
Manipulating the mechanical properties of biomimetic hydrogels with multivalent host–guest interactions†
 *ac
and
Liming
Bian
*abde
*ac
and
Liming
Bian
*abde
Abstract
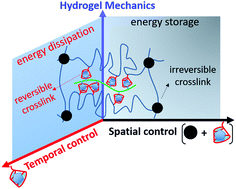
Biomimetic hydrogels with hierarchical network structures are promising biomaterials for tissue engineering due to their unique mechanical properties. One successful biomimetic strategy for facile construction of high-performance hydrogels is to incorporate reversible crosslinks as sacrificial bonds into chemical polymer networks. By mimicking the unfolding–refolding functions of the skeletal muscle protein titin, the reversible crosslinks can reinforce the otherwise weak and brittle hydrogels. However, the contribution of multivalent reversible crosslinks to the overall hydrogel mechanical properties has rarely been investigated. Herein we present the biomimetic hydrogels with multivalent host–guest interactions as reversible crosslinks, which provide not only energy storage capacity, but also elevated energy dissipation capacity to the dually crosslinked networks, therefore leading to the improved hydrogel ductility and tensile strength. Our results also reveal the manner of multivalent host–guest crosslinks contributing to the hydrogel mechanical properties, including gelation rate, energy storage and dissipation, tensile hysteresis, and fast spontaneous recovery.
- This article is part of the themed collection: Hydrogel properties and applications
 Please wait while we load your content...
Please wait while we load your content...

