
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A new approach to very high lithium salt content quasi-solid state electrolytes for lithium metal batteries using plastic crystals†
Danah
Al-Masri
 a,
Ruhamah
Yunis
a,
Haijin
Zhu
a,
Liyu
Jin
b,
Peter
Bruce
bde,
Anthony F.
Hollenkamp
c and
Jennifer M.
Pringle
*a
a,
Ruhamah
Yunis
a,
Haijin
Zhu
a,
Liyu
Jin
b,
Peter
Bruce
bde,
Anthony F.
Hollenkamp
c and
Jennifer M.
Pringle
*a
aDeakin University, Melbourne, Institute for Frontier Materials, Victoria 3125, Australia. E-mail: j.pringle@deakin.edu.au
bDepartment of Materials, University of Oxford, Parks Road, Oxford, OX1 3PH, UK
cCommonwealth Scientific and Industrial Research Organisation (CSIRO), Clayton, 3168, VIC, Australia
dThe Henry Royce Institute, Parks Road, Oxford, OX1 3PH, UK
eThe Faraday Institution, Quad One, Becquerel Avenue, Harwell Campus, Didcot, OX11 0RA, UK
First published on 28th October 2019
Abstract
While the high energy density of lithium metal has long been a strong driver for the development of lithium metal batteries, harnessing the full theoretical capacity in a safe, practical device requires significant advances in electrolyte design. The use of quasi-solid state electrolytes can be greatly beneficial for increasing safety, suppressing the growth of lithium dendrites and prolonging cell lifetime. Organic ionic plastic crystals (OIPCs) are a unique class of disordered solid that can support high ionic conductivities and lithium ion mobility. Until recently, OIPCs were used primarily as matrix materials and incorporated only low dopant concentrations of lithium salts. Here we report a very high lithium content electrolyte containing 90 mol% lithium bis(fluorosulfonyl)imide, Li[FSI], combined with 10 mol% of a conductive pyrrolidinium FSI-based OIPC. The resultant quasi-solid state electrolyte achieves a conductivity of 0.24 mS cm−1 at 30 °C, supports stable lithium electrochemistry and has a very good lithium ion transference number of 0.68. Symmetrical Li|Li cell cycling is demonstrated at 0.1 mA cm−2 for 100 hours. This showcases a new approach for designing safer quasi-solid state electrolytes with high lithium content and excellent electrochemical and transport properties.
Introduction
There is increasing demand for the development of next generation batteries with increased safety, energy density and rate capability in order to meet the tremendous growth in technology, portable devices and electrical vehicles. Despite continued developments in lithium-ion batteries and new emerging battery technologies,1–5 the use of lithium metal anodes, with a theoretical capacity of 3860 mA h g−1 and an electrode potential of −3.04 V (vs. SHE), remains an attractive alternative for greatly improved theoretical energy density.6,7 However, the use of lithium metal is hindered by lithium dendrite growth during repeated battery cycling, leading to short circuits and a fire hazard, particularly when organic solvent-electrolytes are used.8,9 To help address the flammability problem, ionic liquid electrolytes have been investigated. These can support the stable performance of lithium, sodium and other emerging battery technologies,10–13 with the advantage of being non-flammable and possessing negligible vapour pressure. However, as with all liquid electrolytes, concerns remain over the possibility of leakage.Solid-state electrolytes have generated increasing interest in recent years.14,15 The safety and stability advantages of ionic electrolytes can be enhanced by use of organic ionic plastic crystals (OIPCs), the disordered solid-state analogues of ionic liquids. While many classes of solid-state electrolyte are limited by poor ion transport and/or brittleness, the intrinsic disorder and plasticity (soft physical properties) of OIPCs can be a significant advantage for target ion transport and good electrolyte/electrode interfacial contact.16–20 For application as electrolytes, the OIPC matrix is traditionally combined, or “doped”, with a small amount of lithium salt and the efficacy of this approach for lithium metal batteries has been demonstrated. For example, 4 mol% lithium bis(fluorosulfonyl)imide (Li[FSI]) in triisobutyl(methyl)phosphonium bis(fluorosulfonyl)imide ([P1i(444)][FSI]) supports good cycling performance of Li|LiFePO4 cells, with 160 mA h g−1 discharge capacity at 20 °C with 0.1C rate.21
The benefits of using very high lithium salt concentration liquid electrolytes have been recognised in recent years, overcoming prior concerns related to the increased viscosity of such systems, and the advantages can include excellent thermal and electrochemical stability, reduced Al (current collector) corrosion and high Li+ transference numbers.22–30 The transference number, which represents the ratio of current carried by the target metal ions (Li+) compared to the other ionic species, is one of the most important parameters that predict battery performance.31,32 Electrolytes with majority salt content, termed “solvent-in-salt”, have been shown to have high lithium transference number.25,33 The concept of “polymer-in-salt” electrolyte was first reported by Angell et al.33 for a polymer system with a glass transition below room temperature, which allowed the electrolyte to retain its flexibility and good ionic conductivity. The authors predicted a transference number close to unity, as estimated from decoupling index values.33 However, problems such as crystallisation of salts and high glass transition temperatures (Tg) can be a hindrance to the development of these electrolytes. A majority lithium salt content electrolyte (“water-in-salt”) was reported to expand the electrochemical window of water and support high efficiency lithium ion battery cycling up to 1000 cycles.34 Recently, Suo et al.25 reported a lithium ion transference number of 0.73 for a molecular solvent-based “solvent-in-salt” liquid electrolyte. The authors reported high cycling efficiency and significant reduction in dendrite formation. However, the safety concerns of molecular solvent electrolytes, because of their volatility and flammability, remain a concern.
For ionic electrolytes that can address these flammability concerns, Zhou et al. reported a dramatic increase in lithium ion transference number from 0.10 to 0.37 when the Li[FSI] content was increased from 10 mol% to 50 mol% in a [C2mpyr][FSI] composite system solidified with PVDF (where [C2mpyr] = N-ethyl-N-methylpyrrolidinium, and FSI = bis(fluorosulfonyl)imide).22 The benefits of high salt concentrations have also been established in sodium-based batteries utilising ionic liquids,35 and in a mixed-phase electrolyte of 40 mol% Na[FSI] in a phosphonium-based OIPC.24 Here, we introduce this concept to a new class of material, to develop “plastic crystal in salt” quasi-solid state electrolytes.
In this work, with respect to the choice of OIPC for the development of a high Li salt content electrolyte, it was important to utilise a particularly conductive and disordered plastic crystal, in order to mitigate mass transport limitations associated with the majority component, the more crystalline Li[FSI] salt. Pyrrolidinium-based ionic liquids and OIPCs have been shown to support stable lithium electrochemistry and uniform plating/stripping of lithium metal,22,36–38 and we have recently reported the synthesis and characterisation of a highly conductive OIPC based on the symmetrical N,N-diethylpyrrolidinium ([C2epyr]) cation and the FSI anion (ca. 10−5 S cm−1 at 30 °C).39 This is higher ionic conductivity than the more widely known [C2mpyr][FSI] OIPC.40,41 The [C2epyr][FSI] undergoes a solid–solid phase transition at −35 °C, enabling the material to exist in a relatively disordered state at ambient temperature. As a result, high ionic conductivity and mechanical properties favourable for practical application are obtained. With respect to the use of this OIPC as a solid electrolyte, preliminary demonstration of stable lithium electrochemistry and a lithium transference number of 0.27 at 60 °C with a low salt content of 5 mol% Li[FSI] has been demonstrated.40 In our previous study, it was shown that the combination of equimolar amounts of Li[FSI] (3.2 mol kg−1) with ([C2epyr]) results in a liquid electrolyte.42 This 50 mol% Li[FSI] in [C2epyr][FSI] liquid electrolyte exhibited a lithium transference number of 0.39 and cycling efficiency of 96% over 100 cycles in a Li|Li symmetrical cell.42 These preliminary results strongly supported the investigation of [C2epyr][FSI] for the development of very high Li content solid electrolytes.
Thus, here we report a quasi-solid state electrolyte based on [C2epyr][FSI] with 90 mol% Li[FSI]. The performance of the quasi-solid state electrolyte is compared to that of the corresponding liquid electrolyte of [C2epyr][FSI] with 50 mol% Li[FSI].42 The thermal phase behaviour of the new 90 mol% Li[FSI] quasi solid state-electrolyte is discussed, as characterised by DSC, while solid-state NMR spectroscopy shows the relative mobility of the different ionic components. Electrochemical measurements of the ionic conductivity, lithium ion transference number and lithium metal plating/stripping behaviour show the good target-ion transport properties of the electrolyte. Finally, cycling and post-mortem analysis on Li|Li symmetrical cells were performed. These results demonstrate the significant promise of the novel, highly concentrated Li plastic crystal in salt material for use in Li metal batteries.
Experimental
Synthesis and purification of [C2epyr][FSI]
Synthesis and purification of [C2epyr][FSI] was reported in our previous publication.39The 90 mol% Li[FSI] + 10 mol% [C2epyr][FSI] sample was prepared by dissolving Li[FSI] (CoorsTek, Inc., >99.5%) and [C2epyr][FSI] in acetonitrile (>99.9%, Sigma-Aldrich). Acetonitrile was then removed under vacuum (0.01 mbar). Samples were dried prior to all experiments under vacuum, at ambient temperature for 24 h and then 50 °C for 24 h. Samples were then transferred to an argon glove box.Thermal analysis
Thermal analysis was conducted by differential scanning calorimetry (DSC) using a Mettler Toledo DSC 822e or NETSCH DSC 214 Polyma instrument. Samples were enclosed in aluminium pans in an argon-filled glove box. Samples were subject to three heating cycles following a 30 min cooling period at the lowest temperature. The data shown was taken from the second heating cycle.The ionic conductivity and the charge transfer resistance
The ionic conductivity and the charge transfer resistance were measured using a Biologic MTZ-35 impedance analyser. Electrochemical impedance spectroscopy (EIS) was used, where a sinusoidal wave potential of amplitude 10 mV was applied at a frequency range of 7.0 MHz to 0.1 mHz, and the current response and corresponding phase shift were measured. The data was analysed using a Nyquist impedance plot, where typical semi-circles were obtained. The first touch-down was attributed to the solution resistance (Rsol) and the diameter of the semi-circle was attributed to the resistance of the electrolyte–electrode interface (RCT).43 Samples were enclosed in a dip-cell composed of two platinum spheres immersed in a sample vial and sealed from air by a rubber ring. Samples were loaded in the dip-cell and sealed an argon-filled glove box. The conductivity of the neat [C2epyr][FSI] sample was previously reported,42 measured in a barrel cell designed for solid state samples.Diffusion coefficients
Diffusion coefficients of 1H, 19F and 7Li were measured at different temperatures using a Bruker Avance III 300 MHz wide-bore NMR spectrometer equipped with a Diff50 diffusion probe. Pulse gradient stimulated echo was used for all the measurements. Diffusion times for 1H, 19F and 7Li were 20, 10, and 10 ms, respectively. Gradient pulse length was 5 ms, and the gradient strength was varied according to diffusion coefficients in order to cover the entire NMR signal attenuation. Recycle delay was 2 s for all nuclei and temperatures, to allow the spin magnetization to recover to equilibrium, and the gradient coil to dissipate heat. Solid-state NMR measurements were performed on a Bruker Avance III 500 MHz wide bore NMR spectrometer equipped with a 4 mm H/F-X double resonance magic angle spinning (MAS) probe. The spinning rate was 8 kHz for 1H and 19F MAS experiments. The static 1H, 19F and 7Li NMR spectra were recorded using the same probe, and the same sample, but in a stationary state. The static spectra were recorded after the spinning. RF pulse length was 4 μs for all the nuclei, and recycle delays were 10 s for 1H and 19F, and 300 s for 7Li NMR.Lithium transference number (tLi+)
Lithium transference number (tLi+) was measured electrochemically following the Vincent–Bruce method.31 Li|Li symmetrical cells were assembled in an argon-filled glove box using CR 2032-type coin cells. The electrolyte was enclosed in a polyethylene ring used as a spacer with thickness of 0.5 mm, inner diameter of 8 mm and outer diameter of 16 mm. No separator material was used.Lithium transference number was also estimated using the diffusion coefficients of 1H, 19F and 7Li measured by NMR. The transference number was estimated to be the ratio of the diffusion coefficient of 7Li relative to the combined diffusion coefficient of all 1H, 19F and 7Li species present.
Li|Li symmetrical cells
Li|Li symmetrical cells, described above, were cycled by a Biologic VMP3 under an applied current.Post-mortem analysis
Post-mortem analysis of lithium metal electrodes was performed following electrolyte removal and electrode washing with dimethyl carbonate (DMC) in an argon-filled glove box. Images were taken using a JEOL JSM-IT300-LV scanning electron microscope (SEM) with an accelerating voltage of 5 kV.Cyclic voltammetry (CV)
Cyclic voltammetry (CV) was performed using a Biologic SP-200 potentiostat and a three electrode cell in an argon-filled glove box. A platinum disk with surface area of 2.0 mm2, a lithium metal coil and a lithium metal strip were used as working, counter and reference electrode respectively. A 20 mV s−1 scan rate was used.The average cycling efficiency (ACE)
The average cycling efficiency (ACE) was measured in the three electrode cell described above according to literature procedure;44 a known amount of lithium was deposited on the electrode surface (Qp) and a fraction of that deposit (Qc) was cycled until all the lithium initially deposited was depleted (giving a potential spike). ACE is given by 100 × NQc/(NQc + Qp), where N is the number of cycles obtained before depletion of the lithium deposit. Current values used throughout this experiment were gradually increased from 0.1 mA cm−2 to 0.5 mA cm−2, where the highest ACE was obtained at Qp = 1.0 mA h cm−2 and Qc = 0.25 mA h cm−2.Results and discussion
Thermal and transport properties
The plastic phase(s) of OIPCs are reached by heating through one or more solid–solid phase transition, with phase I the most conductive – naming convention designates phase I as the highest temperature solid phase. These solid–solid phase transitions signify the onset of rotational and/or translational motion of the ions within the crystal lattice of the OIPC, creating short-range disorder and defects that support good ionic mobility. Fig. 1(a) shows the DSC traces of neat [C2epyr][FSI], neat Li[FSI] and the mixture with 90 mol% Li[FSI]. The phase behaviour of [C2epyr][FSI] and the mixtures with up to 50 mol% Li[FSI] were reported previously.42 The neat [C2epyr][FSI] undergoes a solid–solid phase transition at −34 °C and melts at 131 °C with a small entropy change (9 J K−1 mol−1), which is a feature indicative of plastic crystal behaviour.45 The 90 mol% Li[FSI] sample displays a small (ca. 1 J K−1 mol−1) solid–solid phase transition at −50 °C, similar to that displayed by the neat Li[FSI]. This suggests the presence of a certain amount of Li[FSI] in the mixture, which is consistent with the NMR discussed below. Broad melting peaks are a feature commonly observed in OIPCs with salt addition, both in single phase and multi-phase systems.13,24,46 Visually, the 90 mol% Li[FSI] sample is a soft solid (Fig. 1, inset). Thus, the combination of 10 mol% OIPC with the Li[FSI] produces a salt that can exist in a quasi-solid state until temperatures well-over ambient, which is beneficial for incorporation into practical devices without leakage.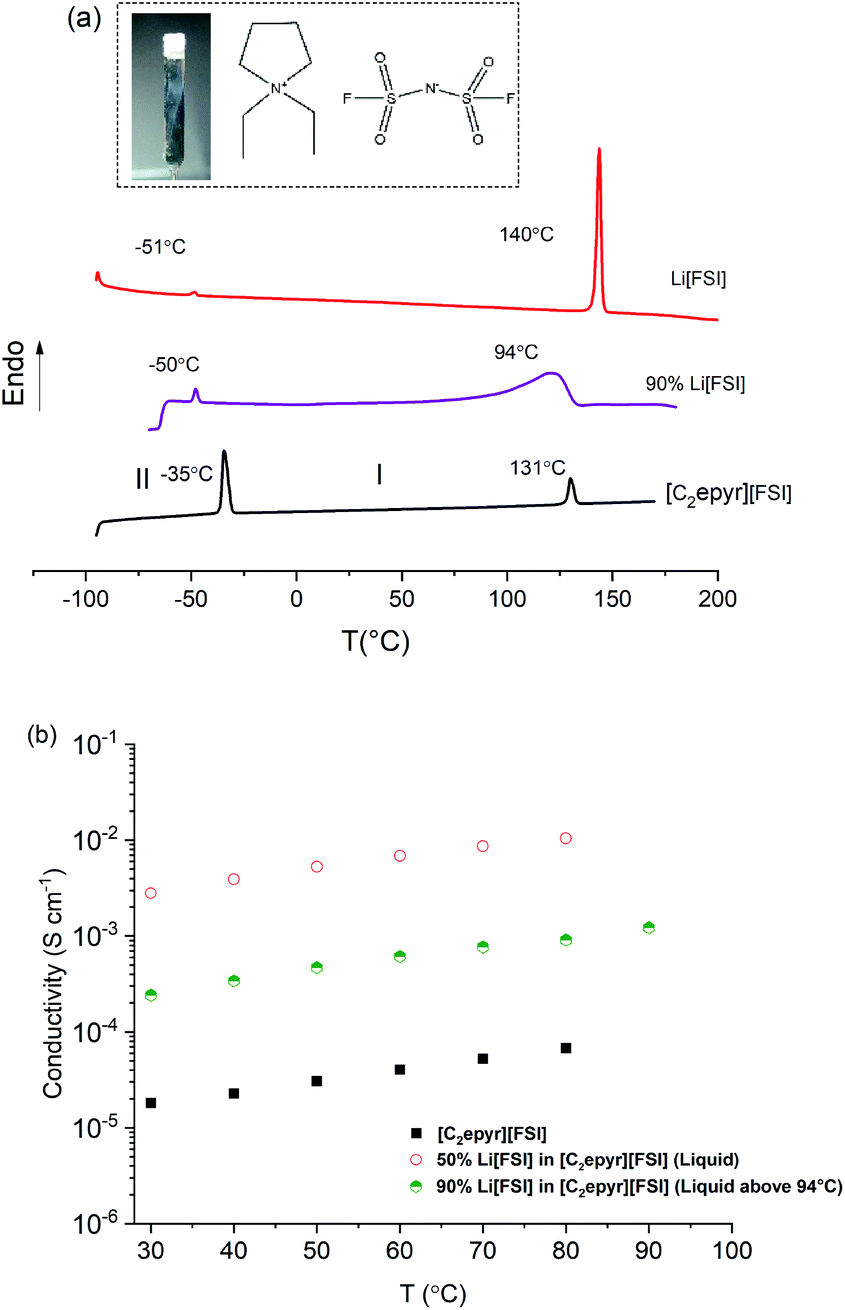 | ||
| Fig. 1 (a) The thermal phase behaviour of the neat plastic crystal [C2epyr][FSI], Li[FSI], and [C2epyr][FSI] combined with 90 mol% Li[FSI], measured by DSC. Onset temperatures shown. An arrow indicates the endothermic heat flow direction. Inset: the structure of [C2epyr][FSI] and a photograph of the new high lithium salt content 90% Li[FSI] in [C2epyr][FSI] quasi-solid state material on a spatula held vertically, demonstrating its solid nature, (b) the ionic conductivity of the neat plastic crystal [C2epyr][FSI] and Li[FSI]-containing materials. Addition of 50 mol% Li[FSI] produces an ionic liquid, while 90 mol% Li[FSI] yields a very conductive quasi-solid state material. | ||
The 90 mol% Li[FSI] quasi-solid state material exhibits an ionic conductivity of ca. 2 × 10−4 S cm−1 at 30 °C (Fig. 1(b)), which is relatively high compared to many other reported solid-state electrolyte materials such as polymers and thin films (conductivity range of ca. 10−3 to 10−7 S cm−1),14 and two orders of magnitude higher than for the previously reported high Li content “polymer-in-salt” systems (ca. 10−6 S cm−1 for 90 mol% lithium salt at room temperature).33
The ionic conductivity of this OIPC-based electrolyte is only about an order of magnitude lower than its liquid counterpart with 50 mol% Li[FSI], but without the associated leakage concerns for device application. The conductivity is also significantly higher than for the neat OIPC, despite the fact that this plastic crystal is one of the most conductive reported to-date and that it is combined with a large proportion of a more crystalline material. This enhanced transport is consistent with the observation of increased dynamics in the mixture, compared to the neat materials, also shown by the NMR analysis discussed below. Thus, the thermal analysis and conductivity suggest the presence of a very mobile phase combined with a Li[FSI]-rich phase. While the material is solid overall, the mobile phase – which has a high Li content – may be a liquid phase that exists within the grain boundaries of the solid Li[FSI] rich phase. As proposed by Forsyth et al.24 for a high Na salt content OIPC system, this morphology could provide the advantages of liquid-like target ion transport within an overall quasi-solid state system, plus a “reservoir” of Li ions that can be utilised during Li cycling. The increased ionic conductivity of OIPC–lithium salt mixtures due to the presence of a liquid phase was also discussed by Henderson et al.47 for a mixture of tetraethylammonium bis(trifluoromethanesulfonyl)imide with lithium bis(trifluoromethanesulfonyl)imide, where the formation of a eutectic composition at low lithium salt concentration allowed a liquid-like conductivity mechanism in the electrolyte.
Lithium transference number
A high transference number of lithium ions (tLi+) is one of the most desirable properties for Li battery electrolytes because it can lower the internal resistance and increase the rate capability of the cell.32 Here, for the new high lithium content electrolyte, a tLi+ of 0.68 was achieved at 25 °C, measured by the Bruce–Vincent electrochemical method.31,46 To the best of our knowledge, this is the highest reported transference number for an OIPC-based electrolyte to date. Previous reports, which have concentrated on lower Li salt concentrations, gave tLi+ ranging from ca. 0.27 to 0.44.24,38,40,41 Single ion conducting polymer electrolytes have been designed that achieve transference numbers closer to unity, such as the 0.85 achieved by Bouchet et al.,33,48,49 but often with a trade-off of relatively low ionic conductivities (e.g. in the 10−5 S cm−2 range at ambient temperature). Thus, the combination of good transference number and high ionic conductivity in the 90 mol% Li[FSI] quasi-solid state OIPC electrolyte are a promising combination for device application.Solid-state and diffusion NMR analysis
The structure of the new electrolyte material and the mobility of the different ions was analysed by multinuclear solid-state NMR spectroscopy. The line widths of static NMR spectra indicate the extent of mobility in materials. OIPCs typically exhibit narrow linewidths as a result of disorder in the matrix allowing higher degrees of motion compared to crystalline solids.20 Thus, this technique can be very valuable for revealing the presence of rotational or translational motion, and any structural heterogeneity in the material.50 For example, in an OIPC-based material the presence of a fraction of mobile ions within a more rigid matrix can be indicated by the presence of a sharp peak superimposed on a broad peak in the static spectra.20,51 Furthermore, 1H, 19F and 7Li NMR analysis can be used to investigate the relative mobilities of the cation, anion and lithium ions, respectively.The line widths of the static 1H NMR spectra for pure [C2epyr][FSI] and the quasi-solid state 90 mol% Li[FSI] + [C2epyr][FSI] electrolyte are shown in Fig. 2(a). The narrower 1H line width of the latter indicates considerably faster [C2epyr] cation dynamics in the quasi-solid state electrolyte compared to the pure OIPC, which is consistent with the higher conductivity.
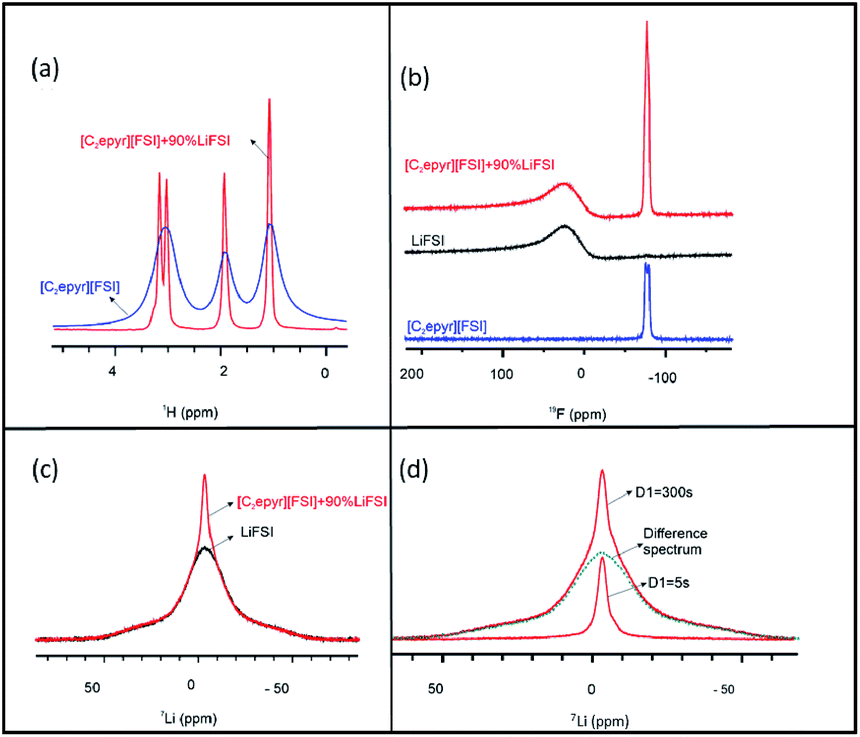 | ||
| Fig. 2 Multi-nuclei solid-state NMR spectra of the new “plastic crystal in salt” material containing 90 mol% Li[FSI] in [C2epyr][FSI], as well as the neat [C2epyr][FSI], demonstrating the ion dynamics. (a) Static 1H NMR spectra of the neat OIPC and the 90 mol% Li[FSI]-containing sample, where the narrower 1H lines indicate faster cation dynamics in the Li[FSI]-containing system as a result of increased disorder in the structure. (b) Static 19F NMR spectra, (c) static 7Li NMR spectra of the 90 mol% Li[FSI] in [C2epyr][FSI], and of Li[FSI], and (d) static 7Li NMR spectra of the 90 mol% Li[FSI] in [C2epyr][FSI] where the lower red solid line was recorded with a 5 second recycle delay (D1) in order to suppress the NMR signal from the longer T1 (crystalline) component; the upper solid red line was recorded with 300 s recycle delay in order to obtain a fully relaxed NMR spectrum from the overall sample; the green dashed line is the difference spectrum that is attributed to the 7Li signal from the more crystalline component. All spectra were acquired at room temperature. | ||
The static 19F NMR spectra of the three different materials is shown in Fig. 2(b). There is a broad peak in the spectrum of the quasi-solid state electrolyte containing 90 mol% Li[FSI] that appears at the same position as the broad peak in the Li[FSI] sample (black line). There is also a sharp peak that appears in the same position as the peak in the pure [C2epyr][FSI] (blue line). Thus, the NMR analysis indicates two different environments for the FSI anions in the new electrolyte system. The position of the broad peak, which is attributed to Li[FSI], is far off the [FSI] peak of the neat [C2epyr][FSI] as a result of the large chemical shift anisotropy (CSA) of the 19F in the more crystalline material, causing significant line-broadening. The isotropic chemical shift of the narrow peak is very close to that of the 19F peak in the neat [C2epyr][FSI], which is also observed in the MAS spectra (Fig. S1†). The static 19F spectrum of the neat [C2epyr][FSI] (Fig. 2(b)) shows line splitting, which is removed by magic angle spinning (Fig. S1†). This suggests that in the static [C2epyr][FSI] sample the two fluorine atoms present on the [FSI] anion are orientated such that they are at different angles relative to the applied magnetic field, causing slightly different chemical shifts. Importantly, this is not observed in the OIPC with 90 mol% Li[FSI], indicating faster reorientation motion of the [FSI] anions in the mobile phase of the new electrolyte that is consistent with the higher conductivity.
Fig. 2(c) shows the static 7Li spectra of the quasi-solid state electrolyte material and of pure Li[FSI]. The spectrum of the new electrolyte shows two peaks – a broad and a narrow component – and the latter is much narrower (and thus due to more mobile Li) than for pure Li[FSI]. This is analogous to the composition previously proposed for a mixed-phase (solid and liquid) mixture of OIPC combined with a sodium salt.24 The two different components were further investigated by assessing the influence of relaxation time, as shown in Fig. 2(d). The fully relaxed static 7Li NMR line, which was obtained with a 300 second relaxation time, shows two peaks: a broad and a narrow component. This narrow component was isolated by a second scan with shorter relaxation time of 5 seconds, which allows the elimination of the crystalline signal that takes much longer to relax. Integration of the narrow peak shows that 16% of lithium is dissolved in the OIPC, and the linewidths show that this results in Li ions that are considerably more mobile than in the solid Li[FSI].
To investigate further the ion mobility, the diffusion coefficients of 7Li, 1H and 19F were measured by pulse-field gradient (PFG) NMR at different temperatures (Fig. 3). This allows comparison of the mobility of the lithium ions in the electrolyte compared to the [C2epyr] cation and [FSI] anion, represented by 1H and 19F respectively.
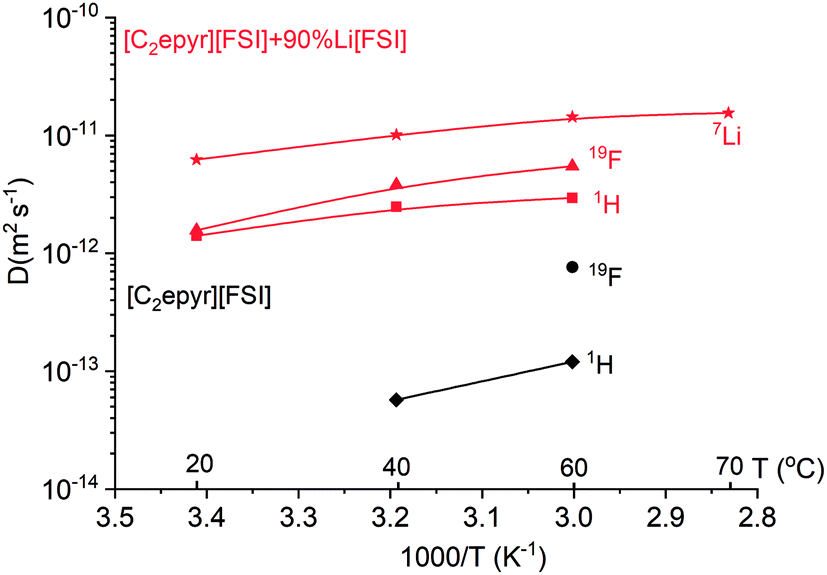 | ||
| Fig. 3 The diffusion coefficients of the different ionic species in the neat [C2epyr][FSI] and 90 mol% Li[FSI] + [C2epyr][FSI] samples, revealing the higher diffusion of 7Li ions compared to the other ionic species. Data was obtained from PFG-NMR experiments. The solid lines are guides to the eye. | ||
The diffusion coefficients for all the ions in the quasi-solid state 90 mol% Li[FSI] mixture are significantly higher than those in the neat [C2epyr][FSI], which is consistent with the linewidths in the static NMR (Fig. 2) and with the higher conductivity (Fig. 1(b)). Importantly, the 7Li is the most diffusive species in the quasi-solid state mixture, giving a transference number of 0.63 by NMR analysis. This is consistent with the high transference number of 0.68 measured electrochemically, discussed above, and an important predictor of good device performance. Typically, transference number values measured electrochemically show lower values than those measured by NMR because of ion pairing.52 The fact that this is not observed in the present system appears to be consistent with the relatively high mobility of Li, shown by NMR, which indicates less coupled ionic mobility. Thus, in summary, the NMR analysis indicates high target ion mobility and also suggests the presence of both a Li[FSI]-rich environment and a second environment of more mobile ions.
Lithium electrochemistry
The highly reactive nature of lithium metal poses a significant practical challenge in the development of next generation battery technologies employing a lithium metal anode. To achieve long cycle-life, stabilisation of the lithium metal is required and the battery electrolyte assumes the principal role through its interaction with lithium. Fundamentally, the electrolyte must form a stable solid-electrolyte interphase (SEI) layer, which modulates the flow of lithium ions to and from the electrode surface, and ideally allows lithium plating and stripping to proceed with high coulombic efficiency.6,7,53 Of the wide range of electrolyte materials that have been investigated for lithium metal battery applications,54,55 many promote the formation of an SEI but ultimately fail to deliver the high cycling efficiencies required. This situation eventually leads to Li dendrite formation, particularly at higher current densities, which remains a significant challenge in the deployment of lithium metal anodes.6,7,14 Cyclic voltammetry (CV) in a three electrode cell is an important first step that provides insight into the electrochemical stability of new electrolytes during lithium cycling, as well as giving an initial estimate of the coulombic efficiency. While coulombic efficiencies obtained from CVs are typically modest due to the fast scan rate compared to cell cycling, this technique can provide fundamental understanding of the lithium plating/stripping kinetics that is important for further development of the electrolyte. Fig. 4(a) shows the cycling voltammetry of the quasi-solid state 90% Li[FSI] + [C2epyr][FSI] sample at room temperature. The material shows stable cycling behaviour, with distinct lithium plating and stripping peaks occurring within the previously reported electrochemical window of [C2epyr][FSI] (>3.6 V).42 Moderate coulombic efficiencies of >70% were obtained at room temperature in the second and third cycle. An increased coulombic efficiency of 84% was achieved using an elevated temperature of 50 °C (Fig. S2†), attributed to an increase in ionic conductivity. The occurrence of a second peak during the stripping cycles is a result of lithium alloying with the platinum working electrode.56 This preliminary assessment of the electrolyte demonstrates that this new class of “plastic crystal in salt” material can stabilise lithium metal and support lithium electrochemistry. | ||
| Fig. 4 (a) Cyclic voltammetry (CV) showing the lithium plating and stripping from the 90 mol% Li[FSI] quasi-solid state electrolyte. Data was obtained using a three-electrode cell: a platinum disc with 2.0 mm2 surface area, a lithium strip and a lithium coil were used as working, reference and counter electrodes respectively. (b) The average cycling efficiency measurement were performed using the same three-electrode cell at the same temperature. | ||
The average cycling efficiency (ACE) was then measured by cycling a fraction of a pre-deposited quantity of lithium in order to gain an accurate estimation of the reversibility of the lithium plating and stripping process. The plot of electrode potential against time for a typical ACE experiment, shown in Fig. 4(b), exhibits a progressive increase in over-potential with cycle number that indicates a build-up of “dead” lithium on the electrode surface, creating a more resistive layer.44 The highest ACE of 85% was obtained at a relatively high current density of 0.5 mA cm−2, where a charge of 1.0 mA h cm−2 was initially deposited on the electrode surface and a charge of 0.25 mA h cm−2 was deposited and stripped during each cycle. The experiment was stopped when the potential value exceeded 2.5 V. High ACEs of >95% are typically reported for liquid electrolytes,44 including the previously reported 50 mol% Li[FSI] in [C2epyr][FSI] liquid electrolyte that produced an ACE of 96%,42 which confirms the good chemical stability of [C2epyr][FSI]. Thus the lower ACE value achieved in the 90% Li[FSI] system is attributed largely to the impact of slower mobility of ions in the solid electrolyte than in the liquid, consistent with the order of magnitude lower ionic conductivity. Additionally, we hypothesise that the formation of multiple electrolyte morphologies at the electrode surface could play a role in reducing the ACE, where the presence of a crystalline Li[FSI] component may have hindered the formation of a stable SEI in some regions. Future work will explore preconditioning of the lithium metal surface to create a favourable morphology, as demonstrated in previous studies.53,57
Li|Li symmetrical cell cycling
The formation of a smooth and stable SEI layer is widely considered essential for long term cycling of lithium metal cells and therefore investigation into the nature and formation of this layer with different electrolytes has been the subject of extensive studies.58–61 Here, the ability of the new quasi-solid state electrolyte to form a stable SEI layer and support lithium cycling was investigated using Li|Li symmetrical cells.Fig. 5(a) shows the cycling data of the quasi-solid state electrolyte obtained at 0.1 mA cm−2 current density with half cycles of one hour. At 25 °C, stable cycling with an over-potential of 200 mV was obtained for the first 28 cycles, followed by an unstable potential trace and ultimately a short circuit at cycle 38 marked by a drop in over-potential. A typical increase in SEI layer resistivity is evident in the increase in over-potential between cycle 1 and cycle 21. This is also apparent in EIS data collected after selected plating cycles, shown in Fig. S3,† where an increase in the interfacial resistance occurs with increasing cycle number – a feature commonly observed in solid-state electrolyte cells.41,62
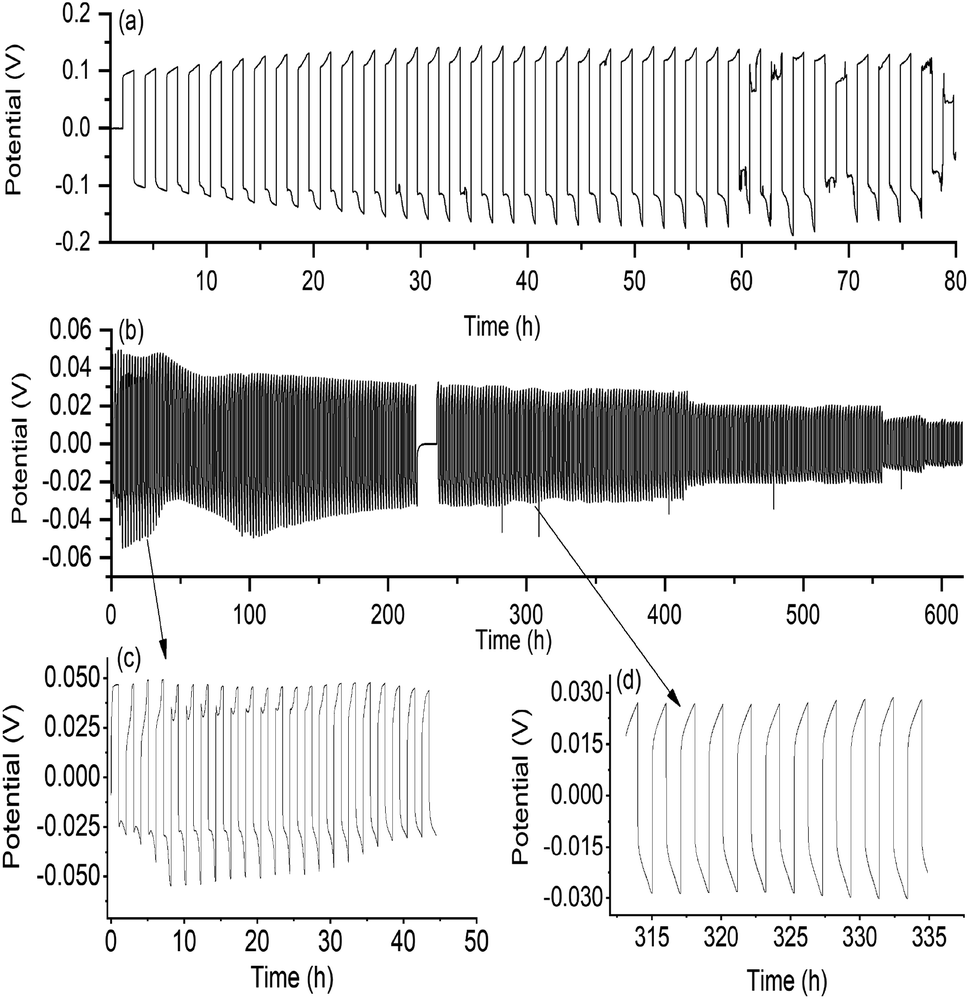 | ||
| Fig. 5 (a) Cycling of Li|Li symmetrical cells at 25 °C with 0.1 mA cm−2 current density and one hour half cycles. The cells contain the (a) quasi-solid state 90 mol% Li[FSI] in [C2epyr][FSI] and (b) the liquid 50 mol% Li[FSI] in [C2epyr][FSI] electrolyte. The apparent gap in the cycling profile in (b) is a result of a pre-set resting period. A close up view of the first 22 cycles of the liquid cell is shown in (c), and at 315 hours shown in (d). As a result of the solid nature of the materials, no separator was needed in the symmetric cell with either electrolyte – only a 0.5 mm thick polyethylene ring spacer. | ||
In order to investigate further the stability of the SEI layer formed with this new cation and anion chemistry, the 50 mol% Li[FSI] in [C2epyr][FSI] liquid electrolyte was cycled in Li|Li symmetrical cells under the same conditions (Fig. 5(b)). Contrary to the quasi-solid state electrolyte, the cycling profile shows decreasing over-potential with cycle number. This is common cycling behaviour for liquid electrolytes, where the growth of dendrites on the electrode surface reduces the inter-electrode spacing and causes the over-potential to drop until the cell short circuits.53,61,63
The decreasing over-potential is in agreement with the corresponding EIS data that shows a decrease in the interface resistance (RCT) over the cycling profile, shown in Fig. S3.† The liquid 50 mol% electrolyte supported stable cycling of lithium for over 300 cycles. This confirms that the chemical components of the electrolyte with the new [C2epyr] cation are suitable for lithium battery applications. It also highlights that the cell behaviour of the 90 mol% Li[FSI] system (Fig. 5(a)) reflects the quasi-solid nature of the material, which changes the transport and interfacial resistances but brings advantages of eliminating leakage and increasing safety. The authors predict that cell design optimisation such as reduction of electrolyte thickness or cycling at elevated temperatures could significantly enhance cycle life.
Post mortem analysis of Li|Li symmetrical cells
The mechanism of dendrite formation on lithium metal surfaces has been the subject of extensive study, and mass transport limitations are widely considered to be a leading cause of the problematic uneven plating and stripping processes.61,64–66 An important potential advantage of increasing the lithium salt concentration is mitigating the mass transport limitations by providing a high concentration of active species near the electrode surface (these being primarily the electroactive Li+, but also the FSI anion for SEI formation).25,67,68 To help identify the failure mechanisms of the symmetrical cells utilising the high salt content liquid and solid electrolytes, SEM analysis was used to assess the electrode morphology post-cycling.The cell containing the liquid electrolyte was cycled for over 300 cycles until it short circuited, shown in Fig. 5. Fig. 6(a) shows an overview image of the lithium metal post-cycling with the liquid electrolyte, revealing a distribution of small dendrites across the metal surface. This is consistent with previous studies where dendritic growth leads to decreasing interfacial resistance due to increased surface area on the lithium metal surface and an ultimate failure of the cell when short circuit occurs.6,43,53,63,69
 | ||
| Fig. 6 SEM images of the lithium electrode surface post-cycling for: (a and b) the liquid 50 mol% Li[FSI] in [C2epyr][FSI], and (c and d) the 90% Li[FSI] quasi-solid state electrolyte. The area marked by an arc and arrows was under the polyethylene ring used as the spacer and thus not in contact with the electrolyte. Circled areas in (c) show reduced dendritic formations for metal cycled using the new quasi-solid state electrolyte. The magnifications used were X18 and X22 for (a) and (c) respectively, X200 for (b) and (d). The scale bars are noted on each figure. | ||
The decreasing resistance is evident in the decreasing over-potential over the cycling profile shown in Fig. 5(b) and in the Nyquist plots obtained after plating the electrode shown in Fig. S3.† The marked electrode area was protected by a polyethylene spacer and thus represents the lithium electrode still in pristine condition.
Fig. 6(c) and (d) shows the surface of the lithium metal post-cycling with the 90 mol% Li[FSI] quasi-solid state electrolyte. The cell was cycled for approximately 100 hours until it short-circuited, shown in Fig. 5(a). Here, the dendrites formed on the electrode surface appear to be smaller compared to those formed with the liquid electrolyte containing a lower concentration of lithium. Some regions on the surface – indicated by white circles – show no significant change to the lithium metal, indicating relatively even plating and stripping processes. The retention of a smooth lithium surface when cycled with highly concentrated electrolyte was demonstrated by Suo et al.,25 where a 7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 salt to solvent system showed a notably smooth surface compared to its lower concentration counterparts. Furthermore, solid-state electrolytes can provide an additional physical barrier to the growth of Li dendrites.43,55 Future studies with the new “plastic crystal in salt” electrolyte will investigate the use of additives or separator materials such as glass or polymer fibres to provide a stronger physical barrier against dendrite formation and further prolong cell lifetime.
:1 salt to solvent system showed a notably smooth surface compared to its lower concentration counterparts. Furthermore, solid-state electrolytes can provide an additional physical barrier to the growth of Li dendrites.43,55 Future studies with the new “plastic crystal in salt” electrolyte will investigate the use of additives or separator materials such as glass or polymer fibres to provide a stronger physical barrier against dendrite formation and further prolong cell lifetime.
Assessment of the stability of the SEI
To investigate the stability of Li cells containing the quasi-solid state 90 mol% Li[FSI] electrolyte, EIS measurements were taken every hour for 32 days to monitor the changes to the cell resistance and SEI. The Nyquist plots, calculated charge transfer resistance (RCT) and solution resistance (Rsol) are shown in Fig. 7.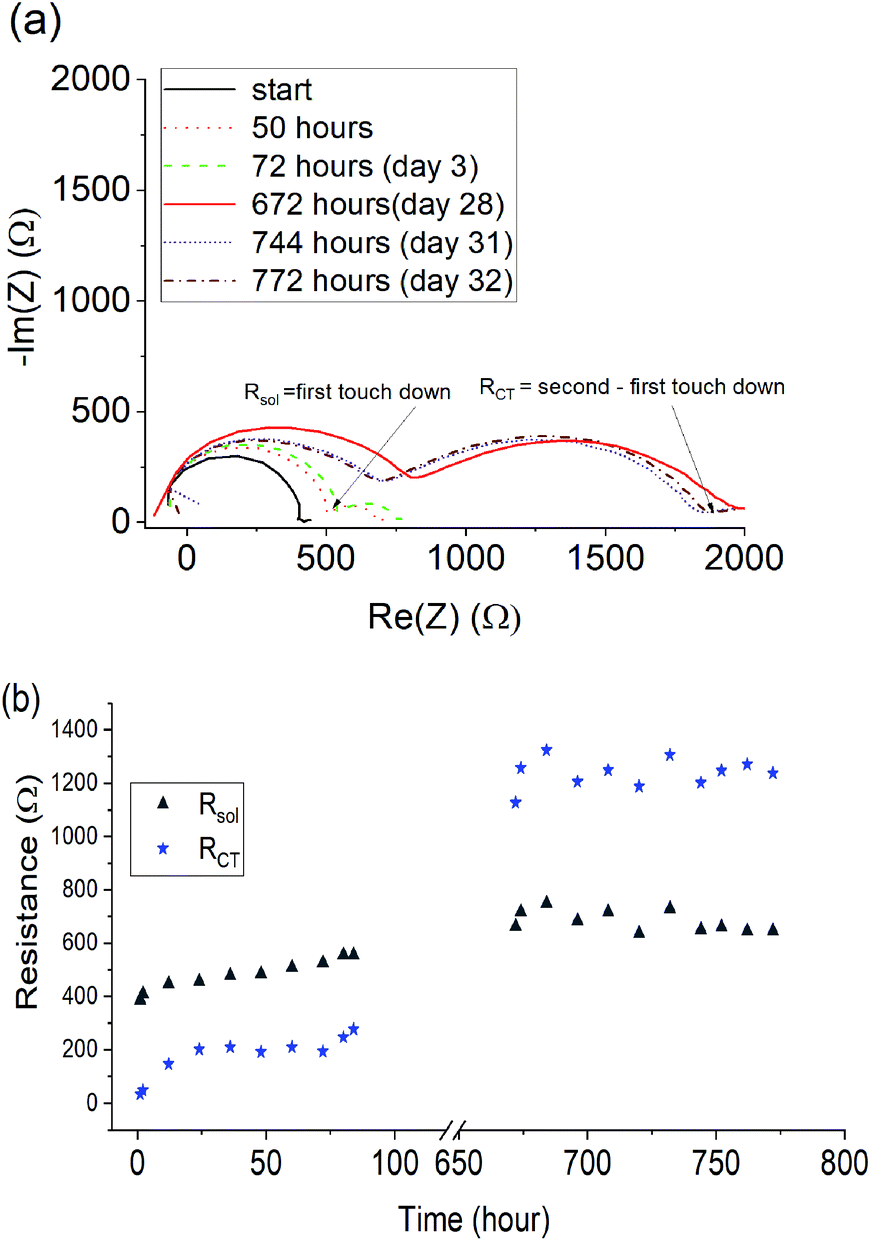 | ||
| Fig. 7 The electrolyte and interfacial resistance in a symmetrical Li|Li cell shown by (a) Nyquist plots obtained every hour for symmetrical cells containing the 90% Li[FSI] quasi-solid state electrolyte resting in a 25 °C oven and (b) the solution resistance (Rsol) and charge transfer resistance (RCT) obtained from the Nyquist plots and plotted vs. time. | ||
A 1 kΩ increase in the RCT occurred over the first 28 days, after which it stabilised, indicating the formation of a stable SEI layer. A less pronounced 200 Ω increase was observed in the solution resistance, attributed to small changes in the composition of the electrolyte as a result of SEI formation. For the high salt content quasi-solid state electrolytes reported here, the stabilization of the interfacial resistance over time suggests that pre-treatment of the lithium metal electrode may be beneficial for forming a stable SEI before cycling. This approach has been demonstrated by Basile et al.53 in ionic liquid-based electrolytes and is proposed here as a future avenue for further improving the cycling behaviour of new high salt content quasi-solid state electrolytes.
Conclusions
The advancement of new solid-state electrolytes is an important strategy for mitigating leakage and the safety concerns associated with the very reactive Li metal. Highly concentrated liquid electrolytes have previously been suggested as a route to improved lithium cycling and reduced dendrite growth, by decreasing the mass transport limitations hampering conventional electrolytes. Here we have combined these two important strategies, reporting a novel “plastic crystal in salt” electrolyte with 90 mol% Li[FSI] for use in lithium metal batteries. The highly conductive organic ionic plastic crystal [C2epyr][FSI] was used at 10 mol% concentration, imparting the predominantly Li[FSI]-based electrolyte with advantageously soft, quasi-solid state properties. Electrochemical analysis of the electrolyte shows fast ion dynamics, where the high ionic conductivity is dominated by lithium ions with a transference number of 0.68. Additionally, stable lithium plating and stripping behaviour was demonstrated, supported by a reduction in dendrite formation on the lithium surface compared to a liquid electrolyte comprised of the same components. NMR analysis suggests the presence of both a Li[FSI]-rich environment and a second environment of very mobile ions – possibly in a liquid phase contained within the grain boundaries of the overall quasi-solid state material – and supports the assessment of high transference number. Thus, the combination of a small amount of high conductivity OIPC with 90 mol% Li[FSI] is proposed as a new route to quasi-solid state electrolytes with fast target ion transport that supports efficient and stable lithium metal plating and stripping. This approach paves the way for the development of other very high salt content quasi solid-state electrolytes utilising plastic crystals, and the advancement of more stable, safer and efficient lithium metal batteries.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Australian Research Council (ARC) through Discovery Grant DP170101087. The authors thank Prof Maria Forsyth for useful discussions. P. G. B. is indebted to the Engineering and Physical Sciences Research Council (EPSRC), including the SUPERGEN Energy Storage Hub [EP/L019469/1], Enabling Next Generation Lithium Batteries [EP/M009521/1], Henry Royce Institute for Advanced Materials [EP/R00661X/1, EP/S019367/1, EP/R010145/1] and the Faraday Institution All-Solid-State Batteries with Li and Na Anodes [FIRG007, FIRG008] for financial support.Notes and references
- J. B. Goodenough and K.-S. Park, J. Am. Chem. Soc., 2013, 135, 1167–1176 CrossRef CAS PubMed.
- C. Ban, Z. Wu, D. T. Gillaspie, L. Chen, Y. Yan, J. L. Blackburn and A. Dillon, Adv. Mater., 2010, 22, E145–E149 CrossRef CAS PubMed.
- D. Ye, K. Ozawa, B. Wang, D. Hulicova-Jurcakova, J. Zou, C. Sun and L. Wang, Nano Energy, 2014, 6, 92–102 CrossRef CAS.
- P. Senguttuvan, S. D. Han, S. Kim, A. L. Lipson, S. Tepavcevic, T. T. Fister, I. D. Bloom, A. K. Burrell and C. Johnson, Adv. Energy Mater., 2016, 6, 1600826 CrossRef.
- A. Singh and A. Chandra, Sci. Rep., 2015, 5, 15551 CrossRef CAS PubMed.
- D. Lin, Y. Liu and Y. Cui, Nat. Nanotechnol., 2017, 12, 194 CrossRef CAS PubMed.
- J.-M. Tarascon and M. Armand, in Materials For Sustainable Energy: A Collection of Peer-Reviewed Research and Review Articles from Nature Publishing Group, World Scientific, 2011, pp. 171–179 Search PubMed.
- J. Qian, W. A. Henderson, W. Xu, P. Bhattacharya, M. Engelhard, O. Borodin and J.-G. Zhang, Nat. Commun., 2015, 6, 6362 CrossRef CAS PubMed.
- A. Mauger, M. Armand, C. Julien and K. Zaghib, J. Power Sources, 2017, 353, 333–342 CrossRef CAS.
- D. R. MacFarlane, N. Tachikawa, M. Forsyth, J. M. Pringle, P. C. Howlett, G. D. Elliott, J. H. Davis, M. Watanabe, P. Simon and C. A. Angell, Energy Environ. Sci., 2014, 7, 232–250 RSC.
- M. Watanabe, M. L. Thomas, S. Zhang, K. Ueno, T. Yasuda and K. Dokko, Chem. Rev., 2017, 117, 7190–7239 CrossRef CAS PubMed.
- A. Best, A. Bhatt and A. Hollenkamp, J. Electrochem. Soc., 2010, 157, A903–A911 CrossRef CAS.
- E. Paillard, Q. Zhou, W. A. Henderson, G. B. Appetecchi, M. Montanino and S. Passerini, J. Electrochem. Soc., 2009, 156, A891–A895 CrossRef CAS.
- A. Manthiram, X. Yu and S. Wang, Nat. Rev. Mater., 2017, 2, 16103 CrossRef CAS.
- C. Masquelier, Nat. Mater., 2011, 10, 649 CrossRef CAS PubMed.
- Y. Abu-Lebdeh, A. Abouimrane, P.-J. Alarco and M. Armand, J. Power Sources, 2006, 154, 255–261 CrossRef CAS.
- D. R. MacFarlane, M. Forsyth, P. C. Howlett, M. Kar, S. Passerini, J. M. Pringle, H. Ohno, M. Watanabe, F. Yan and W. Zheng, Nat. Rev. Mater., 2016, 1, 15005 CrossRef CAS.
- Z.-B. Zhou and H. Matsumoto, Electrochem. Commun., 2007, 9, 1017–1022 CrossRef CAS.
- D. R. MacFarlane and M. Forsyth, Adv. Mater., 2001, 13, 957–966 CrossRef CAS.
- L. Jin, K. M. Nairn, C. M. Forsyth, A. J. Seeber, D. R. MacFarlane, P. C. Howlett, M. Forsyth and J. M. Pringle, J. Am. Chem. Soc., 2012, 134, 9688–9697 CrossRef CAS PubMed.
- L. Jin, P. C. Howlett, J. M. Pringle, J. Janikowski, M. Armand, D. R. MacFarlane and M. Forsyth, Energy Environ. Sci., 2014, 7, 3352–3361 RSC.
- Y. Zhou, X. Wang, H. Zhu, M. Yoshizawa-Fujita, Y. Miyachi, M. Armand, M. Forsyth, G. W. Greene, J. M. Pringle and P. C. Howlett, ChemSusChem, 2017, 10, 3135–3145 CrossRef CAS PubMed.
- Y. Yamada and A. Yamada, J. Electrochem. Soc., 2015, 162, A2406–A2423 CrossRef CAS.
- F. Makhlooghiazad, D. Gunzelmann, M. Hilder, D. R. MacFarlane, M. Armand, P. C. Howlett and M. Forsyth, Adv. Energy Mater., 2017, 7, 1601272 CrossRef.
- L. Suo, Y.-S. Hu, H. Li, M. Armand and L. Chen, Nat. Commun., 2013, 4, 1481 CrossRef PubMed.
- J. Wang, Y. Yamada, K. Sodeyama, C. H. Chiang, Y. Tateyama and A. Yamada, Nat. Commun., 2016, 7, 12032 CrossRef CAS PubMed.
- F. Wu, N. Chen, R. Chen, Q. Zhu, J. Qian and L. Li, Chem. Mater., 2016, 28, 848–856 CrossRef CAS.
- L. Suo, O. Borodin, W. Sun, X. Fan, C. Yang, F. Wang, T. Gao, Z. Ma, M. Schroeder, A. von Cresce, S. M. Russell, M. Armand, A. Angell, K. Xu and C. Wang, Angew. Chem., 2016, 128, 7252–7257 CrossRef.
- M. J. Marczewski, B. Stanje, I. Hanzu, M. Wilkening and P. Johansson, Phys. Chem. Chem. Phys., 2014, 16, 12341–12349 RSC.
- P. Meister, X. Qi, R. Kloepsch, E. Krämer, B. Streipert, M. Winter and T. Placke, ChemSusChem, 2017, 10, 804–814 CrossRef CAS PubMed.
- J. Evans, C. A. Vincent and P. G. Bruce, Polymer, 1987, 28, 2324–2328 CrossRef CAS.
- M. Doyle, T. F. Fuller and J. Newman, Electrochim. Acta, 1994, 39, 2073–2081 CrossRef CAS.
- C. Angell, C. Liu and E. Sanchez, Nature, 1993, 362, 137 CrossRef CAS.
- L. Suo, O. Borodin, T. Gao, M. Olguin, J. Ho, X. Fan, C. Luo, C. Wang and K. Xu, Science, 2015, 350, 938–943 CrossRef CAS PubMed.
- C. Ding, T. Nohira, R. Hagiwara, K. Matsumoto, Y. Okamoto, A. Fukunaga, S. Sakai, K. Nitta and S. Inazawa, J. Power Sources, 2014, 269, 124–128 CrossRef CAS.
- L. Grande, J. von Zamory, S. L. Koch, J. Kalhoff, E. Paillard and S. Passerini, ACS Appl. Mater. Interfaces, 2015, 7, 5950–5958 CrossRef CAS PubMed.
- L. Jin, P. Howlett, J. Efthimiadis, M. Kar, D. Macfarlane and M. Forsyth, J. Mater. Chem., 2011, 21, 10171–10178 RSC.
- Y. Zhou, X. Wang, H. Zhu, M. Armand, M. Forsyth, G. W. Greene, J. M. Pringle and P. C. Howlett, Energy Storage Materials, 2018, 15, 407–414 CrossRef.
- R. Yunis, T. W. Newbegin, A. F. Hollenkamp and J. M. Pringle, Mater. Chem. Front., 2018, 2, 1207–1214 RSC.
- H. Yamada, Y. Miyachi, Y. Takeoka, M. Rikukawa and M. Yoshizawa-Fujita, Electrochim. Acta, 2019, 303, 293–298 CrossRef CAS.
- Y. Zhou, X. Wang, H. Zhu, M. Yoshizawa-Fujita, Y. Miyachi, M. Armand, M. Forsyth, G. W. Greene, J. M. Pringle and P. C. Howlett, ChemSusChem, 2017, 10, 3135–3145 CrossRef CAS PubMed.
- D. Al-Masri, R. Yunis, A. F. Hollenkamp and J. M. Pringle, Chem. Commun., 2018, 54, 3660–3663 RSC.
- N. Schweikert, A. Hofmann, M. Schulz, M. Scheuermann, S. T. Boles, T. Hanemann, H. Hahn and S. Indris, J. Power Sources, 2013, 228, 237–243 CrossRef CAS.
- P. C. Howlett, D. R. MacFarlane and A. F. Hollenkamp, Electrochem. Solid-State Lett., 2004, 7, A97–A101 CrossRef CAS.
- J. M. Pringle, P. C. Howlett, D. R. MacFarlane and M. Forsyth, J. Mater. Chem., 2010, 20, 2056–2062 RSC.
- Y. Lu, M. Tikekar, R. Mohanty, K. Hendrickson, L. Ma and L. A. Archer, Adv. Energy Mater., 2015, 5, 1402073 CrossRef.
- W. A. Henderson, D. M. Seo, Q. Zhou, P. D. Boyle, J. H. Shin, H. C. De Long, P. C. Trulove and S. Passerini, Adv. Energy Mater., 2012, 2, 1343–1350 CrossRef CAS.
- C. Sun, J. Liu, Y. Gong, D. P. Wilkinson and J. Zhang, Nano Energy, 2017, 33, 363–386 CrossRef CAS.
- R. Bouchet, S. Maria, R. Meziane, A. Aboulaich, L. Lienafa, J.-P. Bonnet, T. N. Phan, D. Bertin, D. Gigmes and D. Devaux, Nat. Mater., 2013, 12, 452 CrossRef CAS PubMed.
- I. Nicotera, C. Oliviero, W. A. Henderson, G. B. Appetecchi and S. Passerini, J. Phys. Chem. B, 2005, 109, 22814–22819 CrossRef CAS PubMed.
- Z. Gadjourova, Y. G. Andreev, D. P. Tunstall and P. G. Bruce, Nature, 2001, 412, 520 CrossRef CAS PubMed.
- S. Zugmann, M. Fleischmann, M. Amereller, R. M. Gschwind, H. D. Wiemhöfer and H. J. Gores, Electrochim. Acta, 2011, 56, 3926–3933 CrossRef CAS.
- A. Basile, A. I. Bhatt and A. P. O'Mullane, Nat. Commun., 2016, 7, ncomms11794 CrossRef CAS PubMed.
- N. Chen, H. Zhang, L. Li, R. Chen and S. Guo, Adv. Energy Mater., 2018, 8, 1702675 CrossRef.
- W. Zhou, S. Wang, Y. Li, S. Xin, A. Manthiram and J. B. Goodenough, J. Am. Chem. Soc., 2016, 138, 9385–9388 CrossRef CAS PubMed.
- R. Wibowo, S. E. Ward Jones and R. G. Compton, J. Phys. Chem. B, 2009, 113, 12293–12298 CrossRef CAS PubMed.
- J. M. Pringle, Phys. Chem. Chem. Phys., 2013, 15, 1339–1351 RSC.
- A. Basile, A. I. Bhatt and A. P. O'Mullane, Nat. Commun., 2016, 7, ncomms11794 CrossRef CAS PubMed.
- G. Bieker, M. Winter and P. Bieker, Phys. Chem. Chem. Phys., 2015, 17, 8670–8679 RSC.
- D. Aurbach, E. Zinigrad, Y. Cohen and H. Teller, Solid State Ionics, 2002, 148, 405–416 CrossRef CAS.
- A. Aryanfar, D. Brooks, B. V. Merinov, W. A. Goddard III, A. J. Colussi and M. R. Hoffmann, J. Phys. Chem. Lett., 2014, 5, 1721–1726 CrossRef CAS PubMed.
- A. Manuel Stephan and K. S. Nahm, Polymer, 2006, 47, 5952–5964 CrossRef.
- R. Bhattacharyya, B. Key, H. Chen, A. S. Best, A. F. Hollenkamp and C. P. Grey, Nat. Mater., 2010, 9, 504 CrossRef CAS PubMed.
- Q. Chen, K. Geng and K. Sieradzki, J. Electrochem. Soc., 2015, 162, A2004–A2007 CrossRef CAS.
- K. N. Wood, E. Kazyak, A. F. Chadwick, K.-H. Chen, J.-G. Zhang, K. Thornton and N. P. Dasgupta, ACS Cent. Sci., 2016, 2, 790–801 CrossRef CAS PubMed.
- Z. Li, J. Huang, B. Y. Liaw, V. Metzler and J. Zhang, J. Power Sources, 2014, 254, 168–182 CrossRef CAS.
- S.-K. Jeong, H.-Y. Seo, D.-H. Kim, H.-K. Han, J.-G. Kim, Y. B. Lee, Y. Iriyama, T. Abe and Z. Ogumi, Electrochem. Commun., 2008, 10, 635–638 CrossRef CAS.
- G. M. Girard, M. Hilder, D. Nucciarone, K. Whitbread, S. Zavorine, M. Moser, M. Forsyth, D. R. MacFarlane and P. C. Howlett, J. Phys. Chem. C, 2017, 121, 21087–21095 CrossRef CAS.
- Y. Zhou, X. Wang, H. Zhu, M. Armand, M. Forsyth, G. W. Greene, J. M. Pringle and P. C. Howlett, Phys. Chem. Chem. Phys., 2017, 19, 2225–2234 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Table of entropies and enthalpies obtained from DSC, cyclic voltammetry at 50 °C, MAS 19F NMR and Nyquist plots obtained from cycling of the liquid 50 mol% Li[FSI] in [C2epyr][FSI] sample. See DOI: 10.1039/c9ta11175a. Supporting research data has been deposited in the Oxford Research Archive and is available under this DOI: 10.5287/bodleian:44Q4nX11g |
| This journal is © The Royal Society of Chemistry 2019 |