
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDiffusion-induced in situ growth of covalent organic frameworks for composite membranes†
Priyanka
Manchanda
,
Stefan
Chisca
,
Lakshmeesha
Upadhyaya
,
Valentina-Elena
Musteata
,
Mark
Carrington
and
Suzana P.
Nunes
 *
*
King Abdullah University of Science and Technology (KAUST), Biological and Environmental Science and Engineering (BESE) Division, Advanced Membranes and Porous Materials Center, 23955-6900 Thuwal, Saudi Arabia. E-mail: suzana.nunes@kaust.edu.sa
First published on 29th October 2019
Abstract
Thin layers of a covalent organic framework (COF) have been synthesized on a flexible polymeric support using a new diffusion-induction method under ambient conditions in reaction times as short as 3 hours. The layer is uniform and has a nodular crystalline morphology, evolved after the initial stages of amorphous fiber formation. COF nano-sheets were formed in the internal cavities of the support. The resultant composite membranes had an ultrahigh water vapor permeance and negligible liquid water transport, properties required for application in dehumidification devices.
Two dimensional (2D) covalent organic frameworks (COFs) are an emerging class of porous, highly ordered and covalently linked networks, exclusively constituted by light elements (C, N, O, B, and H).1 The shape and pore size of COFs can be controlled by the judicious selection of appropriate building blocks from a wide portfolio of versatile monomers with different stability levels.2 For instance, imine-based COFs are more stable in water and protic solvents than their boron-linked counterparts. Besides the bonding strength between atoms constituting the 2D COF layer, the interlayer interactions and their stacking and crystallinity are essential for stability.3 Due to the unique qualities of the well-defined, highly ordered, and easily tuneable pore structure, COFs have gained attention in different fields, such as optoelectronics, energy/gas storage, catalysis and molecular separation.4–10 The latter is our main interest. COFs have been previously blended into a polymer matrix as fillers for mixed-matrix membranes.11 However, to take full advantage of the COF potential selectivity, we believe that multilayer composite membranes on polymer supports should be the most convenient approach. Besides the uniform pore control and the lack of defects, we search for the following conditions: (1) mild temperature COF synthesis, (2) COF thermal, chemical and mechanical stability as a selective layer, (3) adequate support for the synthesis medium, and (4) COF-support adhesion.
Since the first report on porous organic frameworks,12 a variety of new compositions have been proposed, but mostly with conventional synthesis methods, leading to microcrystalline powders, using high temperatures and long reaction times. However, the high-temperature normally used for COF synthesis and its insolubility as microcrystalline powder are major hurdles for their processability, especially for application as coatings or membranes. Free standing COF membranes have been developed at high temperature.13 COF membranes have been fabricated by solvothermal synthesis on a ceramic support.14 Flexible polymeric supports would increase the chance of scalability. However, in this case, COF preparation should be done at relatively mild temperature to avoid damage of the polymeric substrate. Ditchel and co-workers15,16 have reported a room temperature synthesis of an imine-linked COF as a powder using metal triflates. As a second consideration to drive COF selection, coatings and membrane applications have a growing demand of materials with high stability. Xu et al.3 proposed the synthesis of an imine-COF in the form of a powder, which is particularly stable under harsh conditions, such as acids, bases and organic solvents. The superior chemical stability of this particular COF is attributed to the presence of dimethoxy groups, which soften the polarization of the imine bond, due to their electron donating induction effect, and stabilize the positively charged phenyl ring through resonance. This further reinforces the interlayer interactions and prevents hydrolysis. This material, a 2D imine COF, synthesized using 1,3,5-tri-(4-aminophenyl) benzene (TPB) and 2,5-dimethoxybenzene-1,4-dicarboxaldehyde (DMTP), was chosen for the membrane development in this work.
Choosing the right chemistry is only the first step in the successful preparation of a multilayer membrane. The material originally prepared as a powder needs to be then engineered as a continuous, defect-free, mechanically stable and highly adherent selective layer on a porous and flexible polymeric support. Nevertheless, the support should resist the solvent medium adopted for the COF synthesis. A few previous approaches have been reported aiming at multilayer membranes. Continuous COF-based membranes have been prepared by in situ growth, layer-by-layer stacking, Langmuir–Blodgett and interfacial polymerization (IP).17 Lately, the room temperature synthesis of freestanding COF thin films by interfacial polymerization (IP) has been explored, which eventually were transferred to a porous substrate.18,19 In most cases, the COF thin film is formed at the interface between the organic and aqueous solutions containing the COF monomers, before transferring it to a ceramic support. While continuous film formation has been successfully demonstrated by this method for a few selected cases, drawbacks are known and justify the search for more adequate processes. First, an inherent limitation is the insolubility of monomers in the classical solvents used for IP. This restricts the portfolio of materials that could be chosen for COF preparation. Second, the adhesion might be an issue under operation, particularly when the film is preformed and transferred to the support. Third, although inorganic supports have been previously used to demonstrate approaches in the lab, due to their high thermal and chemical tolerance, their cost and scalability are disadvantageous when compared to polymeric analogues. In spite of all recent breakthroughs in COF composite membranes, the practical, facile and scalable fabrication as continuous (few cm2) thin films on porous polymeric support is still challenging and has been hardly achieved. We envisioned that membrane fabrication via a contra diffusion methodology could be a possible way to address the challenge of obtaining defect free composite COF membranes. Contra diffusion has been successfully applied to grow ZIF-8 and polydopamine on the surface of porous supports.20–23 However, to our knowledge; it was never applied for COF preparation. In our work, for COF formation, a fundamental change was applied. While for ZIF-8 preparation, the linker was placed in one side of the membrane and a coordinating Zn2+ solution on the other side, we placed a stoichiometric monomer composition in one compartment of the diffusion cell; the other compartment was filled with the catalyst solution. The extent of the reaction could then be controlled by the slow exposure to the catalyst. This is important, since ordered and well-controlled COF formation is highly sensitive to the monomer ratio in the reaction medium. This can be hardly controlled in previous processes, such as IP layer formation. This arrangement had primordial influence on the characteristics of the COF selective layer. We used a crosslinked polyacrylonitrile (XPAN) support, with a gradient of porosity, previously prepared on a polypropylene non-woven support, which would allow reaction in different organic solvents and therefore a broad variety of monomers. The catalyst was scandium triflate (Sc(OTf)3). The rational choice of a Lewis acid catalyst was crucial for the fabrication of the composite membrane, since it allows COF formation through an imine exchange mechanism, rather than a condensation mechanism (employed by Brønsted acids). Previous reports indicate that Lewis acids may facilitate faster COF formation.16,19 With Sc(OTf)3, COF formation takes place in few minutes at room temperature, whereas by using Brønsted acids (acetic acid or p-toluene sulfonic acid) the formation of COF would require a much longer time (12–72 h) and higher temperature (60–120 °C).
We investigated the formation process, crystallinity and morphology of the COF composite membrane by varying the reaction time and amount of the catalyst. Fig. 1 shows a schematic outline of the process and the constituting layers of the final membrane. The COF polymer composite was prepared in situ, by contra-diffusion in a PermeGear diffusion cell (Fig. S1†).
 | ||
| Fig. 1 Schematic outline of the process and the constituting layers of the COF–polymer composite membrane. | ||
In a typical procedure, a coupon of XPAN (4.9 cm2) was placed in between the two compartments of the cell and sealed using Parafilm and a clamp. A solution of the monomers, in a fixed stoichiometric ratio i.e. DMTP (0.120 mmol, 23.3 mg) and TPB (0.080 mmol, 28.1 mg), was solubilized in 5 mL of a mixture of dioxane![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :mesitylene (4:1 v/v) and charged on the non-woven support side. The other side of the support, with pore size in the ultrafiltration range (tens of nanometers), was exposed to a catalyst solution, Sc(OTf)3 (3.5 mg (0.007 mmol) and 7 mg (0.014 mmol)), also in 5 mL of dioxane:mesitylene (4:1 v/v). Since the same solvent mixture was used on both sides, the chemical potential gradient was only due to the reactant and catalyst concentration differences. A slow interdiffusion process led to the formation of a continuous and crystalline COF material on and inside the XPAN support. The assembly was left undisturbed for different time intervals to allow the continuous growth of TPB-DMTP-COF. After the fixed time interval, the solution in each compartment was removed using a pipette, and the membrane was carefully taken and washed three times (3× 20 mL) with a mixture of dioxane:mesitylene (4:1 v/v) to remove the unreacted monomers, followed by washing with tetrahydrofuran and water. To confirm imine formation, Fourier transform infrared (FTIR) spectroscopy was used. The characteristic peaks corresponding to C
:mesitylene (4:1 v/v) and charged on the non-woven support side. The other side of the support, with pore size in the ultrafiltration range (tens of nanometers), was exposed to a catalyst solution, Sc(OTf)3 (3.5 mg (0.007 mmol) and 7 mg (0.014 mmol)), also in 5 mL of dioxane:mesitylene (4:1 v/v). Since the same solvent mixture was used on both sides, the chemical potential gradient was only due to the reactant and catalyst concentration differences. A slow interdiffusion process led to the formation of a continuous and crystalline COF material on and inside the XPAN support. The assembly was left undisturbed for different time intervals to allow the continuous growth of TPB-DMTP-COF. After the fixed time interval, the solution in each compartment was removed using a pipette, and the membrane was carefully taken and washed three times (3× 20 mL) with a mixture of dioxane:mesitylene (4:1 v/v) to remove the unreacted monomers, followed by washing with tetrahydrofuran and water. To confirm imine formation, Fourier transform infrared (FTIR) spectroscopy was used. The characteristic peaks corresponding to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O of DMTP at 1670 cm−1 and N–H stretching of TPB at 3400 cm−1 disappeared with the appearance of a peak in the range of 1600 cm−1, characteristic to the formation of imine bond both for the TPB-DMTP COF powder and the TPB-DMTP COF–XPAN composite membrane (Fig. S2†).
O of DMTP at 1670 cm−1 and N–H stretching of TPB at 3400 cm−1 disappeared with the appearance of a peak in the range of 1600 cm−1, characteristic to the formation of imine bond both for the TPB-DMTP COF powder and the TPB-DMTP COF–XPAN composite membrane (Fig. S2†).
Fig. 2 shows the powder X-ray diffraction (PXRD) patterns and the morphological characterization by scanning electron microscopy (SEM) and transmission electron microscopy (TEM) of the COF-composite membranes obtained with 3.5 mg (0.007 mmol) of Sc(OTf)3 and different reaction times. After 1.5 h reaction time, we observed precipitate formation in the compartment containing the catalyst solution and a fibrillar network on the surface of the XPAN support, as imaged by SEM (Fig. 2b and S3a†). No polymerization was detected in the internal pores of the support, as confirmed by the cross-sectional image in Fig. S3b.† The PXRD profile of the COF–XPAN composite reacted for 1.5 h (Fig. 2a, yellow curve) does not show any diffraction peaks in the 2θ region from 2.5 to 10°. Only the peaks characteristic of the XPAN support can be seen in the region of 2θ (10 to 30°) (analogous to Fig. S4a,† red curve). This indicates that the network formed under these conditions is amorphous.
 | ||
| Fig. 2 Evolution of the morphology and crystallinity of the COF composite membranes with the reaction time. (a) PXRD profiles for the COF composite membranes obtained after 1.5 h, 24 h and 24 h + annealing, by using 3.5 mg of Sc(OTf)3; the SEM surface image of the membranes obtained after (b) 1.5 h, (c) 6 h and (d) 24 h with 24 h of annealing; (e and f) cross-sectional SEM image and (g) TEM image of the COF membrane obtained after 24 h, with 24 h of annealing (inset image in a different area). | ||
By increasing the reaction time to 6 h, the fibrillar morphology evolves into a dense 2D continuous layer with a nodular structure on the surface of the porous support. The nodules have a diameter in the range of 300–400 nm, as shown in Fig. 2c and S3c.† The transition from the fibrillar to nodular morphology can be clearly seen in areas of Fig. S3c† marked by circles. Simultaneously in the bulk of the support, we observed the incipient development of 2D nanosheets, growing orthogonally to the larger finger-like cavity walls (Fig. S3d†).
These nanosheets result from the slow diffusion of the catalyst into the cavities, which were previously filled with the monomer solution. However, after 6 h of reaction, the XPAN surface was not uniformly covered by the COF layer, and the PXRD profiles were still characteristic of an amorphous structure. Therefore, we increased the reaction time to 24 h. The PXRD profiles of the COF–XPAN composite (Fig. 2a) then exhibited sharper peaks in the 2θ region from 2.5 to 10°, indicating crystallinity. The peaks at 2θ = 2.9, 5.0, 5.7, and 7.4 were observed which corroborate well with those previously reported for the TPB-DMTP COF in AA stacking mode prepared using a different procedure.3 The SEM surface images (Fig. S3e†) show that after 24 h, the fibrillar morphology was completely converted to a continuous nodular crystalline COF layer on the XPAN support. Analogous to the reaction conducted for 6 h, 2D nanosheets evolved into a leaf-like architecture with a thickness of around 10 nm, orthogonally to the walls of the larger cavities in the support structure, as shown in Fig. S3f.† The nanosheets are now more prominent.
With the aim of increasing the crystallinity even more and completely closing any potential discontinuity of the COF layer, we subjected the system to an additional annealing step. Annealing in the absence of monomers has been reported to increase ordering in a different COF system.24 We removed the monomer solution after 24 h of diffusion reaction and annealed it in the presence of the catalyst solution for 24 h more. The PXRD profiles of the resulting system indeed exhibited higher crystallinity with sharper peaks at 2θ = 2.7, 5.0, 5.7, and 7.4 (blue curve in Fig. 2a). This is attributed to the improved periodic arrangement of the COF layers. The thin COF layer has now a rougher surface with larger crystalline nodules (Fig. S3g†). Interestingly, the 2D nanosheets in the bulk of the support have now a secondary hierarchical structure assembled into a nano-flower/leaf-like morphology, partially filling the cavities, as shown in Fig. 2e, f and S3h.†
The π–π staking of COF layers could be analysed in detail using high-resolution transmission electron microscopy (TEM). Fig. 2g and S5† show the TEM images of a composite membrane fabricated with 24 h diffusion time and 24 h annealing using 3.5 mg of Sc(OTf)3. The image of the upper part of the cross-section indicates that the COF was grown on the surface and inside the support membrane and they form crystalline domains (Fig. S5a†) as magnified in Fig. S5b.† The Fourier transform of this image (Fig. S5c†) indicates a semi-crystalline morphology. Fig. 2g and S5d, e† show the high magnification image of the stacked COF layers. In Fig. S5d,† the arrows indicate stacking layers with different orientations and different periodicities. The highlighted area 1 in Fig. S5d† has a periodicity with a d-spacing of 0.32 nm, while area 2 in the same image has an interlayer distance of 0.17 nm. The magnified areas are shown in Fig. 2g. This different distance can be seen as a cutting effect, i.e., domains with different orientations could have been dissected under different angles. Xu et al.3 reported an interlayer distance of 3.5215 Å for the AA stacking of an analogous COF, prepared by a different method. This also agrees with our obtained PXRD profiles. Besides the orientation of the domains, for the lower periodicities observed in area 2, we cannot exclude the hypothesis of confinement effects which may constrict crystal growth. The electron diffraction pattern shown in Fig. S5e† also confirms the crystalline structure, with the bright spots describing the reciprocal lattice of the crystal. Therefore, by using 3.5 mg of Sc(OTf)3, we succeeded in obtaining a crystalline COF composite membrane without using any anchor linker25 within 24 h and with even higher crystallinity when we annealed in the presence of the catalyst solution for another 24 h.
To make this process faster and to accelerate the formation of crystalline COF composite membranes, we increased the amount of the catalyst. Fig. 3 shows the characteristics of the powder and COF composite membranes synthesized with 7 mg of Sc(OTf)3 and reaction times of 3 and 6 h. By increasing the catalyst amount from 0.007 mmol to 0.014 mmol, i.e. from 3.5 mg to 7 mg, the PXRD profiles of the composite membranes exhibited higher-order diffraction peaks within only 3 h, as depicted in Fig. 3c. However, when the diffusion time was increased from 3 h to 6 h, slightly broader PXRD features were obtained (Fig. 3c). This indicates that longer time in the presence of a higher concentration of Sc(OTf)3 will lead to a decrease in the crystallinity. A similar behaviour was observed for the PXRD diffraction peaks of analogous powders (Fig. 3c). This might be correlated with the fact that longer times at a higher concentration of Sc(OTf)3 can inhibit imine exchange.16,26 The surface morphology of the COF composite membrane prepared with 7 mg of Sc(OTf)3 and 3 h diffusion reaction time is uniform, and the COF layer is continuous and decorated with regular nodules with a size of around 80 nm, as shown in Fig. 3d, while the morphology of the as synthesized COF powder under the analogous conditions has nodules 10-fold larger, as shown in Fig. 3b. The support internal cavities have 2D nanosheets with leaf-like architectures (Fig. 3e) similar to those observed with 3.5 mg catalyst after 24 h reaction time (Fig. S3f†). Thus, by increasing the amount of the catalyst from 3.5 mg to 7 mg, we can grow COF composite membranes 6 times faster.
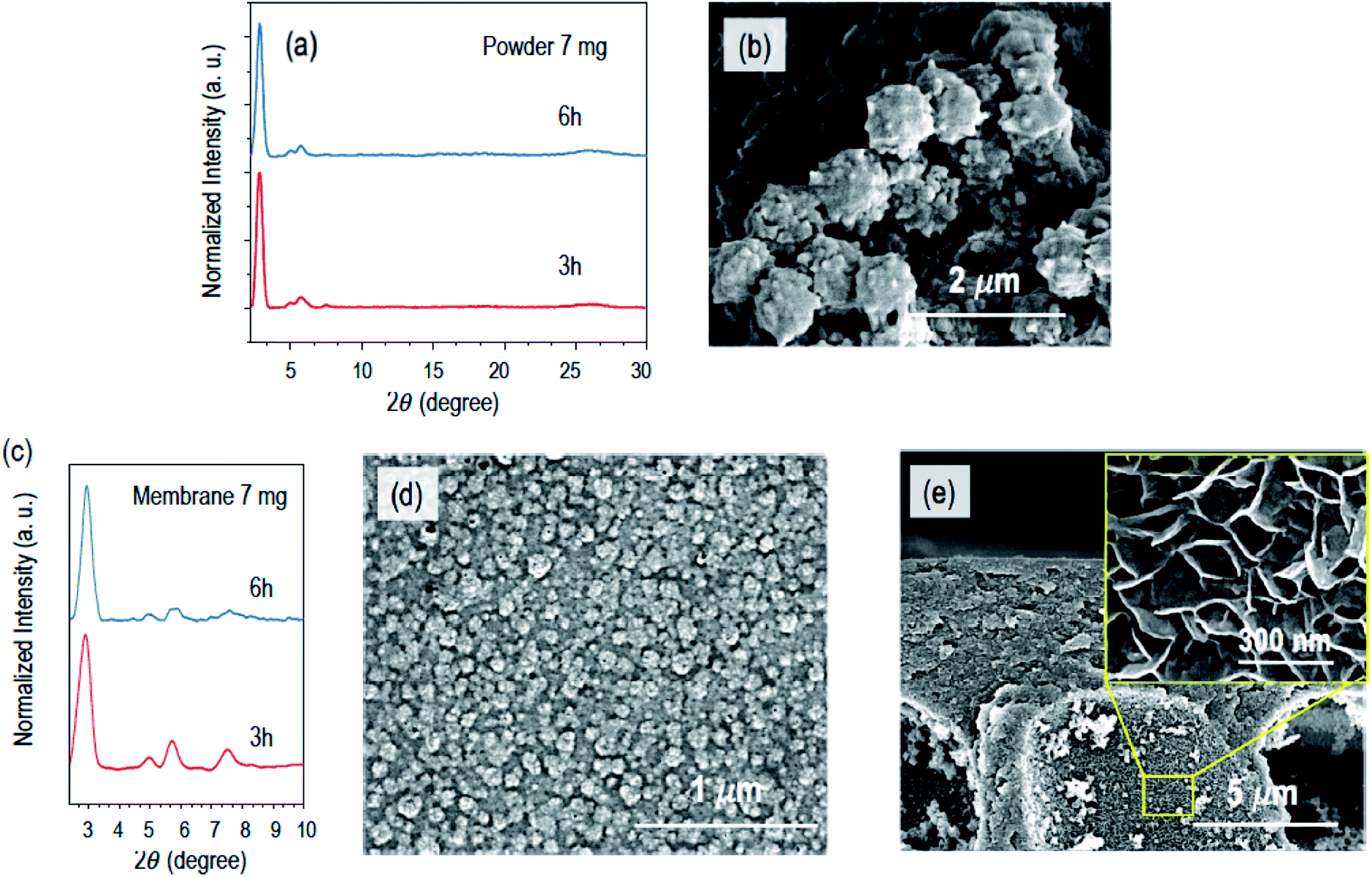 | ||
| Fig. 3 (a) PXRD profiles of the COF powder synthesized in bulk after reaction times of 3 and 6 h, using 7 mg of Sc(OTf)3; (b) SEM image of the COF powder obtained with 3 h of reaction; (c) PXRD profiles of COF membranes obtained with 3 and 6 h reaction time and 7 mg of Sc(OTf)3; (d) surface and (e) cross-sectional SEM images of COF composite membranes prepared with 3 h of reaction. | ||
We tested the permeance of the freeze-dried COF–XPAN composite membranes for liquid and vapor water. Water vapor removal from a humidified stream is one of the critical processes in industry and domestic air conditioning systems.27–29 Technologies like liquid adsorption and desiccant drying systems are in use for dehumidification but are energy-intensive.30–32 However, membrane technology has become an alternative to the traditional process due to energy efficiency and reliability. More and more focus has been dedicated to the development of new membrane materials with a higher water vapor permeation rate to attain desired humidity in the product stream.33 The liquid water permeance for this kind of application should be low to avoid leakage or back flow of the condensed water. The liquid water permeance of the COF–XPAN composite membranes was negligible. Table 1 shows the water vapor permeation rates for the membranes and their comparison with those of other composite membranes from recent reports.
| Membrane | Support | Condition | Temperature (°C) | Water vapor permeance (GPU) | Reference |
|---|---|---|---|---|---|
| Pebax® 1657 | Polyetherimide | Composite flat sheet | 21 | 260 | 35 |
| Polydopamine-TFC | Polyethersulphone | Composite flat sheet | 30 | 1029 | 36 |
| Polyvinyl alcohol/LiCl | Stainless steel scaffold | Composite flat sheet | 1790 | 37 | |
| TPB-DMTP COF–XPAN composite | Polyacrylonitrile on non-woven | Composite flat sheet | 25 | 2973 ± 22 | This work |
The water vapor permeation through the membrane was calculated using eqn (1):
| Qvapor =QN2 × γH20 × Vm/MW,H20 | (1) |
The permeance can be estimated using eqn (2):
| Permeance = Qvapor/(A × ΔP) | (2) |
The composite membranes prepared here have a water vapor permeation rate of 2973 ± 22 GPU (gas permeation rate, 1 GPU = 10 cm3 (STP) cm−2 s−1 cm−1 Hg), which is more than 10 times higher than that of composite membranes reported for Pebax®, which is considered promising for dehumidification.34,35
A modified upright permeability cup method (ASTM E96 standards)38 was used to analyse the water vapor permeance of the composite membrane, as outlined in Fig S6.†
The transport of liquid water thorough the small pores of the COF membrane has a different requirement. Water first needs to effectively wet the membrane to penetrate the pores. The COF surface has a water contact angle of 80 ± 1°, which is much higher than that of the XPAN support, which is 47 ± 3° and therefore makes it more hydrophobic. As a result, the transport of liquid water in the small COF pores would require a much high hydrodynamic pressure.
Conclusions
Diffusion-induced in situ growth leads to a flexible and scalable COF layer grown on a porous polymeric support. The method is advantageous, when compared to previously reported methods for COF sheet formation on/in porous supports. The COF layer is prepared in situ at room temperature with high ordering. There is no need for transferring a pre-formed layer, which would be a sensitive step and a frequent cause of defects. The method is simple and the size of the membrane is limited only by the size of the diffusion cell, which can be scaled up. We demonstrated a potential application in membrane dehumidification and other opportunities can be opened by applying the method to a large variety of materials.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was sponsored by the King Abdullah University of Science and Technology (KAUST), grants BAS/1/1057-01-01 and REP/1/3988-06-01.References
- C. S. Diercks and O. M. Yaghi, Science, 2017, 355, eaal1585 CrossRef PubMed.
- N. Huang, P. Wang and D. L. Jiang, Nat. Rev. Mater., 2016, 1, 1–19 Search PubMed.
- H. Xu, J. Gao and D. L. Jiang, Nat. Chem., 2015, 7, 905–912 CrossRef CAS PubMed.
- M. Calik, F. Auras, L. M. Salonen, K. Bader, I. Grill, M. Handloser, D. D. Medina, M. Dogru, F. Lobermann, D. Trauner, A. Hartschuh and T. Bein, J. Am. Chem. Soc., 2014, 136, 17802–17807 CrossRef CAS PubMed.
- D. D. Medina, T. Sick and T. Bein, Adv. Energy Mater., 2017, 7, 1700387 CrossRef.
- Z. P. Li, X. Feng, Y. C. Zou, Y. W. Zhang, H. Xia, X. M. Liu and Y. Mu, Chem. Commun., 2014, 50, 13825–13828 RSC.
- B. P. Biswal, H. D. Chaudhari, R. Banerjee and U. K. Kharul, Chem.–Eur. J., 2016, 22, 4695–4699 CrossRef CAS PubMed.
- J. R. Fu, S. Das, G. L. Xing, T. Ben, V. Valtchev and S. L. Qiu, J. Am. Chem. Soc., 2016, 138, 7673–7680 CrossRef CAS PubMed.
- S. Y. Ding, M. Dong, Y. W. Wang, Y. T. Chen, H. Z. Wang, C. Y. Su and W. Wang, J. Am. Chem. Soc., 2016, 138, 3031–3037 CrossRef CAS PubMed.
- S. Lin, C. S. Diercks, Y. B. Zhang, N. Kornienko, E. M. Nichols, Y. B. Zhao, A. R. Paris, D. Kim, P. Yang, O. M. Yaghi and C. J. Chang, Science, 2015, 349, 1208–1213 CrossRef CAS PubMed.
- P. H. H. Duong, V. A. Kuehl, B. Mastorovich, J. O. Hoberg, B. A. Parkinson and K. D. Li-Oakey, J. Membr. Sci., 2019, 574, 338–348 CrossRef CAS.
- A. P. Cote, A. I. Benin, N. W. Ockwig, M. O'Keeffe, A. J. Matzger and O. M. Yaghi, Science, 2005, 310, 1166–1170 CrossRef CAS PubMed.
- S. Kandambeth, B. P. Biswal, H. D. Chaudhari, K. C. Rout, H. S. Kunjattu, S. Mitra, S. Karak, A. Das, R. Mukherjee, U. K. Kharul and R. Banerjee, Adv. Mater., 2017, 29, 1603945 CrossRef PubMed.
- H. W. Fan, J. H. Gu, H. Meng, A. Knebel and J. Caro, Angew. Chem., Int. Ed., 2018, 57, 4083–4087 CrossRef CAS PubMed.
- E. Vitaku and W. R. Dichtel, J. Am. Chem. Soc., 2017, 139, 12911–12914 CrossRef CAS PubMed.
- M. Matsumoto, R. R. Dasari, W. Ji, C. H. Feriante, T. C. Parker, S. R. Marder and W. R. Dichtel, J. Am. Chem. Soc., 2017, 139, 4999–5002 CrossRef CAS PubMed.
- D. B. Shinde, G. Sheng, X. Li, M. Ostwal, A. H. Emwas, K. W. Huang and Z. P. Lai, J. Am. Chem. Soc., 2018, 140, 14342–14349 CrossRef CAS PubMed.
- K. Dey, M. Pal, K. C. Rout, H. S. Kunjattu, A. Das, R. Mukherjee, U. K. Kharul and R. Banerjee, J. Am. Chem. Soc., 2017, 139, 13083–13091 CrossRef CAS PubMed.
- M. Matsumoto, L. Valentino, G. M. Stiehl, H. B. Balch, A. R. Corcos, F. Wang, D. C. Ralph, B. J. Marinas and W. R. Dichtel, Chem, 2018, 4, 308–317 CAS.
- J. F. Yao, D. H. Dong, D. Li, L. He, G. S. Xu and H. T. Wang, Chem. Commun., 2011, 47, 2559–2561 RSC.
- H. T. Kwon and H. K. Jeong, J. Am. Chem. Soc., 2013, 135, 10763–10768 CrossRef CAS PubMed.
- Y. Du, W. Z. Qiu, Y. Lv, J. Wu and Z. K. Xu, ACS Appl. Mater. Interfaces, 2016, 8, 29696–29704 CrossRef CAS PubMed.
- A. Karimi, V. Vatanpour, A. Khataee and M. Safarpour, Ind. Eng. Chem. Res., 2019, 73, 95–105 CrossRef CAS.
- B. J. Smith, A. C. Overholts, N. Hwang and W. R. Dichtel, Chem. Commun., 2016, 52, 3690–3693 RSC.
- J. H. Sun, A. Klechikov, C. Moise, M. Prodana, M. Enachescu and A. V. Talyzin, Angew. Chem., Int. Ed., 2018, 57, 1034–1038 CrossRef CAS PubMed.
- N. Giuseppone, J. L. Schmitt, E. Schwartz and J. M. Lehn, J. Am. Chem. Soc., 2005, 127, 5528–5539 CrossRef CAS PubMed.
- H. Sijbesma, K. Nymeijer, R. van Marwijk, R. Heijboer, J. Potreck and M. Wessling, J. Membr. Sci., 2008, 313, 263–276 CrossRef CAS.
- K. L. Wang, S. H. McCray, D. D. Newbold and E. L. Cussler, J. Membr. Sci., 1992, 72, 231–244 CrossRef CAS.
- K. Kneifel, S. Nowak, W. Albrecht, R. Hilke, R. Just and K. V. Peinemann, J. Membr. Sci., 2006, 276, 241–251 CrossRef CAS.
- G. Q. Chen, S. Kanehashi, C. M. Doherty, A. J. Hill and S. E. Kentish, J. Membr. Sci., 2015, 487, 249–255 CrossRef CAS.
- M. Strand, J. Pagels, A. Szpila, A. Gudmundsson, E. Swietlicki, M. Bohgard and M. Sanati, Energy Fuels, 2002, 16, 1499–1506 CrossRef CAS.
- Y. H. Zurigat, M. K. Abu-Arabi and S. A. Abdul-Wahab, Energy Convers. Manage., 2004, 45, 141–155 CrossRef CAS.
- F. H. Akhtar, H. Vovushua, L. F. Villalobos, R. Shevate, M. Kumar, S. P. Nunes, U. Schwingenschlogl and K. V. Peinemann, J. Membr. Sci., 2019, 572, 641–649 CrossRef CAS.
- F. H. Akhtar, M. Kumar and K. V. Peinemann, J. Membr. Sci., 2017, 525, 187–194 CrossRef CAS.
- H. Q. Lin, S. M. Thompson, A. Serbanescu-Martin, J. G. Wijmans, K. D. Amo, K. A. Lokhandwala and T. C. Merkel, J. Membr. Sci., 2012, 413, 70–81 CrossRef.
- S. H. Yun, P. G. Ingole, K. H. Kim, W. K. Choi, J. H. Kim and H. K. J. C. E. J. Lee, Chem. Eng. J., 2014, 258, 348–356 CrossRef CAS.
- D. T. Bui, A. Nida, K. C. Ng and K. J. Chua, J. Membr. Sci., 2016, 498, 254–262 CrossRef CAS.
- Standard test methods for water vapor transmission of materials, Annual book of ASTM standards. Designation, 1989, vol. E96–E80, pp. 730–739 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ta11191c |
| This journal is © The Royal Society of Chemistry 2019 |