2018, 9, 5987–5993 RSC.
- J.-A. Yang, J. Yeom, B. W. Hwang, A. S. Hoffman and S. K. Hahn, Prog. Polym. Sci., 2014, 39, 1973–1986 CrossRef CAS.
- S. Mura, J. Nicolas and P. Couvreur, Nat. Mater., 2013, 12, 991 CrossRef CAS.
- E. S. Lee, Z. Gao and Y. H. Bae, J. Controlled Release, 2008, 132, 164–170 CrossRef CAS.
- G. M. Kim, Y. H. Bae and W. H. Jo, Macromol. Biosci., 2005, 5, 1118–1124 CrossRef CAS.
- E. K. M. Ueda, P. W. Gout and L. Morganti, J. Chromatogr., 2003, 988, 1–23 CrossRef CAS.
- E. Hochuli, H. Döbeli and A. Schacher, J. Chromatogr., 1987, 411, 177–184 CrossRef CAS.
- Y.-T. Lai, Y.-Y. Chang, L. Hu, Y. Yang, A. Chao, Z.-Y. Du, J. A. Tanner, M.-L. Chye, C. Qian, K.-M. Ng, H. Li and H. Sun, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 2948–2953 CrossRef CAS.
- R. J. Peters, M. Nijemeisland and J. C. van Hest, Angew. Chem., Int. Ed., 2015, 127, 9750–9753 CrossRef.
- J. Cabanas-Danés, E. D. Rodrigues, E. Landman, J. Van Weerd, C. van Blitterswijk, T. Verrips, J. Huskens, M. Karperien and P. Jonkheijm, J. Am. Chem. Soc., 2014, 136, 12675–12681 CrossRef.
- Q. Tang, D. Zhao, Q. Zhou, H. Yang, K. Peng and X. Zhang, Macromol. Rapid Commun., 2018, 39, 1800109 CrossRef.
- D. E. Fullenkamp, L. He, D. G. Barrett, W. R. Burghardt and P. B. Messersmith, Macromolecules, 2013, 46, 1167–1174 CrossRef CAS PubMed.
- C. Wang, R. J. Stewart and J. KopeČek, Nature, 1999, 397, 417 CrossRef CAS.
- D. M. Loveless, S. L. Jeon and S. L. Craig, Macromolecules, 2005, 38, 10171–10177 CrossRef CAS.
- Y. Chujo, K. Sada and T. Saegusa, Macromolecules, 1993, 26, 6315–6319 CrossRef CAS.
- Y. Chujo, K. Sada and T. Saegusa, Macromolecules, 1993, 26, 6320–6323 CrossRef CAS.
- C. Chittasupho, Ther. Delivery, 2012, 3, 1171–1187 CrossRef CAS.
- A. Mulder, J. Huskens and D. N. Reinhoudt, Org. Biomol. Chem., 2004, 2, 3409–3424 RSC.
- S. Lata, A. Reichel, R. Brock, R. Tampé and J. Piehler, J. Am. Chem. Soc., 2005, 127, 10205–10215 CrossRef CAS PubMed.
- C. You and J. Piehler, Anal. Bioanal. Chem., 2014, 406, 3345–3357 CrossRef CAS PubMed.
- S. V. Wegner, F. C. Schenk, S. Witzel, F. Bialas and J. P. Spatz, Macromolecules, 2016, 49, 4229–4235 CrossRef CAS.
- A. A. Watrelot, D. T. Tran, T. Buffeteau, D. Deffieux, C. Le Bourvellec, S. Quideau and C. M. Renard, Appl. Surf. Sci., 2016, 371, 512–518 CrossRef CAS.
- N. Illy, M. Robitzer, R. Auvergne, S. Caillol, G. David and B. Boutevin, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 39–48 CrossRef CAS.
- D. L. Elbert and J. A. Hubbell, Biomacromolecules, 2001, 2, 430–441 CrossRef CAS.
- K. Jang, K. Miura, Y. Koyama and T. Takata, Org. Lett., 2012, 14, 3088–3091 CrossRef CAS.
- D.-W. Lee and R. H. Baney, Biomacromolecules, 2004, 5, 1310–1315 CrossRef CAS.
- J. Wan, K. Zhang, C. Li, Y. Li and S. Niu, Sens. Actuators, B, 2017, 246, 696–702 CrossRef CAS.
- J. Shao, Y. Yang and Q. Zhong, Polym. Degrad. Stab., 2003, 82, 395–398 CrossRef CAS.
- S. Tang and B. D. Olsen, Macromolecules, 2016, 49, 9163–9175 CrossRef CAS.
- N. E. Good, G. D. Winget, W. Winter, T. N. Connolly, S. Izawa and R. M. Singh, Biochemistry, 1966, 5, 467–477 CrossRef CAS.
- G. Weng, S. Thanneeru and J. He, Adv. Mater., 2018, 30, 1706526 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8tb02360c |
| This journal is © The Royal Society of Chemistry 2019 |