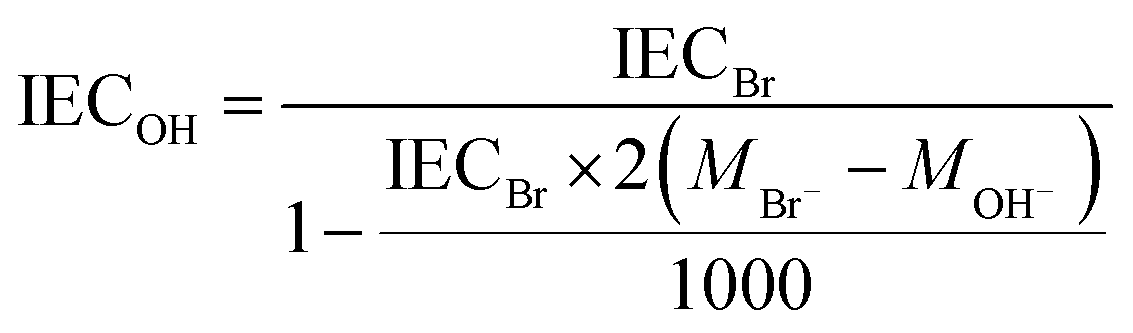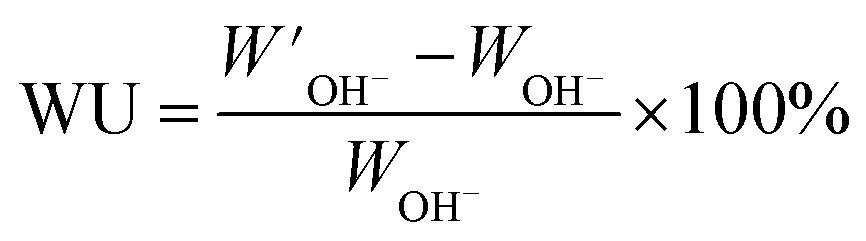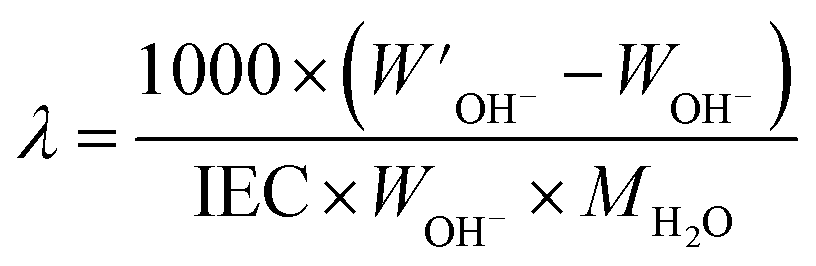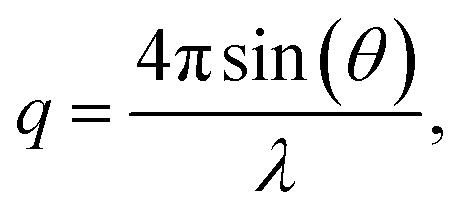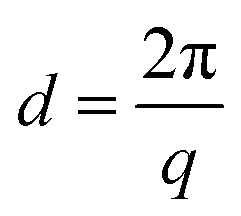DOI:
10.1039/C9TA09213G
(Paper)
J. Mater. Chem. A, 2019,
7, 27164-27174
Ether-free polyfluorenes tethered with quinuclidinium cations as hydroxide exchange membranes†
Received
21st August 2019
, Accepted 11th November 2019
First published on 11th November 2019
Abstract
We report on aryl ether-free 2,7-diphenylfluorene-based copolymers tethered with quinuclidinium (Qui) cations via hexyl spacers, prepared through superacid catalyzed Friedel–Crafts polycondensation and quaternization reactions. The 2,7-diphenylfluorene monomers were synthesised by Suzuki coupling and were employed to increase polymer backbone stiffness. Corresponding copolymers and anion-exchange membranes (AEMs) tethered with piperidinium (Pip) and trimethylalkyl ammonium (TMA) cations were prepared as reference materials. At a given water content, the AEM functionalized with Qui cations was the most efficient hydroxide conductor and its OH− conductivity reached 100 mS cm−1 at 80 °C at an ion exchange capacity of 2.0 mequiv. g−1. Moreover, this membrane showed the highest thermal and alkaline stability in the series. 1H NMR analysis of AEMs stored in 2 M aq. NaOH at 90 °C over 672 h revealed the complete absence of any ring-opening β-elimination in the bicyclic cage-like Qui structure, and less than 2% β-elimination in the hexyl spacer. In contrast, the Pip cations were found to degrade via β-elimination in both the monocyclic ring structure and the hexyl spacer. Results on the Pip-modified AEM implied that a β-hydrogen in the linear alkyl spacer chain was approximately 4 times more vulnerable to elimination than a β-hydrogen in the 6-membered ring. In addition, all the cations degraded via substitution reactions to some degree, and the total loss of Qui, Pip and TMA cations over the period was estimated to be 4, 12 and 9%, respectively. The overall findings demonstrate that the combination of aryl ether-free backbone polymers and Qui cations results in durable and high-performance AEMs suitable for use in alkaline electrochemical energy conversion and storage devices.
1. Introduction
The alkaline membrane fuel cell (AEMFC) has emerged as an attractive and commercially viable technology for efficient and environmentally benign energy conversion. For example, the high pH operating conditions enable, e.g., the utilization of inexpensive platinum-group metal-free catalysts and less expensive metallic materials in the fuel cell system. This offers distinct advantages over the proton-exchange membrane fuel cell, which operates under acidic conditions. In general, the performance of fuel cells depends on the many operating parameters and the system components. One of the most critical components in AEMFCs is the polymeric OH− conducting anion-exchange membrane (AEM), and perhaps the main challenge in the development of this technology is to chemically design and synthesize AEMs with sufficient long-term alkaline stability under the harsh conditions in the fuel cell.1–6 Most studies of alkaline stability have so far been carried out with fully hydrated AEMs. However, more recent studies have shown that there is a significant increase in the alkaline degradation rate when the hydration state of the AEM is reduced, which may occur in the AEMFC under some operating conditions.7,8
Aromatic polymers functionalized with quaternary ammonium (QA) cations belong to the most studied class of AEM materials because of their availability, relative ease of synthesis, and generally high thermal, oxidative and hydrolytic stability.5,6 The vast number of studies on AEMs based on different aromatic polymer backbones, including polyethers, polysulfones, polyphenylenes, polybenzimidazoles, etc., has shown that aryl ether links are sensitive to hydrolysis under alkaline conditions, especially if this reaction is activated by nearby electron-withdrawing groups such as sulfone links.9–20 Hence, AEMs based on ether-free aromatic backbones, such as polyphenylenes,9,21–23 poly(arylene alkylene)s24–32 and sterically protected polyimidazoliums,33–36 as well as on aliphatic backbones such as poly(diallyldialkyl ammonium)37 and polynorbornenes,38,39 have shown excellent alkaline stability.
Because of the high reactivity of OH−, QA cations may degrade via a large number of different pathways depending on their molecular structure. These include Hofmann β-eliminations, nucleophilic substitution at α-carbons, and various rearrangement reactions.6,40–43 QA cations placed in benzylic positions on the polymer backbone are especially prone to degradation by substitution reactions and many studies have shown that the alkaline stability can be significantly enhanced if the QA cations instead are attached to the backbone via flexible alkyl spacer chains.9,17 Kreuer and Marino have carried out model studies of low-molecular weight compounds and reported that certain alicyclic QAs have an outstanding alkaline stability.42 For example, the monocyclic N,N-dimethylpiperidinium cation showed a 21 times longer halftime than the conventional benzyltrimethyl QA cation in 6 M NaOH at 160 °C. The spirocyclic 6-azonia-spiro[5.5]undecane-6-ium cation displayed an even longer halftime, 26 times longer than the benzyltrimethyl QA cation under the same conditions.42 The high stability of the alicyclic QAs has been rationalized by a low ring strain combined with the constrained conformations imposed by the ring structure, which increase the transition state energy of both substitution and elimination reactions.42 Later studies by us and others on AEMs functionalized with alicyclic cations such as piperidinium25,26,44 and different spirocyclic QAs32,37,45–51 have confirmed a high alkaline stability. However, the results also indicate that the stability is highly dependent on precisely how the cations are attached to the polymer backbone.32,47,49,51 If the alicyclic cations are placed in positions where their ring conformation can be deformed, e.g., in a rigid polymer backbone, there is a tendency for the stability to decrease.25,32
In the present work we have prepared and studied AEMs based on aryl ether-free fluorene-based copolymers tethered with quinuclidinium (Qui) cations placed on flexible hexyl spacers. Qui is a bicyclic QA cation consisting of 6-membered rings in a rigid cage-like arrangement. This provides a low ring strain and results in a severely constrained conformation, which is likely to increase the transition state energy of β-elimination and substitution reactions beyond that of, e.g., the monocyclic piperidinium (Pip) cation. First, a precursor copolymer was prepared by triflic acid catalyzed Friedel–Crafts polycondensation of trifluoroacetophenone and a balanced mixture of dialkyl- and dibromoalkyl-functionalized 2,7-diphenylfluorene monomers. The latter monomers were synthesized and employed to increase backbone stiffness. Next, the precursor copolymer was quaternized to produce samples functionalized with Qui, Pip and trimethylalkyl ammonium (TMA) cations, respectively. AEMs were cast from solution and their structure–property relationships were compared on the basis of thermal and alkaline stability, morphology, water uptake, and OH− conductivity.
2. Experimental
2.1 Materials
2,7-Dibromofluorene (97%, Sigma-Aldrich), 9,9-dioctyl-2,7-dibromofluorene (96%, Sigma-Aldrich), 1,6-dibromohexane (96%, Sigma-Aldrich), phenylboronic acid (95%, Sigma-Aldrich), tetrabutylammonium bromide (TBAB, 98%, Sigma-Aldrich), KOH (100% pellets, VWR), toluene (99.9%, VWR), 2,2,2-trifluoroacetophenone (TFAp, 99%, Sigma-Aldrich), trifluoroacetic acid (TFA, 99%, Acros), trifluoromethanesulfonic acid (TFSA, 99%, Acros), tetrakis(triphenylphosphine)palladium(0) ((Pd[PPh3]4), 99%, Sigma-Aldrich), Na2CO3 (VWR), DMSO-d6 (99.96 atom% D, Sigma-Aldrich), chloroform-d (99.8 atom% D, Sigma-Aldrich), trimethylamine (7.3 M aq. solution, Acros), N-methyl piperidine (99% Sigma-Aldrich), quinuclidine (97%, Sigma-Aldrich), diethyl ether (99+%, Sigma-Aldrich), NaNO3 (99%, Sigma-Aldrich), methanol (MeOH, HPLC grade, Honeywell) and AgNO3 (99.995%, Sigma-Aldrich) were all used without further purification. Dichloromethane (DCM) was dried using an MBraun dry solvent dispenser system MB-SPS 800.
2.2 Synthesis of 2,7-dibromo-9,9-bis(6-bromohexyl)-fluorene
The compound was synthesized by a modified published procedure.52 2,7-Dibromofluorene (5 g, 15.4 mmol), TBAB (0.49 g, 1.52 mmol) and 1,6-dibromohexane (52 ml, 82 g, 338 mmol) were added to a two-neck round bottom flask. Next, 15.4 ml of 45 wt% aq. KOH was added and the mixture was stirred at 75 °C for 1 h. After extracting the mixture three times with DCM, the organic layer was washed three times with brine and dried over MgSO4. Subsequently, the organic mixture was evaporated under vacuum to remove the excess 1,6-dibromohexane. Finally, the crude product was purified by dry vacuum column chromatography, using a mixture of heptane and toluene as the eluent.53 The product was isolated as a white powder (7.2 g, 72% isolated yield).
2.3 Synthesis of 2,7-diphenyl-9,9-bis(6-bromohexyl)-fluorene (DPBHF)
2,7-Dibromo-9,9-bis(6-bromohexyl)-fluorene (7.1 g, 10.9 mmol) was dissolved in toluene (135 ml) in a two-neck round bottom flask. Next, phenylboronic acid (4.0 g, 32.8 mmol) and 135 ml of 1 M Na2CO3 solution (135 ml) were added. The mixture was degassed by 4 vacuum/argon cycles. Pd(PPh3)4 (13.6 mg, 0.08 mmol) was added to the solution and the reaction mixture was refluxed under argon for 48 h. The resulting mixture was extracted with DCM, washed with brine and dried over MgSO4. The organic phase was passed through a silica plug, and evaporated until dryness to yield the target compound as yellow crystals (6.75 g, 96% isolated yield).
2.4 Synthesis of 9,9-dioctyl-2,7-diphenyl-9H-fluorene (DODPF)
2,7-Dibromo-9,9-dioctyl-9H-fluorene (2.0 g, 3.5 mmol) and toluene (35 ml) were added to a two-neck round bottom flask. After addition of phenylboronic acid (1.33 g, 10.5 mmol) and 35 ml of 1 M Na2CO3, the mixture was degassed by 4 vacuum/argon cycles. Subsequently, Pd(PPh3)4 (4.2 mg, 0.10 mmol) was added and the reaction mixture was refluxed under argon for 48 h. The resulting mixture was extracted with DCM, washed with brine, and then dried over MgSO4. Next, the mixture was purified by dry vacuum column chromatography, using a volumetric gradient mixture of heptane and toluene as the eluent, to give the product as white crystals (1.18 g, 60% isolated yield).
2.5 Polycondensation
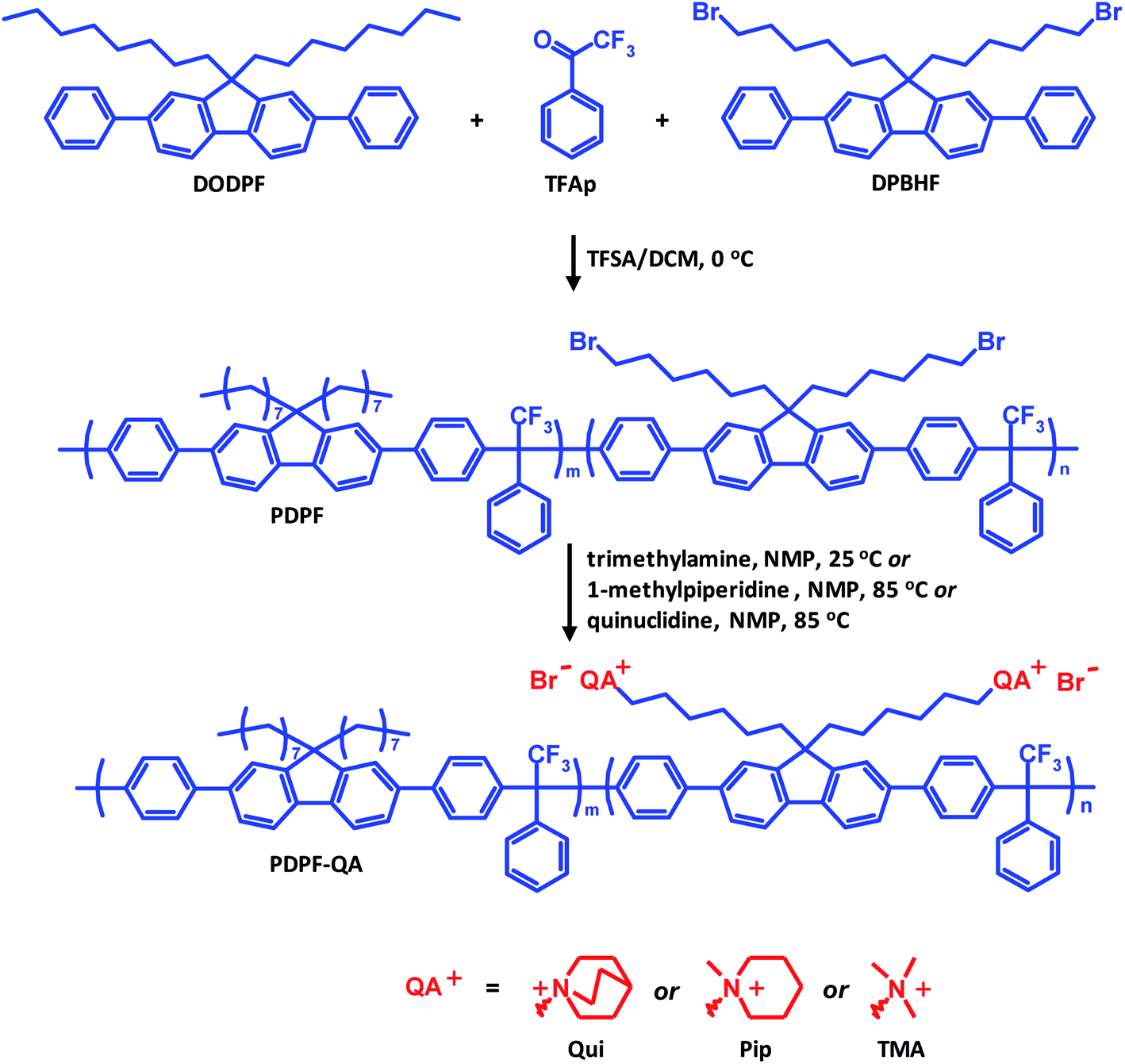
A bromoalkylated precursor copolymer based on DPBHF, DODPF and TFAp was prepared using a Friedel–Crafts type polycondensation reaction under nitrogen (Scheme 1). DPBHF (2.5 g, 3.88 mmol) and DODPF (0.26 g, 0.48 mmol) were dissolved in dry DCM (17 ml) in a Schlenk round-bottom flask. After addition of TFAp (0.67 ml, 4.8 mmol) the solution was cooled in an ice bath. Next, TFSA (2.9 ml, 33 mmol) was added dropwise. After 90 min when the viscosity had increased drastically, the reaction was terminated by dilution with chloroform followed by precipitation of the product in methanol. The precipitate was dried under vacuum to give the precursor polymer, designated as PDPF, as a white fibrous material.
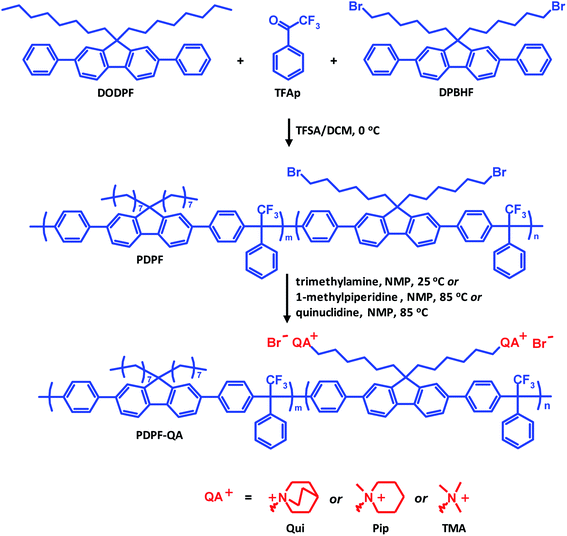 |
| | Scheme 1 Preparation of the PDPF copolymer by superacid catalyzed polycondensation of DODPF, DPBHF and TFAp, followed by quaternization reactions to attach Qui, Pip and TMA cations, respectively. | |
2.6 Quaternization
The precursor copolymer PDPF was functionalized in Menshutkin reactions with three different quaternary amines, quinuclidine, 1-methylpiperidine and trimethylamine to form the cationic copolymers designated as PDPF–Qui, PDPF–Pip and PDPF–TMA, respectively. The preparation of PDPF–TMA is given as an example. Trimethylamine (1.08 ml, 7.9 mmol, 5 equiv., 400% excess) was added to a solution of the precursor polymer (0.7 g, 1 equiv.) in NMP (20 g) and the reaction was stirred at room temperature for 7 days. The polymer was precipitated in 200 ml of diethyl ether, washed with fresh diethyl ether three times, and then dried under vacuum at room temperature. The Menshutkin reactions involving 1-methylpiperidine and quinuclidine followed the same procedure, but were performed at 80 °C because of their lower reactivity.
2.7 Structural characterization
1H NMR spectroscopy was carried out on a Bruker DRX400 spectrometer at a proton frequency of 400.13 MHz. The solvents used were DMSO-d6 (δ = 2.50 ppm) and chloroform-d (δ = 7.26 ppm). Size exclusion chromatography (SEC) was carried out using a Malvern Viscotek instrument with a 2×PL-Gel Mix-B LS column set (2 × 30 cm) equipped with OmniSEC Triple Detectors (refractive index, viscosity and light scattering) with chloroform as the eluent at 35 °C. A polystyrene standard (Mn = 96 kDa and PDI = 1.03, Polymer Laboratories Ltd.) was used for calibration. The precursor PDPF copolymer was dissolved in chloroform for 24 h prior to the measurement and passed through a Teflon filter with a pore diameter of 0.2 μm before injection.
2.8 Membrane preparation
AEMs were prepared by solvent casting at 80 °C for 48 h from 5 wt% polymer solutions in NMP. The polymers were dissolved overnight at room temperature and then passed through a Teflon filter (0.45 μm) onto a Petri dish (Ø = 5 cm). After the casting process, the membranes were carefully peeled from the dishes and washed several times with deionized water. The AEMs were stored in deionized water at room temperature for at least 48 h prior to any measurement.
2.9 Determination of ion exchange capacity and water uptake
The experimental ion exchange capacity (IECtitr) was determined by Mohr titration. At least 0.05 g of membrane sample in the Br− form was dried under vacuum at 50 °C for 48 h. Next, the sample was immediately weighed and immersed in 25.0 ml of 0.2 M NaNO3 at 40–50 °C for 5 days to achieve full exchange of the Br− ions. A 5 ml solution was titrated with 0.01 M aq. AgNO3 using K2CrO4 as the indicator. The end point of the titration was observed by a color change from yellow to reddish brown, as well as by precipitation of Ag2CrO4. The titration was repeated 3 more times for each sample to obtain the average value of IECtitr. Finally, the IEC of the AEMs in the OH− form (IECOH) was calculated as:| | 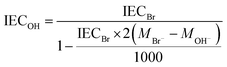 | (1) |
In order to measure the water uptake of the AEMs, the dry weight of the membranes in the Br− form (WBr) was first measured after drying under vacuum at 50 °C for 48 h. The values of WBr and IECtitr were both used to calculate the dry weight of the AEMs in the OH− form (WOH). The dry membranes in the Br− form were ion-exchanged to the OH− form by immersion in degassed 1 M aq. NaOH (60 ml) solution for 5 days under a nitrogen atmosphere. During this period, the degassed 1 M aq. NaOH solution was replaced at least three times with fresh solution to ensure complete conversion. Next, the membranes were quickly washed five times with degassed de-ionized water until the rinse water was neutral. The membranes were then transferred to deionized degassed water and kept for 24 h under nitrogen at 20 °C. Finally, the samples were wiped dry with tissue paper and quickly weighed to obtain W′OH. The same procedure was applied at 40 °C for 10 h and at 60 and 80 °C for 6 h. The water uptake was calculated as:
| | 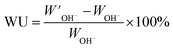 | (2) |
The hydration number (
λ), defined as the number of water molecules per functional group, was calculated as:
| | 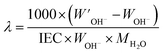 | (3) |
2.10 Thermal characterization
The thermal decomposition of the AEMs was analyzed by thermogravimetric analysis (TGA) using a TA instruments TGA Q5000. Before the analysis, the samples were dried at room temperature for 7 days under vacuum. All membranes in the Br− form were preheated to 150 °C for 10 min in the furnace to remove traces of water. Data were collected in the temperature range 50–600 °C at a heating rate of 10 °C min−1 under a nitrogen atmosphere. The decomposition temperature (Td,95) is defined as the temperature where 5% weight loss is noted. A differential scanning calorimeter (DSC) model Q2000 analyzer from TA Instruments was employed to measure the glass transition temperature (Tg) of the precursor copolymer. Data from a heating/cooling/heating cycle were collected between 50 and 280 °C and the Tg values were determined from the second heating cycle.
2.11 Conductivity measurements
The OH− conductivity of the AEMs was measured by using a Novocontrol high resolution dielectric analyzer V 1.01 S at 50 mV, in a frequency range of 10−1 to 107 Hz. First, the samples were ion-exchanged to the OH− form, following the same procedure as for the water uptake measurements described above. The OH− conductivity was subsequently measured during the cycle 20 → 80 → 20 → 80 °C under fully hydrated (immersed) conditions using a two-probe method.
2.12 Small angle X-ray scattering
Small angle X-ray scattering (SAXS) measurements were performed to study any ionic clustering in the dry AEMs in the Br− form. The analysis was carried out using a SAXLAB ApS system (JJ-Xray, Denmark) combined with a Pilatus detector. The scattering vector (q) is related to the scattering angle according to:| | 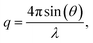 | (4) |
where λ is the wavelength of the Cu K(α) radiation (1.542 Å) and 2θ is the scattering angle. The average distance between ionic clusters (d) is calculated by Bragg's law| | 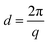 | (5) |
2.13 Alkaline stability measurements
The thermochemical stability of the AEMs under alkaline conditions was studied using 1H NMR spectroscopy before and after storage in alkaline solutions at elevated temperature. Pieces of membranes were stored in 2 M aq. NaOH for 28 days and in 5 M aq. NaOH for 7 days at 90 °C, respectively. After the storage, the membranes were washed with deionized water and ion-exchanged to the Br− form, followed by thorough washing with deionized water to remove salt. After drying, the samples were dissolved in DMSO-d6 and analyzed by 1H NMR spectroscopy. TFA (20 vol%) was added to NMR sample solutions to shift the water signal from 3.33 to ∼10 ppm, and expose the sample signals in the 3–3.5 ppm region.
3. Results and discussion
3.1 Polymer synthesis and characterization
A series of three anion-conducting copolymers functionalized with Qui, Pip and TMA cations, respectively, were prepared based on the same aryl ether-free fluorene-containing backbone, as shown in Scheme 1. Two 2,7-diphenylfluorene monomers were synthesized by Suzuki coupling and alkylation reactions (Scheme S1†). DPBHF carries two bromohexyl chains which allowed for the introduction of the QA cations by Menshutkin reactions with the corresponding tertiary amine. DODPF is tethered with two octyl chains to increase the solubility of the final polymer, and was employed as a co-monomer to control the IECs and enhance the solubility of the final AEMs. The use of the diphenylfluorene monomers instead of the corresponding non-phenylated ones facilitates the polycondensation reaction, and produces polymers with higher chain rigidity that likely reduces swelling and water uptake of the AEMs. DPBHF and DODPF were obtained in total yields of 70 and 60%, respectively, and their structure and purity were confirmed by 1H NMR spectroscopy (Fig. S1–S3†).
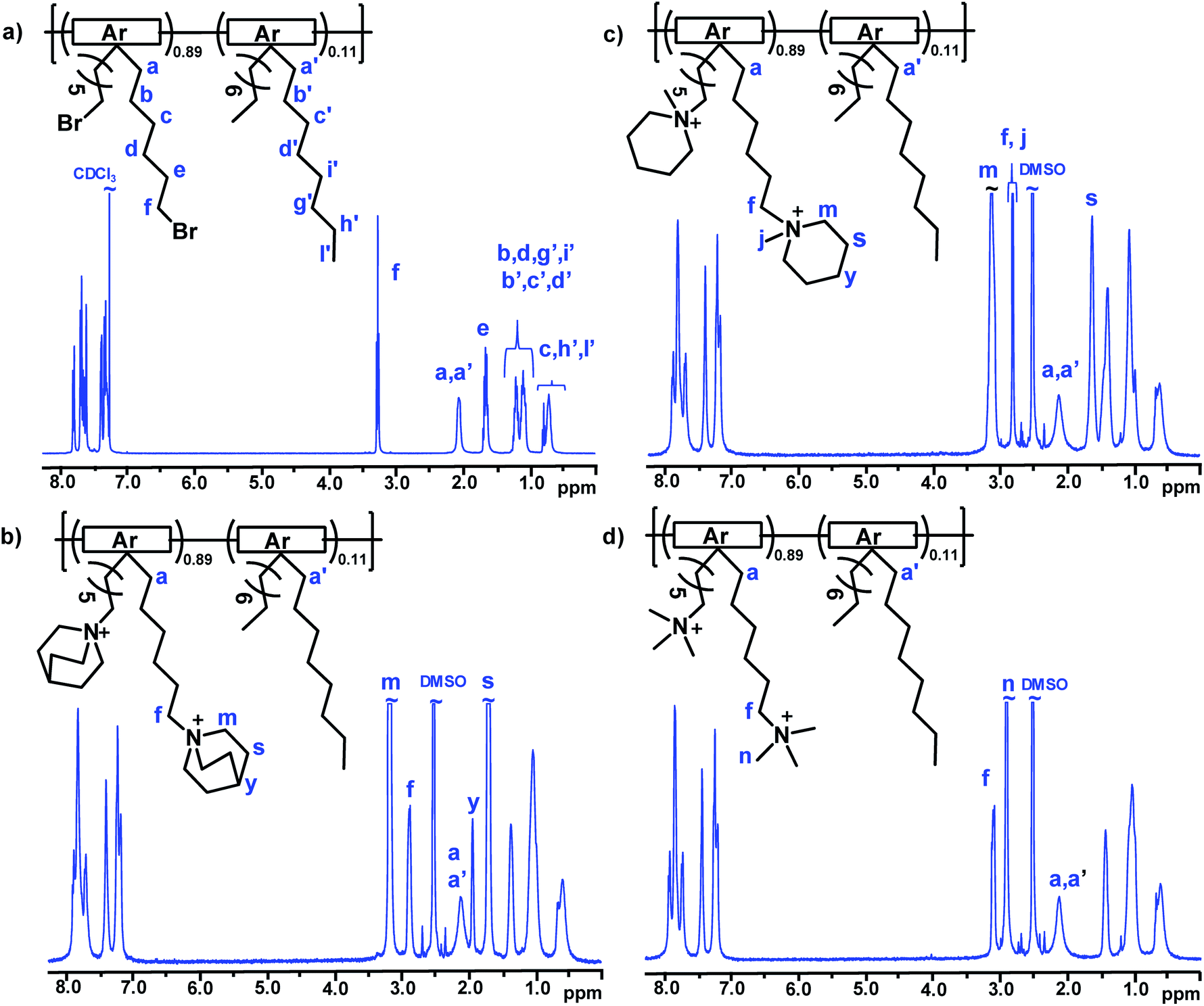
The precursor copolymer was synthesized by a Friedel–Crafts type polycondensation reaction employing the two 2,7-diphenylfluorene monomers in combination with TFAp (Scheme 1). Moreover, TFAp was kept in 10% excess in relation to the two diphenylfluorene monomers to facilitate the polycondensation.54 This is in sharp contrast to typical polycondensations where ensuring a strict stoichiometric balance of the monomers is crucial in order to obtain high molecular weight polymers. To prevent side reactions, the reaction was run at 0 °C and with low monomer concentration (0.20 mol L−1). The present polycondensation was effective and had a very high reaction rate. SEC analysis of the resulting precursor copolymer showed a molecular weight of Mn = 63 kDa and a polydispersity of MwMn−1 = 1.47 (Fig. S4†). 1H NMR spectroscopy verified the molecular structure of the copolymer (Fig. 1a). The signal of the methylene bromide (–CH2Br) protons appeared at 3.25 ppm, and the signals of the additional methylene (–CH2–) protons in the alkyl chains were found between 0.7 and 2.05 ppm. Because of their structural similarity, the two diphenylfluorene monomers can be expected to have a comparable reactivity and solubility in the polycondensation. Indeed, the resulting copolymer had a composition very similar to the monomer feed, as confirmed by 1H NMR (Fig. 1a). Hence, the copolymer was determined to be composed of 89 and 11 mol% DPBHF and DODPF units, respectively, by comparing the intensity of the methylene bromide signal (f) arising at 3.25 ppm with those of signals a and a′ at 2.05 ppm, which corresponds to the Hα of the fluorene units (Fig. 1a). Analysis by TGA and DSC indicated a thermal decomposition temperature of Td,95 = 325 °C (Fig. 4) and glass transition temperature of Tg = 213 °C (Fig. S6†).
 |
| | Fig. 1
1H NMR spectra of the bromoalkylated PDPF precursor copolymer (a), recorded in CDCl3, and the cationic copolymers PDPF–Qui (b), PDPF–Pip (c) and PDPF–TMA (d) in the Br− form recorded in DMSO-d6. | |
In the next step, the bromine atoms of the precursor copolymer were substituted in Menshutkin reactions with quinuclidine, 1-methylpiperidine and trimethylamine to form the cationic PDPF–Qui, PDPF–Pip and PDPF–TMA copolymers, respectively (Scheme 1). These reactions were highly effective and complete conversion from the alkyl bromide to the respective QA was achieved, as confirmed by both 1H NMR data and Mohr titrations (Table 1). As expected, the cationic copolymers were soluble in DMSO and NMP, but not in chloroform or water. After quaternization the 1H MNR signals of the –CH2Br protons completely disappeared while new signals belonging to the newly introduced QA cations emerged (Fig. 1b–d, respectively). For example, the methyl protons of the TMA cation and the α-protons on the alkyl spacers gave rise to new signals at 2.9 and 3.1 ppm, respectively. Moreover the α-protons of the Pip and Qui rings, and the α-protons on the spacers gave rise to signals at 3.1 and 2.9 ppm, respectively. The signals arising from the other methylene protons appeared in the region between 1.1 and 1.8 ppm and were overlapped (Fig. 1).
Table 1 Properties of the AEMs tethered with QA groups
| AEM |
IEC (mequiv. g−1) |
WU80c (wt%) |
λ
|
σ
OH
(mS cm−1) |
T
d95
(°C) |
| From NMRa data |
From Mohr titrationsb |
|
IEC in the OH− form calculated from 1H NMR (IEC of the Br− form within parentheses).
IEC in the OH− form calculated from the titration of AEMs in the Br− form (IEC of the Br− form within parentheses).
OH− form, in the fully hydrated state at 80 °C.
Analyzed by TGA 10 °C min−1 under N2.
|
| PDPF–Qui |
2.01 (1.78) |
1.99 (1.77) |
154 |
43 |
100 |
267 |
| PDPF–Pip |
2.06 (1.82) |
2.03 (1.80) |
137 |
37 |
75 |
200 |
| PDPF–TMA |
2.24 (1.96) |
2.22 (1.95) |
178 |
44 |
107 |
203 |
3.2 Membrane preparation and morphology
AEMs were cast at 80 °C from 5 wt% solutions of the cationic polymers dissolved in NMP. The AEMs were transparent, yellow-brown and flexible. Although all the AEMs were based on the same precursor copolymer, the IEC values were influenced by the size of the attached cation. Hence, PDPF–TMA had the highest IEC value of 2.22 mequiv. g−1, followed by PDPF–Pip and PDPF–Qui with 2.03 and 1.99 mequiv. g−1, respectively. As can be seen in Table 1, the IEC values obtained from the Mohr titrations were very close to the theoretical values calculated from NMR data of the precursor copolymer, which indicated high yields in the Menshutkin reactions. SAXS measurements were performed to study any ionic clustering and phase separation of the AEMs. However, no distinct ionomer peaks were observed for any of the samples (Fig. S6†), probably due to the high stiffness of the backbone. Previously, different poly(arylene piperidinium)s32 and poly(arylene alkylene)s26 with polymer backbones similar to that of the present AEMs have also shown none or only very weak ionomer peaks.
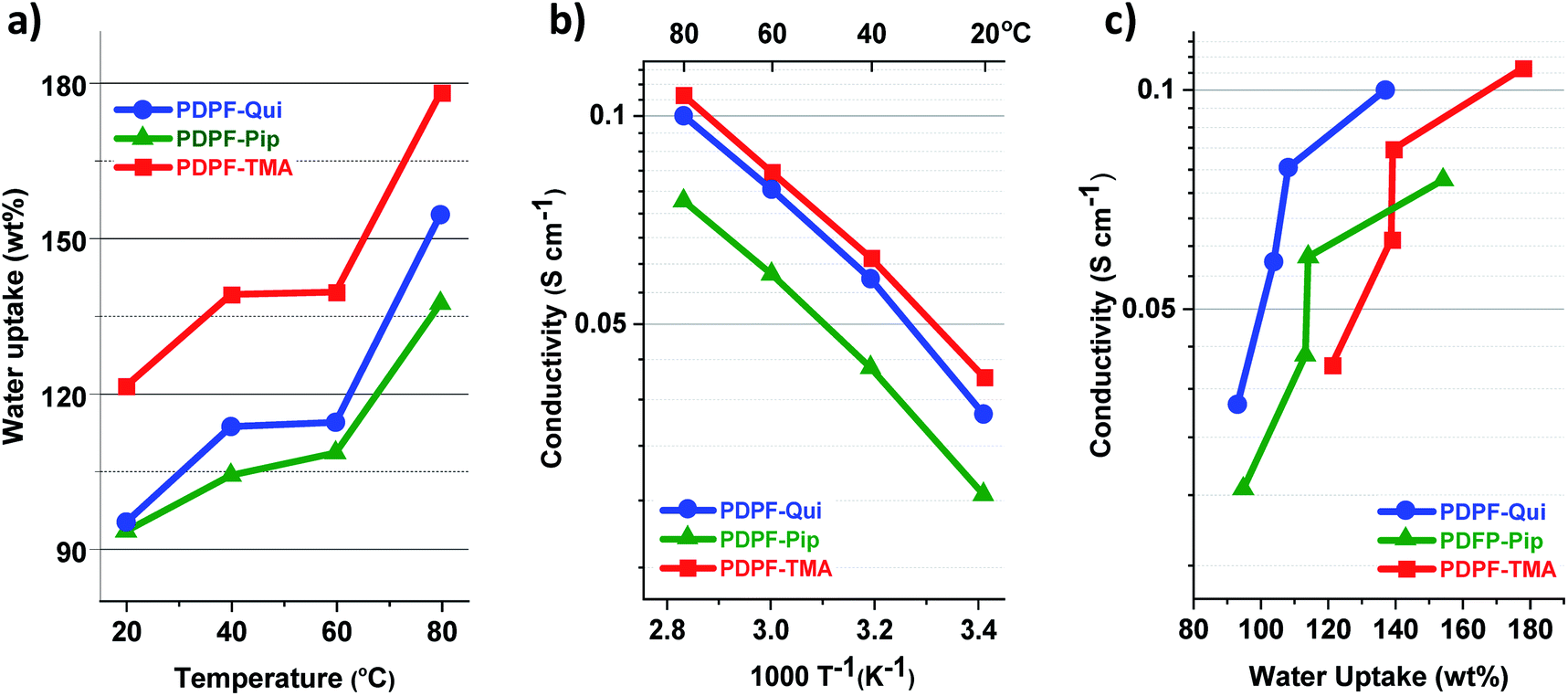
3.3 Water uptake and hydroxide conductivity
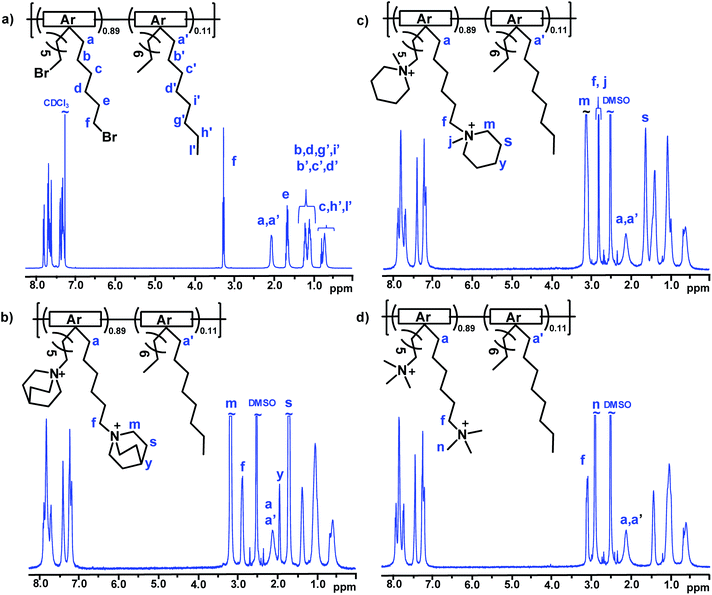
Sufficient water uptake of AEMs is crucial for both ion dissociation, i.e., formation of charge carriers, and transportation of ions by creating a percolating hydrated phase domain.55 However, too high water uptake not only decreases the ion conductivity by dilution effects, but also deteriorates the mechanical properties of the AEMs. Fig. 2a shows the water uptake of fully hydrated AEMs immersed in water between 20 and 80 °C. As expected, the water uptake increased with both IEC and temperature. PDPF–TMA with the highest IEC reached the highest water uptake of 121 and 178 wt% at 20 and 80 °C, respectively. PDPF–Qui and PDPF–Pip (with similar IEC values) had comparable water uptake, 95 and 93 wt%, respectively at 20 °C. However, at 80 °C PDPF–Qui had a higher water uptake that reached up to 155%, while PDPF–Pip showed a lower water uptake of 137 wt% at this temperature.
 |
| | Fig. 2 Water uptake of fully hydrated (immersed) AEMs in the OH− form as a function of temperature (a), the OH− conductivity of fully hydrated (immersed) AEMs as a function of T−1 (b), and OH− conductivity of fully hydrated AEMs as a function of water uptake (c). | |
The OH− conductivity of fully hydrated AEMs was measured by electrochemical impedance spectroscopy using a two-probe cell. To avoid the formation of carbonates via uptake of CO2, both the sample preparation and measurements were performed under a strict N2 atmosphere. As expected, the OH− conductivity increased with temperature and essentially followed the water uptake of the AEMs. Hence, PDPF–TMA displayed the highest conductivity of 107 mS cm−1 followed by PDPF–Qui and PDPF–Pip with 100 and 75 mS cm−1 at 80 °C (Fig. 2b). The low value of the latter AEM may be due to the combination of both insufficient ionic clustering and water percolation. Between 20 and 80 °C, all samples showed an Arrhenius-like behavior with an apparent activation energy of Ea = 13.4, 14.2, and 14.1 kJ mol−1 for PDPF–TMA, PDPF–Qui and PDPF–Pip, respectively. This was close to Ea values previously measured by Pan et al. for AEMs based on poly(arylene ether sulfone)s functionalized with benzyltrimethyl ammonium cations (13 kJ mol−1)56Fig. 2c shows the relationship between the OH− conductivity and water uptake. As shown, PDPF–Qui reached the highest conductivity at a given water content and can thus be regarded as the most efficient OH− conductor.
On the basis of their IEC values and water uptake, the conductivity of the present membranes compared very favorably with that of previously reported AEMs,57 including AEMs having TMA cations attached to fluorene-based polymer backbones via alkyl spacers.23 For example, Miyatake et al. prepared copolymers containing perfluoroalkylene and fluorenyl groups tethered with pendant TMA cations through a Ni-promoted polycondensation reaction.30 AEMs based on these copolymers reached a OH− conductivity of 86 mS cm−1 at 80 °C. (IEC = 1.47 meq. g−1, water uptake 105 wt%).30 Moreover, Bae and co-workers synthesized fluorene-based copolymers by a polycondensation reaction using a mixture of dibromoalkylated and dimethylated fluorene monomers.29 The resulting AEMs showed a OH− conductivity of 120 mS cm−1 (IEC = 2.5 meq. g−1, water uptake 180 wt%) at 80 °C. The very high conductivity reached by the present PDPF–Qui and PDPF–TMA membranes most probably resulted from the presence of an efficient percolating water-rich phase which was induced by the high local mobility of the cations attached via flexible alkyl spacers, in connection with the absence of dilution effects (despite the high water uptake).
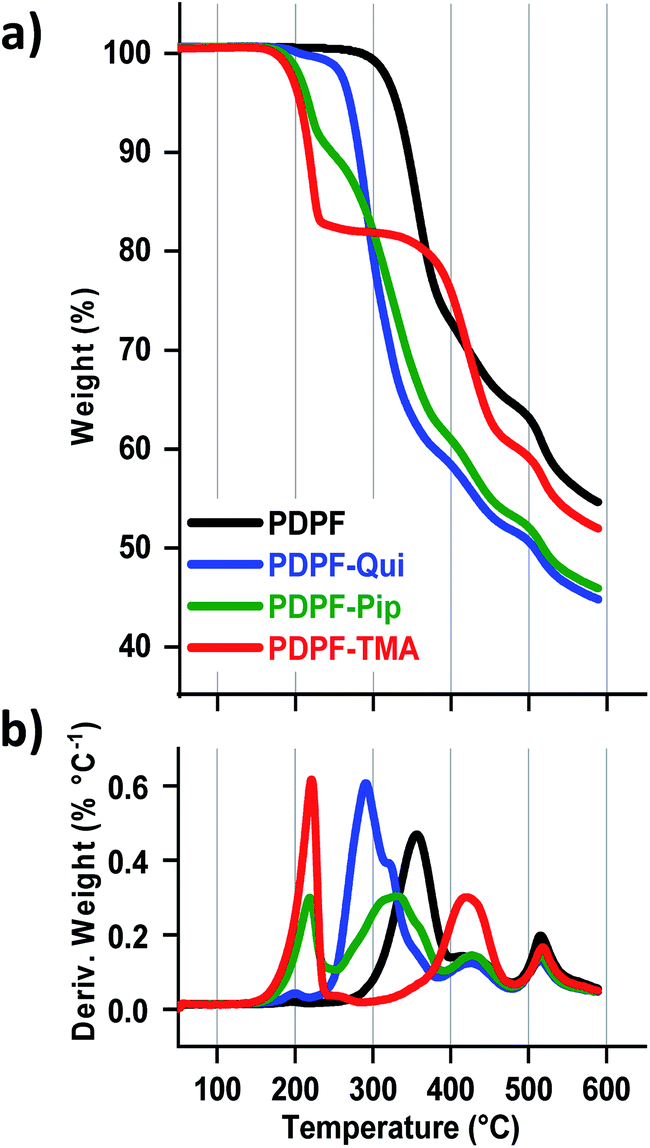
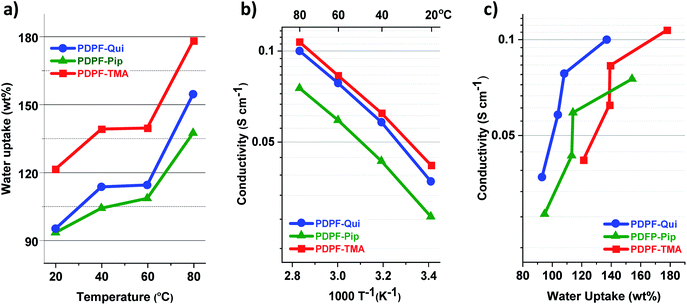
3.4 Thermal stability
The thermal stability of the AEMs in the Br− form was analyzed by TGA under N2 flow at 10 °C min−1 (Fig. 3) and the thermal decomposition temperatures (Td,95) are presented in Table 1. The AEMs decomposed in three distinct steps with the cations decomposing first, followed by the decomposition of the alkyl chains, and finally the aromatic backbone at ∼500 °C. PDPF–Qui clearly possessed the highest thermal stability with Td,95 = 267 °C, while PDPF–TMA and PDPF–Pip performed similarly, with Td,95 = 203 and 207 °C, respectively. We have previously studied the thermal stability of poly(phenylene oxide) tethered with different alicyclic cations via alkyl spacers and found that the Qui cations were exceptionally stable with Td,95 = 283 °C. Presumably, the cage-like and symmetrical structure significantly contributes to the excellent thermal stability of this cation.58
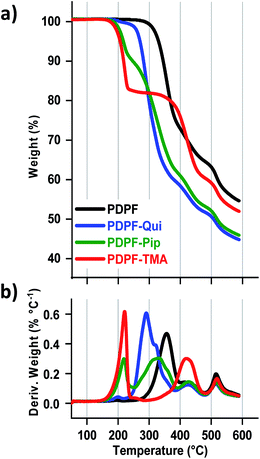 |
| | Fig. 3 TGA data measured under N2 flow at 10 °C min−1 (a) and the corresponding first derivative of the TGA curve (b). | |
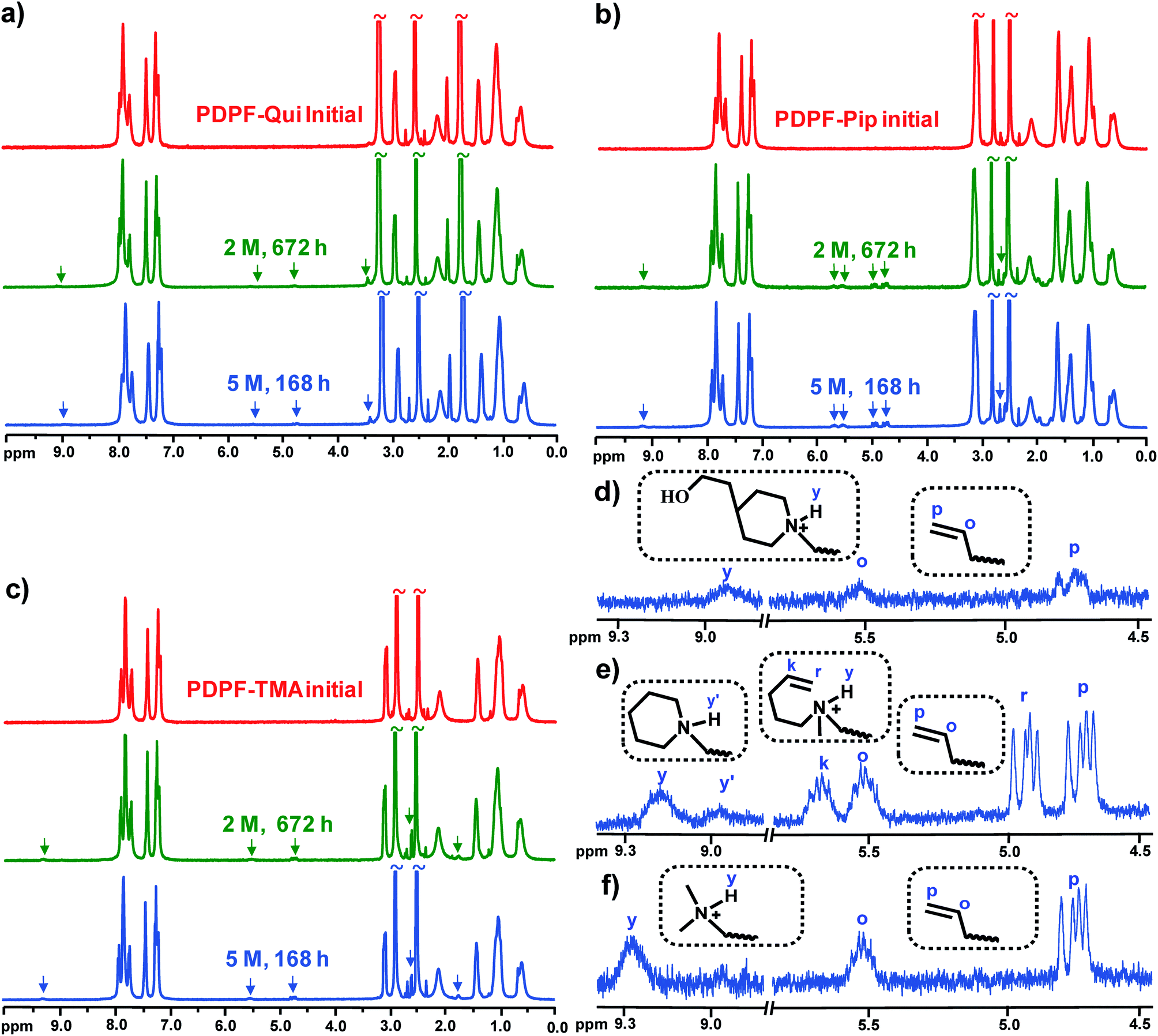
3.5 Alkaline stability
The alkaline stability is a very important characteristic of AEMs that currently limits their applicability for, e.g., fuel cells.2 NMR spectroscopy has previously been shown to be a powerful tool to assess the degree of membrane degradation and to identify degradation mechanisms.13 In the present case, the alkaline stability was studied by 1H NMR spectroscopy of sample pieces before and after storage in 2 and 5 M aq. NaOH, respectively, at 90 °C. Prior to the NMR analysis, the alkali-treated sample pieces were ion-exchanged to the Br− form, thoroughly washed, dried, and subsequently dissolved in DMSO-d6. By this procedure, any degradation product that is not covalently attached to the polymer structure will be washed away and will thus not be detected. In order to facilitate the analysis of the samples, 20 vol% TFA was added to shift the water signal downfield from 3.5 ppm to above 10 ppm, and to protonate any present tertiary amines formed in degradation reactions. As can be seen in Fig. 4a–c, new signals appeared between 4.5 and 5.8 ppm and at around 9 ppm for all AEMs after exposure to 2 M and 5 M aq. NaOH, respectively. The position and relative intensity of the former two signals suggested the presence of alkene (![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2) and alkenyl (–CHCH2) protons, most probably through the formation of vinylic groups by way of Hofmann β-elimination reactions. Moreover, signals emerging at ∼9 ppm corresponded to protonated tertiary amines formed in degradation reactions via either nucleophilic substitution or ring-opening β-elimination reactions. As expected, additions of D2O caused an immediate proton-to-deuterium exchange and the disappearance of these signals (Fig. S7†).
CH2) and alkenyl (–CHCH2) protons, most probably through the formation of vinylic groups by way of Hofmann β-elimination reactions. Moreover, signals emerging at ∼9 ppm corresponded to protonated tertiary amines formed in degradation reactions via either nucleophilic substitution or ring-opening β-elimination reactions. As expected, additions of D2O caused an immediate proton-to-deuterium exchange and the disappearance of these signals (Fig. S7†).
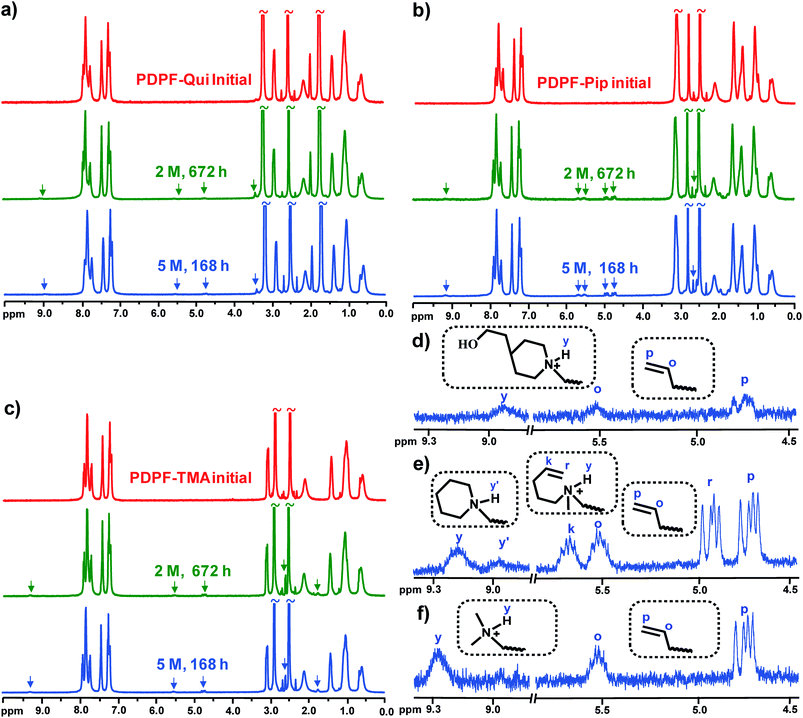 |
| | Fig. 4
1H NMR spectra of PDPF–Qui (a), PDPF–Pip (b) and PDPF–TMA (c) before and after immersion in 2 and 5 M aq. NaOH at 90 °C for different periods of time. Emerging signals between 4.5–5.8 and 9.0–9.4 ppm after storage in 5 M aq. NaOH for 168 h are marked by arrows and enlarged in panels (d), (e) and (f) for PDPF–Qui, PDPF–Pip and PDPF–TMA, respectively. | |
Fig. 4d–f show an expansion of the region around the new signals and reveal further details of the membrane degradation mechanisms. In the case of PDPF–TMA, there can only be β-elimination in the alkyl spacer chain due to the absence of additional β-hydrogens and a single set of vinylic signals was seen at ∼4.7 (p) and ∼5.5 (o) ppm in Fig. 4f. In addition, a signal arising from a protonated tertiary amine was observed at ∼9.3 ppm (y). This indicated a second degradation pathway, most probably through nucleophilic substitution on the methyl group. As can be seen in Fig. 4e, PDPF–Pip displayed two sets of vinylic signals: one at ∼4.7 (p) and ∼5.5 (o) ppm and another at ∼4.9 (r) and ∼5.7 (k) ppm. The first set had approximately the same positions as observed for PDPF–TMA, strongly implying β-elimination in the alkyl spacer chain. The second set was then attributed to β-elimination in the Pip ring. In addition, two different signals from protonated amines were detected at ∼9.2 (y) and ∼8.95 (y′) ppm, respectively. The intensity of the former was close to the intensity of signal k and was ascribed to ring opening β-elimination. The latter signal was consequently attributed to methyl substitution (Fig. 4e). Fig. 4d shows one set of vinylic signals for PDPF–Qui at the same shifts as observed for PDPF-TMA, hence implying β-elimination in the alkyl chain. However, these signals were significantly weaker than in the case of both PDPF–Pip and PDPF–TMA. Notably, there were no detectable vinylic signals from any ring-opening β-elimination reaction, suggesting that the cage-like bicyclic configuration of the Qui cation is highly resistant against this degradation reaction under the conditions investigated. A small signal from a protonated tertiary amine was detected at ∼8.9 ppm (y), hinting that a second degradation reaction was active, presumably a nucleophilic ring-opening substitution reaction producing hydroxyl groups (Fig. 4d).
The degree of ionic loss by each degradation pathway was estimated by integration of the corresponding 1H NMR signals and comparison with the respective aromatic signals which were unaffected after the alkaline treatments. As can be seen in Table 2 for the data recorded after storage in 5 M aq. NaOH at 90 °C, PDPF–Qui had the highest stability with no detectable β-elimination in the Qui ring, a mere ∼1.6% loss by β-elimination in the alkyl spacer unit, and ∼3% by ring-opening substitution. This resulted in a total ionic loss of only approximately 5%. The degradation of the Pip cations by both substitution and elimination reactions was significantly higher and the total ionic loss reached ∼23% under the same conditions. The loss of Pip cations by β-elimination in the alkyl spacer and the ring was ∼11 and ∼5%, respectively. Because each Pip cation contains four β-hydrogens in the ring and two β-hydrogens in the alkyl spacer, these results implied that a β-hydrogen in the linear alkyl spacer chain was about 4 times more vulnerable to elimination compared to a β-hydrogen in the 6-membered cationic Pip ring. The observed higher alkaline stability of the latter was in line with the assumption that the ring configuration imposes conformational restrictions which increase the transition state energy of both substitution and elimination reactions.59 The degradation of the non-cyclic TMA cations by both substitution on α-carbons and elimination reactions on the alkyl spacers was ∼4.4%, and the total ionic loss thus reached ∼9% under both alkaline conditions investigated.
Table 2 Ionic loss after alkaline treatment of AEMs as evaluated by 1H NMR spectroscopya
| Treatment |
AEM |
Ionic loss by Hofmann β-eliminationb (%) |
Ionic loss by substitutionc (%) |
Total ionic loss (%) |
| Alkyl spacer |
Alicyclic ring |
|
Ionic loss of the AEMs via different degradation mechanisms calculated from relevant 1H NMR signals and compared to the aromatic signals.
Hoffman β-elimination in the alkyl spacer and in the alicyclic ring of PDPF–Pip.
Nucleophilic attack at an α-carbon leading to methyl or ring-opening substitution (n.a. = not applicable).
|
| 2 M aq. NaOH, 90 °C, 672 h |
PDPF–TMA |
4.4 |
n.a. |
4.4 |
8.8 |
| PDPF–Pip |
7.7 |
4.0 |
0.5 |
12.2 |
| PDPF–Qui |
1.2 |
0 |
3.0 |
4.2 |
| 5 M aq. NaOH |
PDPF–TMA |
4.4 |
n.a. |
4.4 |
8.8 |
| 90 °C, 168 h |
PDPF–Pip |
11 |
5.0 |
7.2 |
23.2 |
| PDPF–Qui |
1.6 |
0 |
3.2 |
4.8 |
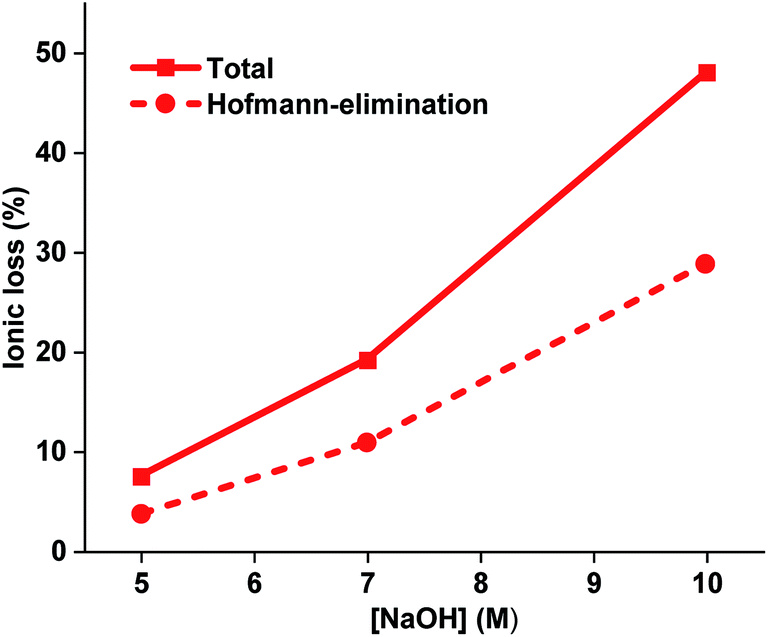
The alkaline stability of all the AEMs was also studied after immersion in 7 and 10 M aq. NaOH for 168 h at 90 °C. However, after these harsh treatments the PDPF–Qui and PDPF–Pip membranes were no longer soluble, which may imply the formation of crosslinks due to the presence of a very small fraction of residual unsubstituted Br atoms in the respective polymer structure. In contrast, PDPF–TMA remained soluble in DMSO-d6, which enabled NMR analysis (Fig. S8 and S9†). The results showed that the total ionic loss of PDPF–TMA after storage in 10 M at 90 °C reached ∼47% and the main degradation pathway was β-elimination in the alkyl spacer chain. As expected, the degradation by β-elimination increased dramatically from 4.4 to 29% as the NaOH concentration was increased from 5 to 10 M during 168 h at 90 °C (Fig. 5). In 5 and 10 M aq. NaOH, the loss by β-elimination was ∼49 and ∼60%, respectively, of the total ionic loss. This hinted that the relative rates of the degradation reactions changed with the alkali concentration.
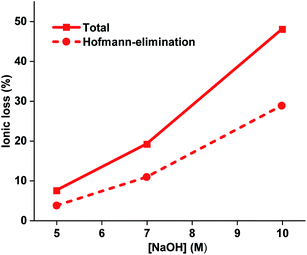 |
| | Fig. 5 Total ionic loss and ionic loss by Hofmann β-elimination, as calculated from 1H NMR spectra of PDPF-TMA recorded after immersion in aq. NaOH of different concentrations for 168 h at 90 °C. | |
4. Conclusions
Aryl ether-free 2,7-diphenylfluorene-based copolymers tethered with Qui cations on hexyl spacers were successfully synthesized via superacid catalyzed Friedel–Crafts polycondensation and subsequent quaternization reactions. Compared with the performances of membranes based on corresponding copolymers carrying TMA and Pip cations, the Qui-functionalized AEMs were efficient OH− conductors and in addition showed the highest thermal and alkaline stability in the series. Careful analysis of protonated membrane polymers by 1H NMR spectroscopy proved to be an efficient method to identify ionic loss mechanisms and to quantify degradation products after the alkaline treatments. This revealed that the Qui cation had a very high resistance against β-elimination under the conditions investigated, with no detectable elimination in the ring structure and to a significantly lower degree in the alkyl spacer chain as compared with the corresponding TMA and Pip cations. The high alkaline stability of the bicyclic Qui cation may be attributed to the low ring strain of the 6-membered rings, in combination with the very strong conformational restrictions imposed by the cage-like structure which most likely contribute to a very high transition state energy of the elimination reactions. In addition, no degradation of the aryl ether-free copolymer backbones was detected, even after storage of AEMs in 10 M NaOH at 90 °C. Hence, the overall findings demonstrate that the strategy of combining aryl ether-free backbone polymers and Qui cations results in durable and high-performance AEMs suitable for use in, e.g., alkaline fuel cells and water electrolyzers.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We thank the Swedish Energy Agency (grants 45057-1 and 37806-3), the Swedish Research Council (grants 45397-1 and 2015-04820) and the Swedish Foundation for Strategic Research, SSF (grant EM16-0060), for financial support. We are also grateful to Peter Holmqvist for assistance with SAXS measurements.
References
- E. J. Park and Y. S. Kim, J. Mater. Chem. A, 2018, 6, 15456–15477 RSC.
- D. R. Dekel, J. Power Sources, 2018, 375, 158–169 CrossRef CAS.
- S. Gottesfeld, D. R. Dekel, M. Page, C. Bae, Y. S. Yan, P. Zelenay and Y. S. Kim, J. Power Sources, 2018, 375, 170–184 CrossRef CAS.
- M. A. Hickner, A. M. Herring and E. B. Coughlin, J. Polym. Sci., Part B: Polym. Phys., 2014, 52, 347 CrossRef CAS.
- J. R. Varcoe, P. Atanassov, D. R. Dekel, A. M. Herring, M. A. Hickner, P. A. Kohl, A. R. Kucernak, W. E. Mustain, K. Nijmeijer, K. Scott, T. W. Xu and L. Zhuang, Energy Environ. Sci., 2014, 7, 3135–3191 RSC.
- C. G. Arges and L. Zhang, ACS Appl. Energy Mater., 2018, 1, 2991–3012 CrossRef CAS.
- D. R. Dekel, M. Arnar, S. Willdorf, M. Kosa, S. Dhara and C. E. Diesendruck, Chem. Mater., 2017, 29, 4425–4431 CrossRef CAS.
- K. D. Kreuer and P. Jannasch, J. Power Sources, 2018, 375, 361–366 CrossRef CAS.
- M. R. Hibbs, J. Polym. Sci., Part B: Polym. Phys., 2013, 51, 1736–1742 CrossRef CAS.
- T. H. Pham and P. Jannasch, ACS Macro Lett., 2015, 4, 1370–1375 CrossRef CAS.
- A. D. Mohanty, S. E. Tignor, J. A. Krause, Y. K. Choe and C. Bae, Macromolecules, 2016, 49, 3361–3372 CrossRef CAS.
- S. Miyanishi and T. Yamaguchi, Phys. Chem. Chem. Phys., 2016, 18, 12009–12023 RSC.
- C. G. Arges and V. Ramani, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 2490–2495 CrossRef CAS PubMed.
- J. J. Han, H. Q. Peng, J. Pan, L. Wei, G. W. Li, C. Chen, L. Xiao, J. T. Lu and L. Zhuang, ACS Appl. Mater. Interfaces, 2013, 5, 13405–13411 CrossRef CAS PubMed.
- N. Li, Q. Zhang, C. Wang, Y. M. Lee and M. D. Guiver, Macromolecules, 2012, 45, 2411–2419 CrossRef CAS.
- C. Vogel and J. Meier-Haack, Desalination, 2014, 342, 156–174 CrossRef CAS.
- H. S. Dang, E. A. Weiber and P. Jannasch, J. Mater. Chem. A, 2015, 3, 5280–5284 RSC.
- J. Yan and M. A. Hickner, Macromolecules, 2010, 43, 2349–2356 CrossRef CAS.
- A. D. Mohanty, C. Y. Ryu, Y. S. Kim and C. Bae, Macromolecules, 2015, 48, 7085–7095 CrossRef CAS.
- R. Akiyama, N. Yokota and K. Miyatake, Macromolecules, 2019, 52, 2131–2138 CrossRef CAS.
- M. R. Hibbs, C. H. Fujimoto and C. J. Cornelius, Macromolecules, 2009, 42, 8316–8321 CrossRef CAS.
- R. Janarthanan, J. L. Horan, B. R. Caire, Z. C. Ziegler, Y. Yang, X. B. Zuo, M. W. Liberatore, M. R. Hibbs and A. M. Herring, J. Polym. Sci., Part B: Polym. Phys., 2013, 51, 1743–1750 CrossRef CAS.
- W. H. Lee, A. D. Mohanty and C. Bae, ACS Macro Lett., 2015, 4, 453–457 CrossRef CAS.
- W. H. Lee, E. J. Park, J. Han, D. W. Shin, Y. S. Kim and C. Bae, ACS Macro Lett., 2017, 6, 566–570 CrossRef CAS.
- J. S. Olsson, T. H. Pham and P. Jannasch, Adv. Funct. Mater., 2018, 28, 1702758 CrossRef.
- J. S. Olsson, T. H. Pham and P. Jannasch, J. Membr. Sci., 2019, 578, 183–195 CrossRef CAS.
- W. H. Lee, Y. S. Kim and C. Bae, ACS Macro Lett., 2015, 4, 814–818 CrossRef CAS.
- A. M. A. Mahmoud and K. Miyatake, J. Mater. Chem. A, 2018, 6, 14400–14409 RSC.
- S. Maurya, S. Noh, I. Matanovic, E. J. Park, C. N. Villarrubia, U. Martinez, J. Han, C. Bae and Y. S. Kim, Energy Environ. Sci., 2018, 11, 3283–3291 RSC.
- H. Ono, T. Kimura, A. Takano, K. Asazawa, J. Miyake, J. Inukai and K. Miyatake, J. Mater. Chem. A, 2017, 5, 24804–24812 RSC.
- M. Ozawa, T. Kimura, K. Otsuji, R. Akiyama, J. Miyake, M. Uchida, J. Inukai and K. Miyatake, ACS Omega, 2018, 3, 16143–16149 CrossRef CAS PubMed.
- T. H. Pham, J. S. Olsson and P. Jannasch, J. Mater. Chem. A, 2019, 7, 15895–15906 RSC.
- O. D. Thomas, K. J. W. Y. Soo, T. J. Peckham, M. P. Kulkarni and S. Holdcroft, J. Am. Chem. Soc., 2012, 134, 10753–10756 CrossRef CAS PubMed.
- A. G. Wright, T. Weissbach and S. Holdcroft, Angew. Chem., Int. Ed., 2016, 55, 4818–4821 CrossRef CAS PubMed.
- J. T. Fan, A. G. Wright, B. Britton, T. Weissbach, T. J. G. Skalski, J. Ward, T. J. Peckham and S. Holdcroft, ACS Macro Lett., 2017, 6, 1089–1093 CrossRef CAS.
- J. T. Fan, S. Willdorf-Cohen, E. M. Schibli, Z. Paula, W. Li, T. J. G. Skalski, A. T. Sergeenko, A. Hohenadel, B. J. Frisken, E. Magliocca, W. E. Mustain, C. E. Diesendruck, D. R. Dekel and S. Holdcroft, Nat. Commun., 2019, 10, 2306 CrossRef PubMed.
- J. S. Olsson, T. H. Pham and P. Jannasch, Macromolecules, 2017, 50, 2784–2793 CrossRef CAS.
- C. Wang, B. M. Mo, Z. F. He, Q. Shao, D. Pan, E. Wujick, J. Guo, X. L. Xie, X. F. Xie and Z. H. Guo, J. Membr. Sci., 2018, 556, 118–125 CrossRef CAS.
- C. Wang, B. M. Mo, Z. F. He, X. F. Xie, C. X. Zhao, L. Q. Zhang, Q. Shao, X. K. Guo, E. K. Wujcik and Z. H. Guo, Polymer, 2018, 138, 363–368 CrossRef CAS.
- A. D. Mohanty and C. Bae, J. Mater. Chem. A, 2014, 2, 17314–17320 RSC.
- J. B. Edson, C. S. Macomber, B. S. Pivovar and J. M. Boncella, J. Membr. Sci., 2012, 399, 49–59 CrossRef.
- M. G. Marino and K. D. Kreuer, ChemSusChem, 2015, 8, 513–523 CrossRef CAS PubMed.
- S. Chempath, B. R. Einsla, L. R. Pratt, C. S. Macomber, J. M. Boncella, J. A. Rau and B. S. Pivovar, J. Phys. Chem. C, 2008, 112, 3179–3182 CrossRef CAS.
- J. H. Wang, Y. Zhao, B. P. Setzler, S. Rojas-Carbonell, C. Ben Yehuda, A. Amel, M. Page, L. Wang, K. Hu, L. Shi, S. Gottesfeld, B. J. Xu and Y. S. Ya, Nat. Energy, 2019, 4, 392–398 CrossRef CAS.
- H. S. Dang and P. Jannasch, J. Mater. Chem. A, 2017, 5, 21965–21978 RSC.
- N. Chen, C. Lu, Y. Li, C. Long and H. Zhu, J. Membr. Sci., 2019, 572, 246–254 CrossRef CAS.
- T. H. Pham, J. S. Olsson and P. Jannasch, J. Am. Chem. Soc., 2017, 139, 2888–2891 CrossRef CAS PubMed.
- D. J. Strasser, B. J. Graziano and D. M. Knauss, J. Mater. Chem. A, 2017, 5, 9627–9640 RSC.
- X. M. Chu, Y. Shi, L. Liu, Y. D. Huang and N. W. Li, J. Mater. Chem. A, 2019, 7, 7717–7727 RSC.
- F. X. Gong, R. Q. Wang, X. B. Chen, P. Chen, Z. W. An and S. B. Zhang, Polym. Chem., 2017, 8, 4207–4219 RSC.
- Y. F. Yang, Y. X. Xu, N. Y. Ye, D. J. Zhang, J. S. Yang and R. H. He, J. Electrochem. Soc., 2018, 165, F350–F356 CrossRef CAS.
- S. H. Oh, S. I. Na, Y. C. Nah, D. Vak, S. S. Kim and D. Y. Kim, Org. Electron., 2007, 8, 773–783 CrossRef CAS.
- D. S. Pedersen and C. Rosenbohm, Synthesis, 2001, 2431–2434, DOI:10.1055/s-2001-18722.
- D. A. Klumpp, M. Garza, A. Jones and S. Mendoza, J. Org. Chem., 1999, 64, 6702–6705 CrossRef CAS PubMed.
- G. Gebel and O. Diat, Fuel Cells, 2005, 5, 261–276 CrossRef CAS.
- J. Pan, S. F. Lu, Y. Li, A. B. Huang, L. Zhuang and J. T. Lu, Adv. Funct. Mater., 2010, 20, 312–319 CrossRef CAS.
- Y. W. Zheng, U. Ash, R. P. Pandey, A. G. Ozioko, J. Ponce-Gonzalez, M. Handl, T. Weissbach, J. Varcoe, S. Holdcroft, M. W. Liberatore, R. Hiesgen and D. R. Dekel, Macromolecules, 2018, 51, 3264–3278 CrossRef CAS.
- L. Liu and P. A. Kohl, J. Polym. Sci., Part A: Polym. Chem., 2018, 56, 1395–1403 CrossRef CAS.
- G. Cospito, G. Illuminati, C. Lillocci and H. Petride, J. Org. Chem., 1981, 46, 2944–2947 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ta09213g |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ,
Joel S.
Olsson
,
Joel S.
Olsson