
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInsights into the electrochemical processes of rechargeable magnesium–sulfur batteries with a new cathode design†
Bhaghavathi P.
Vinayan
 *a,
Holger
Euchner
a,
Zhirong
Zhao-Karger
b,
Musa Ali
Cambaz
a,
Zhenyou
Li
b,
Thomas
Diemant
c,
R. Jürgen
Behm
ac,
Axel
Gross
ad and
Maximilian
Fichtner
ab
*a,
Holger
Euchner
a,
Zhirong
Zhao-Karger
b,
Musa Ali
Cambaz
a,
Zhenyou
Li
b,
Thomas
Diemant
c,
R. Jürgen
Behm
ac,
Axel
Gross
ad and
Maximilian
Fichtner
ab
aHelmholtz Institute Ulm (HIU) Electrochemical Energy Storage, Helmholtzstr. 11, D-89081 Ulm, Germany. E-mail: vinayan.parambath@kit.edu
bInstitute of Nanotechnology, Karlsruhe Institute of Technology (KIT), P.O. Box 3640, D-76021 Karlsruhe, Germany
cInstitute of Surface Chemistry and Catalysis, Ulm University, Albert-Einstein-Allee 47, D-89081 Ulm, Germany
dInstitute of Theoretical Chemistry, Ulm University, Albert-Einstein-Allee 11, D-89081 Ulm, Germany
First published on 22nd October 2019
Abstract
The present study shows the electrochemical performance of a room-temperature magnesium/sulfur (Mg/S) battery with a newly designed sulfur (3–0.5 mgsulfur cm−2) composite cathode. Operando Raman spectroscopy is employed to investigate the formation of polysulfide species at the cathode of Mg/S cells during the charge/discharge process, while density functional theory (DFT) calculations are used to correlate the Raman modes with a series of polysulfide species (Sxn−, x = 1–8). Operando Raman spectroscopy proves the chemical transformation from elemental sulfur (S8) via long and short chain polysulfides to magnesium sulfide upon discharge and conversion back to elemental sulfur during charging. Furthermore, the spectral measurements indicate the formation of a nanocrystalline magnesium sulfide with a cubic zinc blende phase at the end of discharge. The changes in the open circuit potential of the Mg/S cell during the resting period are investigated with the help of various spectroscopic techniques. These studies conclude that cell impedance is dominated by the anode side impedance due to the formation of a passivation layer under static conditions. Finally, the impedance studies under dynamic conditions verify that the applied electrode potential plays a significant role on the evolution of the anode interfacial impedance.
1. Introduction
Amongst the several battery chemistries possible, the most appealing alternatives for Li involve the use of multivalent cations like magnesium (Mg). This has several reasons. The primary one is the abundance of Mg raw materials (Mg is the 8th most abundant element in the Earth's crust, vs. Li being the 25th), making them 20 to 50 times cheaper than Li, e.g., $265 per ton vs. $5000 per ton for MgCO3 and Li2CO3, respectively.1 Performance-wise, the low cost alternative Mg yields a standard reduction potential of −2.37 V (Mg/Mg2+) vs. SHE (standard hydrogen electrode) and a large theoretical electrochemical capacity (∼3832 mA h cm−3).2 In spite of recent achievements in case of Mg based rechargeable batteries, especially in the field of electrolyte and cathode development,1,3 a fundamental understanding of the battery chemistry is still missing and needs further exploration. The main challenges in Mg batteries come from the slow kinetics of the multivalent (MV) ions (Mg2+) at the cathode and the identification of appropriate electrolytes, which should be compatible with cathode/anode interfaces. Electrolytes capable of efficient and reversible stripping/plating of MV ions at the anode, and also supporting reversible intercalation against a high voltage cathode, remain a significant and fundamental scientific challenge. In this regard, high energy density conversion type cathode materials like sulfur (S) are promising in combination with Mg anodes because of the high theoretical capacity (1672 mA h g−1 or 3459 mA h cm−3), as well as their abundance and low cost.4Table 1 compares the theoretical energy density of a sulfur cathode with that of various alkali/alkaline earth metal anodes.5–9 The combination of a Mg anode and a sulfur cathode yields a high energy density of ∼1722 W h kg−1, assuming a two-electron conversion reaction (Mg2+ + S + 2e− → MgS). This is three times higher than the energy density of a Li ion battery with graphite as the anode and cobalt oxide as the cathode.10| Electrochemical reactions | ΔG (kJ mol−1) | Voltage (V) | Energy density (W h kg−1) | Energy density (W h L−1) |
|---|---|---|---|---|
| Mg + S ⇄ MgS | −341.8 | 1.77 | 1722 | 3200 |
| 2Li + S ⇄ Li2S | −432.6 | 2.24 | 2500 | 2780 |
| 2Na + S ⇄ Na2S | −357.8 | 1.85 | 1273 | 2363 |
| 2Al + 3S ⇄ Al2S3 | −213.3 | 1.11 | 1340 | 2676 |
| Zn + S ⇄ ZnS | −201.3 | 1.04 | 574 | 2162 |
| CoO2 + LiC6 ⇄ LiCoO2 + C6 | −347.4 | 3.60 | 568 | 1901 |
High energy density cathode materials, such as sulfur (electrophilic in nature) in Mg rechargeable batteries, need a non-nucleophilic electrolyte to guarantee the stability of the latter. Electrolytes, which are simultaneously Mg-ion-conducting and compatible with sulfur, represent a particular challenge. Facing this challenge, our group has recently developed two new working electrolytes for the Mg/S system. The first one is based on Mg-bishexamethyl disilazide (Mg(HMDS)2), which was treated with aluminum chloride in different solvents.11 The second one is a non-corrosive (Cl− free) fluorinated alkoxyborate magnesium electrolyte (Mg[B(hfip)4]2) in glyme solvent,12 being compatible with currently used Mg electrode materials. In the meantime, Cui et al. developed a boron-centered anion-based magnesium electrolyte by facile one-step mixing of tris(2H-hexafluoroisopropyl)borate and MgF2 in 1,2-dimethoxyethane.13 The same group also synthesized organic magnesium borate electrolyte that mainly contains tetrakis(hexafluoroisopropyl)borate anions [B(HFP)4]− and [Mg4Cl6(DME)6]2+ solvated cations.14 Both of these electrolytes showed promising electrochemical results in magnesium–sulfur/selenium batteries. Chilin Li et al. proposed a Mg battery with a dual-salt Mg2+/Li+ electrolyte and FeSx conversion cathode, and this cell showed a reversible capacity of more than 200 mA h g−1 after 200 cycles.15
The electrochemical performances of sulfur cathodes in Mg/S batteries, especially with high sulfur loading (>1 mgsulfur cm−2), usually show the large overpotential between charge and discharge, rapid capacity fading, and poor cycling efficiency, etc.10,16 Most of these issues strongly correlate with the formation of soluble polysulfide species at the cathode and their shuttling to the anode, and the sluggish redox kinetics of Mg with sulfur.10 Recent studies proved that the presence of small amounts of sulfur species at the Mg anode enhances the cell resistance and overpotential considerably and may lead to large hysteresis between charging and discharging.5,17 In addition, the loss of sulfur active material from the cathode to the anode via dissolution also causes capacity fading. In both cases, a good confinement of polysulfide species at the cathode is indeed very important. At the cathode of a Mg/S battery, the interfacial electrochemical kinetics depend on two factors. First, the adequate binding affinity of sulfur/Sxn− (x = 1–8) with the host matrix allows the adsorption of these species with sufficient surface coverage on the adsorbent surface. Second, the efficient charge transfer at the cathode/electrolyte interface requires the fast transport of the electrons, generated during the redox reactions of the adsorbed sulfur species, through the cathode. This can be achieved using a polar conductive host matrix which provides a large surface area for a more efficient binding of the polar polysulfide species at the cathode and, moreover, maintains an efficient electron transport.18 Methods for blocking the undesired Mg polysulfide (Mg PS) transport to the anode side include the use of additional carbon coated or chemically modified separators and special binders.5,19
To investigate the redox processes of Mg/S cells, various characterization techniques have been reported previously.20–22 Recently, Zhao-Karger et al. showed by XPS that a Mg/S cell with Mg[B(hfip)4]2 electrolyte undergoes many step process, transforming from bulk sulfur to Sx−.5 Nakayama et al. investigated the charge/discharge reaction mechanism of a Mg/S cell with sulfone-based magnesium electrolytes by various ex situ techniques like XPS, X-ray absorption near edge structure (XANES) spectroscopy, XRD, high-energy XRD (HE-XRD), transmission X-ray microscopy (TXM) and solid-state NMR.23 These studies conclude that MgS with the zinc blende (ZB) structure type forms as the final discharge product on the cathode side. In another report, Robba et al. investigated the reduction processes in Mg/S cells with Mg(TFSI)2–MgCl2 electrolyte by operando XRD and S K-edge X-ray absorption (XANES) studies.20 From this study, they concluded that the electrochemically formed MgS phase has a different local structure than chemically synthesized MgS. Elemental sulfur as well as different polysulfide species in Li/S batteries, which are formed as reaction intermediates at different stages of the discharge/charge cycle, are Raman active. Hence, the operando Raman analysis of the sulfur cathode in a Mg/S cell can provide additional information about the redox reaction mechanism. Based on the above-discussed findings, we have designed new cathodes for Mg/S batteries with different sulfur loadings (3–0.5 mgsulfur cm−2), using a nitrogen doped hybrid nanocomposite of multiwall carbon nanotubes (MWCNTs) and graphene as the host matrix, and investigated the formation of various polysulfide species during the discharging/charging of a Mg/S cell by operando Raman spectroscopy and XPS. Furthermore, we have studied the evolution of the Mg/S cell impedances at the cathode and anode sides separately under static and dynamic conditions and correlated these results with the findings of operando Raman spectroscopy. The combination of operando spectral measurements with DFT calculations allows us to gain a deeper understanding of the reaction mechanisms in the Mg/S system and may help to develop new design strategies and improvements for Mg/S batteries.
2. Experimental section
2.1. Synthesis of materials
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and further stirred in deionized (DI) water for proper mixing. The suspension was then dried in a vacuum oven (at 50 °C). The final suspension was loaded into a quartz tube and flushed with argon gas for 15 min at room temperature. Subsequently, hydrogen (99.99%) gas was admitted and the temperature was increased to 500 °C for 2 h. After that, heating was stopped and the sample was cooled back to room temperature. In the following, the hybrid structure of graphene and MWCNTs will be labeled as G-MWCNT. Nitrogen plasma treatment of the G-MWCNT sample was performed using a sputtering system with nitrogen gas (99.999%) as a plasma source.26 About 500 mg of sample was taken in a glass Petri dish and kept at the substrate side of the plasma chamber for 30 min. The chamber pressure and plasma power were maintained at 0.1 mbar and 130 W, respectively. During this experiment, graphite was used as the sputtering target to avoid the presence of any other impurities within the sample. For the rest of the manuscript, the final nitrogen doped carbon support will be referred to as NC. Finally, sulfur was loaded into the NC host matrix by melt infiltration at 155 °C in an argon gas atmosphere (from now on labeled as ‘S/NC’).
:1. The ball milling was carried out at 300 rpm for 32 h in a planetary ball mill (Fritsch PULVERISETTE 6) with an 80 mL silicon nitride vial and silicon nitride balls. The ball-to-powder ratio was 1:20. All samples were loaded into the ball mill vial inside an argon filled glovebox.
:1 and further stirred in deionized (DI) water for proper mixing. The suspension was then dried in a vacuum oven (at 50 °C). The final suspension was loaded into a quartz tube and flushed with argon gas for 15 min at room temperature. Subsequently, hydrogen (99.99%) gas was admitted and the temperature was increased to 500 °C for 2 h. After that, heating was stopped and the sample was cooled back to room temperature. In the following, the hybrid structure of graphene and MWCNTs will be labeled as G-MWCNT. Nitrogen plasma treatment of the G-MWCNT sample was performed using a sputtering system with nitrogen gas (99.999%) as a plasma source.26 About 500 mg of sample was taken in a glass Petri dish and kept at the substrate side of the plasma chamber for 30 min. The chamber pressure and plasma power were maintained at 0.1 mbar and 130 W, respectively. During this experiment, graphite was used as the sputtering target to avoid the presence of any other impurities within the sample. For the rest of the manuscript, the final nitrogen doped carbon support will be referred to as NC. Finally, sulfur was loaded into the NC host matrix by melt infiltration at 155 °C in an argon gas atmosphere (from now on labeled as ‘S/NC’).
:1. The ball milling was carried out at 300 rpm for 32 h in a planetary ball mill (Fritsch PULVERISETTE 6) with an 80 mL silicon nitride vial and silicon nitride balls. The ball-to-powder ratio was 1:20. All samples were loaded into the ball mill vial inside an argon filled glovebox.
2.2. Physico-chemical characterization
The X-ray diffraction (XRD) measurements were conducted using a Stadi P diffractometer (STOE & Cie) with a MYTHEN detector using a Cu Kα X-ray source. Thermogravimetric analysis (TGA) of the samples was carried out along with differential scanning calorimetry (DSC) using a Setaram thermal analyzer SENSYS evo instrument. The measurements were conducted from room temperature to 450 °C under a helium flow (20 mL min−1) with a heating rate of 10 °C min−1. Brunauer–Emmett–Teller (BET) surface area analysis of the samples was performed with a Micromeritics ASAP 2020 MP system. The morphology of the samples was studied using scanning electron microscopy (SEM) and transmission electron microscopy (TEM).The chemical state of the sample surfaces was determined by X-ray photoelectron spectroscopy (XPS) measurements using monochromatized Al Kα (1486.6 eV) radiation (PHI 5800 MultiTechnique ESCA System, Physical Electronics). The measurements were conducted with a detection angle of 45°, using pass energies of 93.9 and 29.35 eV at the analyzer for survey and detailed spectra, respectively. To avoid surface contamination, the samples were transferred in an inert gas atmosphere to the sample load lock of the XPS system. During XPS measurements the samples were neutralized with electrons from a flood gun (current 3 μA) to compensate for charging effects at the surface. For binding energy calibration the C 1s peak of graphene was set to 284.6 eV. Peak fitting was done with CasaXPS using Shirley-type backgrounds and Gaussian–Lorentzian peak profiles. For the S 2p peaks, doublets with a peak area ratio of 2:1 and a spin–orbit splitting of 1.2 eV were used.
2.3. Electrochemical experiments
The electrode slurry for the cathode was prepared by mixing S/NC powder (90 wt%) with a PVDF binder (10 wt%, Kynar) using NMP as the solvent. Next, the obtained slurry was cast onto Al foil by the doctor blade technique and thereafter dried at 60 °C for 24 h. The electrodes were prepared with two different sulfur loadings of ∼3 mg cm−2 and 0.5 mg cm−2. All cells were assembled inside an argon filled glovebox (MBraun GmbH) and consisted of a sulfur cathode, Mg anode, and polypropylene separator (EL-Cell). The electrolyte was 0.4 M Mg[B(hfip)4]2 in DME solvent. 100 μL electrolyte was used in each assembled cell. All electrochemical measurements were performed using a three-electrode cell (PAT-Cell from EL-CELL) with an Mg ring as the reference electrode (MgRE) and Mg metal as the counter electrode (MgCE).The electrochemical performances of the cells were evaluated by galvanostatic and voltammetric experiments using a battery tester (Bio-Logic VMP-3) at room temperature (∼25 °C). Cyclic voltammetry (CV) experiments were carried out at a scan rate of 0.5 mV s−1. The charge/discharge specific capacities mentioned in this paper were calculated based on the mass of sulfur (1C = 1672 mA gsulfur−1) by excluding the carbon content. Electrochemical impedance spectroscopy (EIS) experiments were conducted using a three-electrode cell (EL-CELL® GmbH) with an applied sinusoidal excitation voltage of 10 mV in the frequency range from 500 kHz to 0.7 Hz.
3. Results and discussion
3.1. Structural and morphological study
A nitrogen doped graphene and multiwall carbon nanotube hybrid nanocomposite (NC) was used as a host matrix for the sulfur cathode. The morphology of the nitrogen doped G-MWCNT sample was verified by TEM and SEM as shown in Fig. 1(a) and S1.† As expected, the overview images indicate the presence of coupled one- and two-dimensional carbon nanostructures within the sample.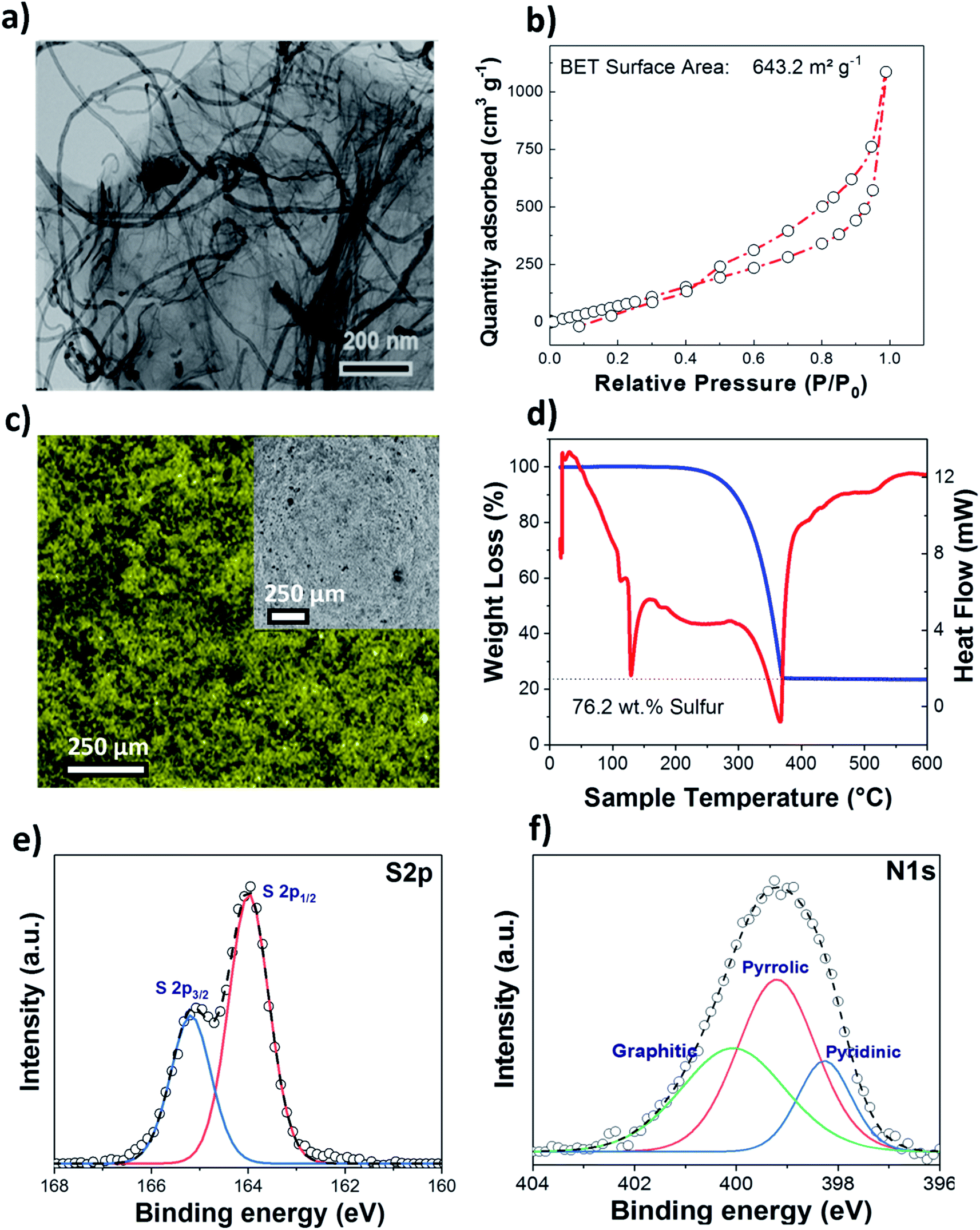 | ||
| Fig. 1 (a) TEM image of the nitrogen doped G-MWCNT nanocomposite. (b) BET surface area of NC. (c) EDX mapping of the sulfur impregnated in NC. (d) TGA results of the sulfur impregnated NC. (e) S 2p and (f) N 1s detailed XP spectra of sulfur loaded NC. | ||
The presence of MWCNTs between graphene sheets prevents them from restacking, thus retaining the large surface area for the adsorption of the sulfur active material during the redox process; simultaneously, MWCNTs enhance the electronic conductivity of the whole cathode composite. The surface area and pore size distributions of the as-prepared NC samples were determined by nitrogen adsorption/desorption measurements (Fig. 1(b)), and a high BET surface area of ∼644 m2 g−1 was obtained, as compared to that of pure MWCNTs (∼283 m2 g−1). The total pore volume for the nitrogen doped carbon matrix was ∼1.65 cm3 g−1, with the sample containing both micro- and mesopores. SEM and EDX (Fig. 1(c)) images of the S/NC sample show the uniform distribution of sulfur. TGA (Fig. 1(d)) moreover gives a high sulfur loading of ∼76.2 wt% for the S/NC sample. Fig. 1(e) and (f) show the S 2p and N 1s XPS detailed spectra of the as-prepared S/NC sample. The S 2p spectrum shows a peak doublet at 164.0/165.2 eV, which is assigned to elemental sulfur.27 The N 1s spectrum (Fig. 1(f)) features three different peaks at 398.5 eV, 399.5 eV and 400.6 eV, which are ascribed to pyridinic N (17%), pyrrolic N (59%) and graphitic N (24%), respectively.28 The total nitrogen content at the surface of the S/NC sample amounts to ∼5 at%, and it is clearly dominated by pyrrolic nitrogen functional groups. During the functionalization of the carbon surface, the type of nitrogen functional group is indeed very important. The doping of nitrogen into a graphene lattice can typically create the three types of bonding configurations mentioned above, pyridinic N, pyrrolic N and graphitic N (quaternary N).29 Pyridinic N binds with two C atoms at the edges or defects of graphene and donates one p electron into the π system.29 Pyrrolic N atoms donate two p electrons to the π system, while graphitic N substitutes for C atoms in the graphene hexagonal ring. A previous theoretical study of N-doped carbon nanotubes with pyrrolic nitrogen showed that these functional groups result in a comparatively higher positive charge on nearby carbon atoms than other nitrogen functional groups.30 Similarly, another study from our group proved that the addition of nitrogen dopants into the graphene lattice can result in a polar nature of the host matrix, which increases the binding strength of polysulfides to the N-doped graphene.27 The C 1s spectrum of S/NC (Fig. S2†) is dominated by a peak at 284.6 eV, which is assigned to graphene (sp2-hybridized C). Furthermore, the peak at 285.1 eV is related to carbon in sp3-hybridized C–C, C–H or C–S bonds, while the other two and rather small peaks at higher binding energies (286.6 eV and 287.9 eV) are due to carbon in C–O and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds.27
O bonds.27
3.2. Electrochemical studies
The electrochemical performance of the S/NC cathode in the Mg/S cell was investigated by cyclic voltammetry (CV) and galvanostatic discharge/charge cycling. The experiments were carried out using a three-electrode cell setup with a Mg ring as the reference electrode (MgRE), Mg metal as the counter electrode (MgCE), and a S/NC cathode as the working electrode (WE). Fig. 2(a) shows the CV curves of the S/NC cathode (3 mgsulfur cm−2) with respect to the MgRE and MgCE electrodes. During the CV scan, the S/NC cathode showed a main reduction peak at 1.51 V, while the oxidation peak appeared at ∼2 V with respect to the MgRE electrodes. It is interesting to note that the reduction peak potential of S/NC shifted slightly to 1.41 V with respect to MgCE, while the oxidation peak moved to 2.39 V, indicating a much higher overpotential in the Mg/S cell during the cathode oxidation. Fig. 2(b) shows the MgCE potential (vs. MgRE) in the Mg/S cell with respect to the scan time, together with the cathode current density. It demonstrates that the contribution to the overall Mg/S cell overpotential is dominated by the anode side. Moreover, it is evident that this overpotential at the anode is higher during the cathode oxidation (∼381 mV) as compared to the cathode reduction (∼100 mV). This indicates that the energy barrier for the reduction of Mg2+ ions to Mg0 and further deposition on the Mg anode is comparatively high in the Mg/S cell, which could have been augmented by the presence of dissolved polysulfide species on the Mg anode side.17 This is corroborated by the fact that the overpotential at the anode side is directly proportional to the amount of polysulfide species dissolved in the electrolyte from the sulfur cathode. To investigate this in detail, we first synthesized Mg polysulfide (Mg PS) in glyme solvent as described in our previous report,5 and then added different concentrations of Mg PS to the electrolyte. The amount of sulfur in the polysulfide solution was determined by inductively coupled plasma atomic emission spectroscopy (ICP-AES), yielding a concentration of 68 mmolsulfur L−1. Afterwards, galvanostatic stripping/plating of symmetrical Mg/Mg cells (∼±100 μA cm−2, tcharge/tdischarge = 30 minutes) was carried out with pure Mg electrolyte in DME solvent, with Mg electrolyte containing low amounts of polysulfide additives (sulfur content: ∼14 mmol L−1), and with Mg electrolyte containing high amounts of polysulfide (sulfur content: ∼54 mmol L−1). The results of these experiments (see Fig. S3†) showed that the overpotential at the Mg electrode increased from 100 mV for pure electrolyte, to 340 mV for low sulfur containing electrolyte. It further increased to 1400 mV when the same electrolyte containing high amounts of sulfur species was used. This clearly proves the negative impact of dissolved polysulfide species at the Mg anode side.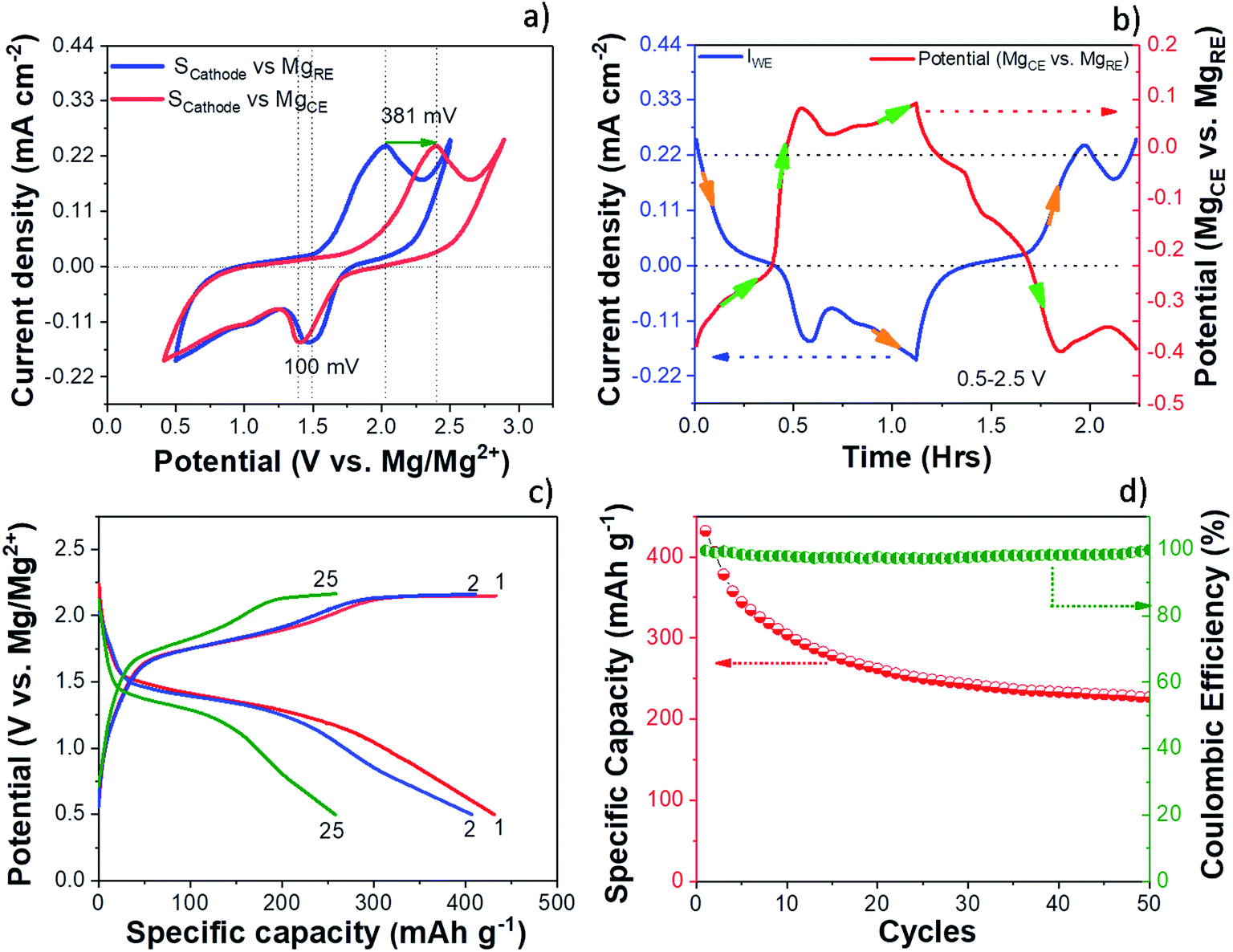 | ||
| Fig. 2 (a) CVs of the S/NC cathode in an Mg/S cell using a three-electrode cell at a scan rate of 0.5 mV s−1. (b) Variation of the cathode (WE) current density and the MgCE potential (vs. MgRE) with respect to the scan time. (c) Charge/discharge voltage profiles of the S/NC cathode at a current rate of C/50. (d) Cycling performance of the S/NC cathode at a current rate of C/50. | ||
Fig. 2(c) and (d) show the galvanostatic charge/discharge voltage profiles and cycling performances of the Mg/S cell with a S/NC cathode (3 mgsulfur cm−2), cycled at a current rate of C/50 in the potential range of 2.25–0.5 V (vs. MgRE). The discharge capacities for the 1st, 2nd and 50th cycles were 431, 406 and 228 mA h g−1, i.e., the electrode retains 94.2% and 53% of its initial capacity after the 2nd and the 50th cycle, respectively. The decrease in the discharge capacity during the cycling could be due to the high sulfur loading at the cathode and the slow kinetics of cathode redox reactions, which prevent the complete utilization of the active material. As we decreased the cathode sulfur loading to ∼0.5 mgsulfur cm−2 (20 wt% sulfur loaded NC), the Mg/S cell capacity increased to ∼1384 mA h g−1 after the first cycle and 1117 mA h g−1 after the 5th cycle (see Fig S4†). We noticed that there was an increase of the cell voltage hysteresis as we deep-discharged the Mg/S cell with high sulfur loading (3 mgsulfur cm−2) to 0.2 V. The high sulfur loading on the cathode can augment the loss of polysulfide species to the anode side via dissolution during the cycling, thus reducing the cycling performance. The XPS spectra of the surface of the cycled Mg anode for the Mg/S cell with high sulfur loading (3 mgsulfur cm−2) indeed show the presence of a small amount of sulfur species (0.23 at%) (Fig. S5a†). These species can form a highly resistive passivation layer on the Mg anode surface by the formation of an electrochemically inactive MgS and augment the detrimental side reactions of the electrolyte with the electrode materials.14,31 In addition to the sulfur species, the XPS spectra of the cycled Mg anode also show the presence of species like fluorine, carbon, and oxygen on the surface layer (see Fig. S5†). The deconvoluted F 1s spectrum gives rise to two peaks at the positions 685.3 eV and 688.4 eV. The first low intensity peak may be due to the formation of some MgFx, while the high intensity second peak is possibly due to the formation of some organic fluorine species (–CF3).32 The XPS study of the cycled Mg anode points to the formation of an interface passivation layer with organic/in-organic species.
3.3. Mechanistic studies
The charge/discharge reaction mechanism or more precisely the change of the chemical state of sulfur during the cycling of the Mg/S cell was investigated in detail by operando Raman spectroscopy. The high sulfur loading (∼3 mgsulfur cm−2) and the use of the same electrolyte amount (100 μL) as that for previous electrochemical experiments were maintained in the operando Raman cell also. Fig. 3(a) and 4(a) show the operando Raman spectra for the S/NC cathode of the Mg/S cell during the discharge and charge processes.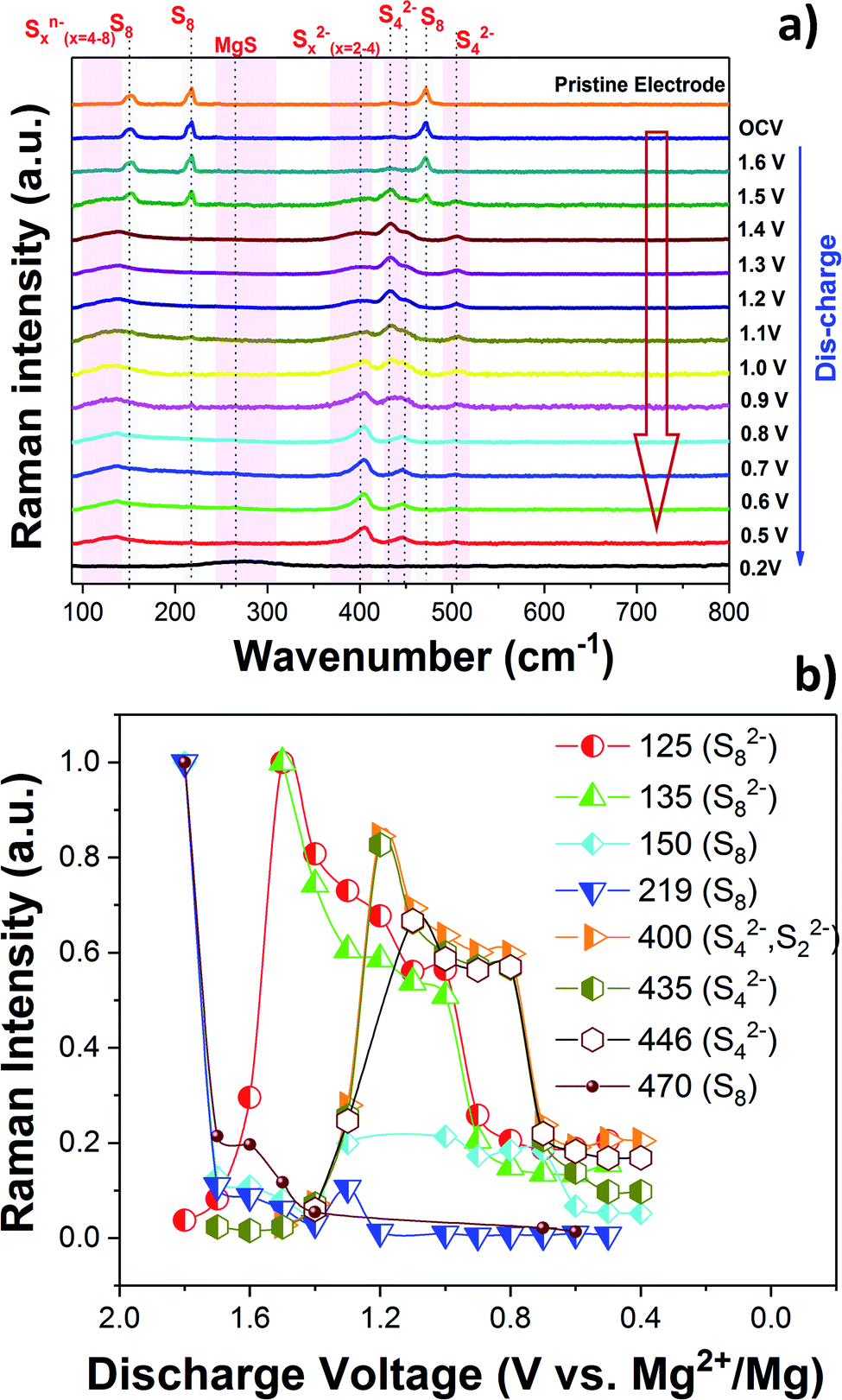 | ||
| Fig. 3 (a) Operando Raman spectra of the S/NC cathode in an Mg/S cell during the discharge. (b) Formation of various polysulfide species during the discharge. | ||
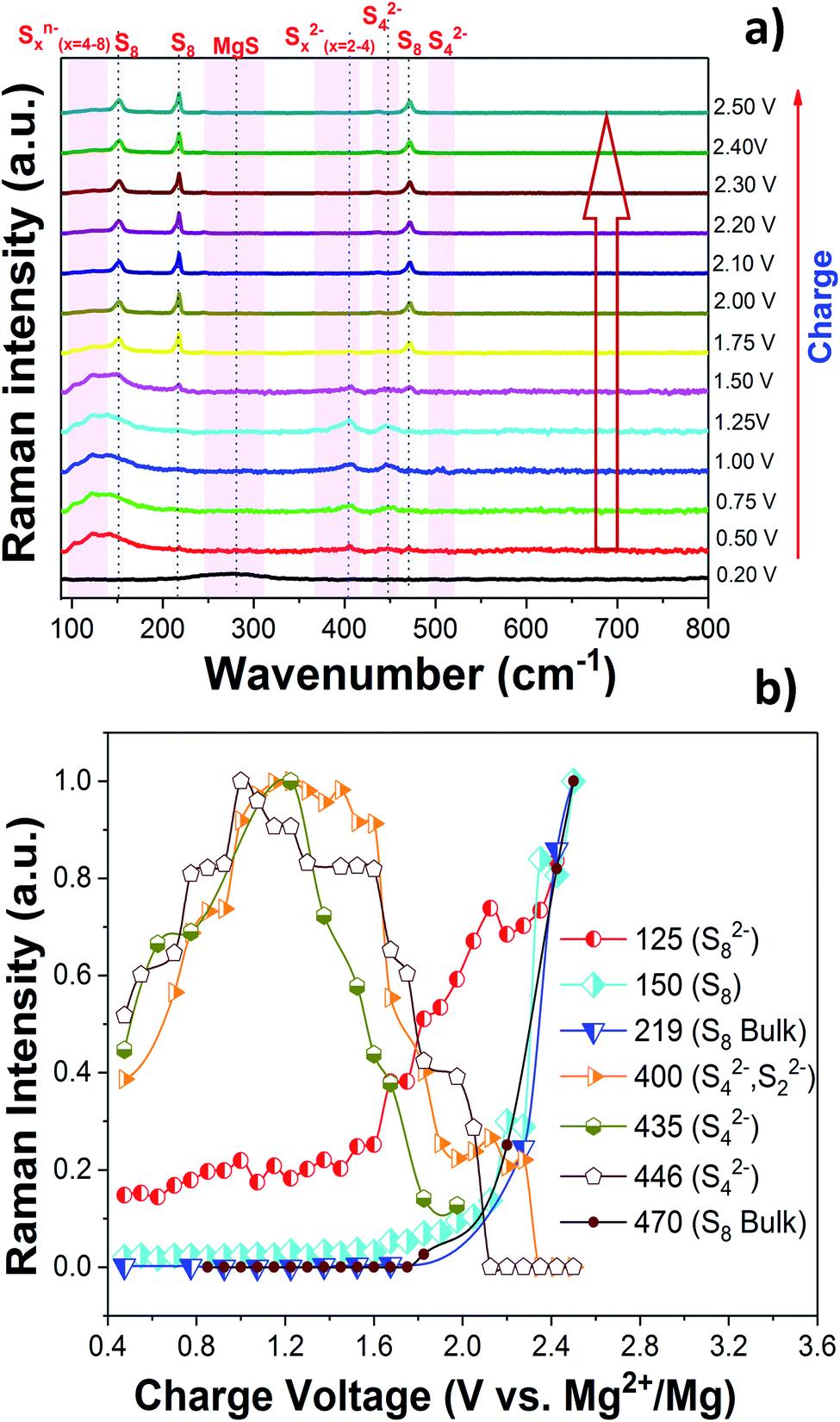 | ||
| Fig. 4 (a) Operando Raman spectra of the S/NC cathode in an Mg/S cell during the charge. (b) Formation of various polysulfide species during the charge. | ||
Before starting the operando Raman experiment, Raman spectra and a Raman map of the pristine S/NC cathode were obtained as shown in Fig. 5(a) and (b). The spectrum of pristine S/NC (Fig. 5(a)) shows peaks at 153 cm−1, 219 cm−1, and 473 cm−1, which correspond to the signals of elemental sulfur with an orthorhombic crystal structure, as also supported by XRD (Fig. S6†). The Raman mapping of sulfur in the S/NC cathode (Fig. 5(b)) was carried out using the intensity of the band at ![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) = 219 cm−1, which again indicates the high sulfur loading on the cathode. Since the pure electrolyte does not show any Raman peaks (see Fig. S7†) in the frequency range of 100–600 cm−1, its contribution can be neglected in the analysis of the sulfur and polysulfide spectra. With the help of density functional theory (DFT) calculations, the observed peaks in the measured spectra were assigned to different polysulfide species.
= 219 cm−1, which again indicates the high sulfur loading on the cathode. Since the pure electrolyte does not show any Raman peaks (see Fig. S7†) in the frequency range of 100–600 cm−1, its contribution can be neglected in the analysis of the sulfur and polysulfide spectra. With the help of density functional theory (DFT) calculations, the observed peaks in the measured spectra were assigned to different polysulfide species.
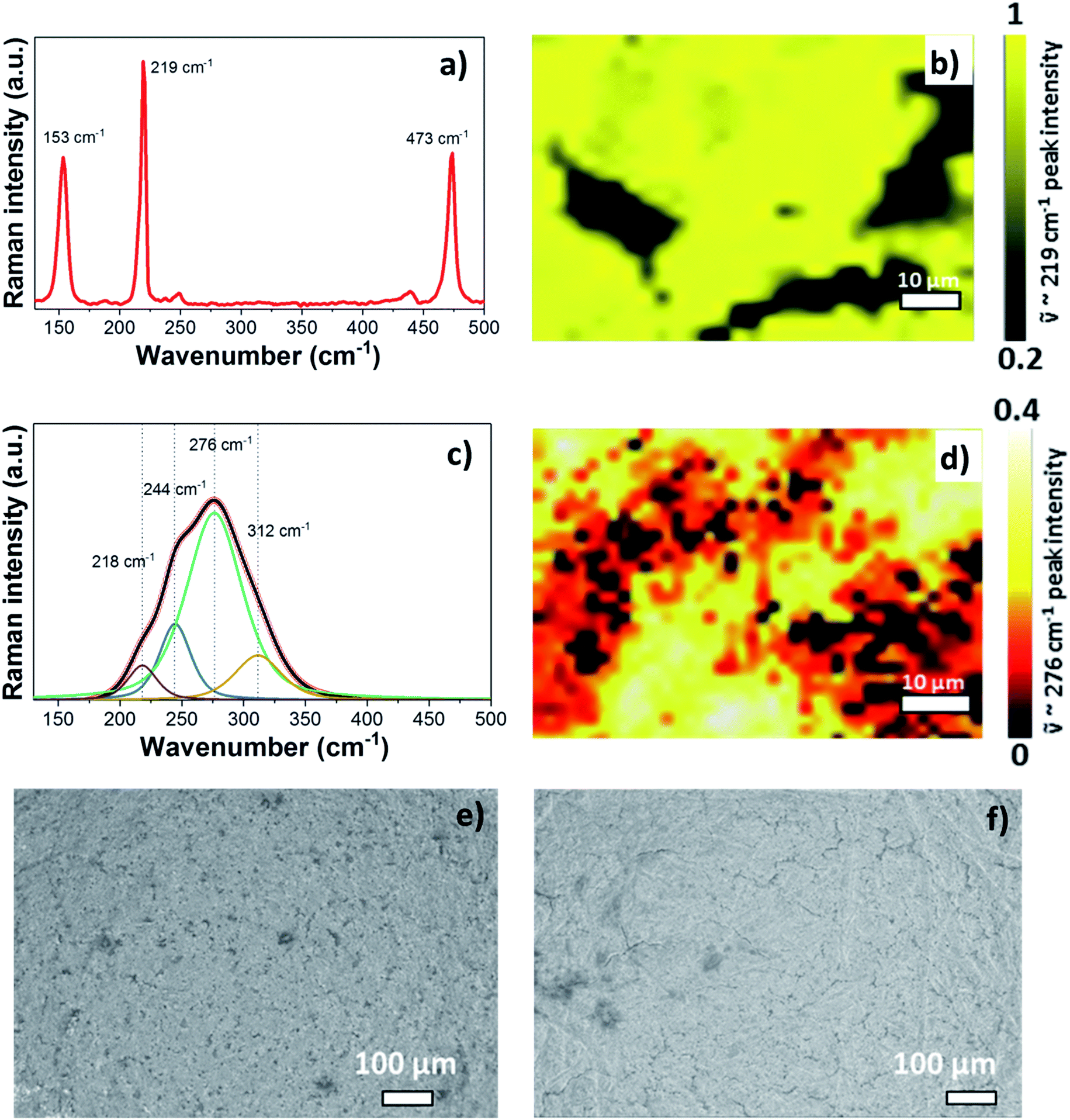 | ||
| Fig. 5 Raman spectra and Raman mapping images of the (a and b) pristine cathode electrode, and (c and d) discharged cathode electrode at 0.2 V. SEM images of the cathode electrode (e) before and (f) after the cycling of the Mg/S cell. | ||
Consequently, DFT calculations were conducted for a series of polysulfide di-anions Sn2− and radical mono-anions Sn− (n = 2–8), applying the Gaussian 09 code,33 with the B3LYP functional and a 6-311G(2df,p) basis set. In addition, solvation effects were taken into account using the polarizable continuum model (PCM), mimicking the impact of the electrolyte. For both scenarios, with and without solvation effects, the molecular geometries for polysulfides of different lengths were optimized and Raman spectra were obtained. Fig. S8 and S9† show the calculated Raman spectra for different polysulfide di-anions and mono-anions, elemental sulfur and bulk MgS (zinc blende). In this study, we first optimized the structures in a vacuum to then use the resulting equilibrium configuration as a starting point for calculations including solvation effects within the PCM. This approach models the solvent as a polarizable continuum, which surrounds a cavity with the shape of the respective solvated molecule. To simulate conditions close to the experimental setup, an adapted electrolyte with a dielectric constant of ε = 7 was used in the PCM. Here it should be noted that the main impact of solvation in our study is to shift the Raman peaks to slightly higher frequencies, while the overall appearance of the spectra is not strongly altered. Polysulfides typically form unbranched chains or ring-like molecules, including a number of conformers for a given chain length. For our Raman study, we only investigated the conformer that was found to have the lowest energy for the respective chain length. In addition, periodic DFT calculations, using the VASP code and the projector augmented wave (PAW) method,34,35 were applied to determine the vibrational spectra of bulk sulfur and bulk MgS. The periodic calculations were conducted with the exchange–correlation energy being described by the generalized gradient approximation in the formulation of Perdew, Burke and Ernzerhof.36 On the sulfur side, the rhombic modification is the energetically most stable one (at 0 K), while the monoclinic phase is energetically slightly unfavorable. Due to the presence of S8 like rings in both phases the vibrational spectra are, however, very similar.
Fig. 3(b) and 4(b) summarize the progress of various polysulfide species formation during discharge and charge experiments. The Raman intensities of the bands corresponding to bulk sulfur and possibly S8 rings (at 150, 219 and 470 cm−1) decreased as the discharge proceeded from the OCV and almost disappeared at ∼1.6 V. This indicates that bulk sulfur is initially reduced, leading to the opening of S8 rings and the formation of long-chain polysulfides. S8n− (n = 1, 2) species may be formed while S8 rings are still present, explaining the coexistence of bands at ∼125 and 135 cm−1, which can be assigned to S8n− chains, with the bands of S8 rings at 150, 219, and 470 cm−1 during the initial stage of discharge (voltages > 1.4 V). The bands associated with the S8n− chains are more visible in the self-discharge study and will therefore be discussed in the corresponding section. The Raman spectra obtained during the initial stage of discharge indicate that the first electrochemical reaction is
| S8 + ne− → S8n− (n = 1, 2) |
Earlier Li/S cell studies show that the sulfur reduction and the formation of polysulfides occur in higher discharge voltage ranges in ether based electrolyte solvents.27,37 Solvent properties such as the dielectric constant and the donor number can, however, have a major influence on the redox reactions of sulfur.38 In low dielectric constant solvents like DME (ε ∼ 7), the amount of polysulfide species formed through chain-growth and disproportionation reactions is expected to be significant because of the low stabilization of these species in such types of solvents.39 High dielectric constant solvents on the other hand can lead to the formation of metastable species like S3˙− radicals.40 It is interesting to note that radicals like S3˙− were not identified in the present Raman experiment.
As the discharge continued, Raman bands at 200, 400, 435 and 446 cm−1 started to appear and their intensities increased below ∼1.4 V. According to our DFT calculations, these bands can be associated with the vibrations of polysulfide species in different configurations as shown in Fig. S8 and S9.† The band at 201 cm−1 was previously assigned to the bending mode of S42−,41,42 and the one at ∼446 cm−1 was identified as the stretching mode of S42−, which agrees with our computational findings. The intensity of the S42− species reaches its maximum (Fig. 3(b)) in the voltage range of 1.2–1.0 V, after which it starts to decrease possibly due to the reduction of S42− to S22−. Short-chain polysulfides like S42− to S22− were observed together at low discharge voltage (1–0.5 V). The formation of short chain polysulfides on the discharged cathode at 0.5 V was further confirmed by XPS. Fig. S10† compares the S 2p XP spectra for the pristine sulfur cathode and the discharged cathode at ∼0.5 V. The S 2p spectrum of the pristine cathode is dominated by a peak doublet at 164.0/165.2 eV, which can be assigned to bulk sulfur. In addition to the main sulfur peak, additional low intensity peaks at 169.1 eV and a doublet peak at 162.3/163.3 eV were observed, indicating the formation of small amounts of sulfate and reduced sulfur species at the electrode.43 The S 2p XPS spectrum recorded after discharge gave mainly two peak doublets at 161.8/163.0 eV and 160.4/161.6 eV, corresponding to S42−/S22− and S2−, respectively.18
In order to reach complete conversion of the short polysulfides to magnesium sulfide (MgS), a deep discharge of the Mg/S operando Raman cell to 0.2 V and additional keeping of the cell at this low potential for a longer time was necessary. Then, the disappearance of all the peaks attributed to the polysulfide species and the formation of broad peaks in the spectral range of 180–350 cm−1 were observed. Fig. 5(c) shows the deconvolution of the broad band of the MgS Raman spectrum of the discharged cathode at 0.2 V. The intensity based Raman mapping of the band centered at 276 cm−1 is illustrated in Fig. 5(d), which proves the transformation of polysulfide species to MgS in the cathode. To confirm the phase of electrochemically formed MgS, we also synthesized MgS by a mechano-chemical procedure. XRD (Fig. S6d and e†) confirms the cubic rock salt structure (space group: Fm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m) of mechano-chemically synthesized MgS. However, the Raman spectra do not show any bands for the cubic rock salt MgS phase, as one would expect from the symmetry of the crystal structure (Fig. S11a†). The rock salt modification is known to be the equilibrium phase of MgS, which we could also confirm by our DFT calculations. Yet, the XRD of the discharged cathode (see Fig. S6f†) shows an amorphous background, thus pointing to nano-crystalline MgS as the final discharge product. The Raman spectra of the discharged cathode indeed point to the formation of the zinc blende (sphalerite) phase (space group: F
m) of mechano-chemically synthesized MgS. However, the Raman spectra do not show any bands for the cubic rock salt MgS phase, as one would expect from the symmetry of the crystal structure (Fig. S11a†). The rock salt modification is known to be the equilibrium phase of MgS, which we could also confirm by our DFT calculations. Yet, the XRD of the discharged cathode (see Fig. S6f†) shows an amorphous background, thus pointing to nano-crystalline MgS as the final discharge product. The Raman spectra of the discharged cathode indeed point to the formation of the zinc blende (sphalerite) phase (space group: F![[4 with combining macron]](https://www.rsc.org/images/entities/char_0034_0304.gif) 3m). They show a broad peak in the range of ∼180–350 cm−1, which nearly matches the calculated Raman band of the zinc blende phase at about 320 cm−1 as shown in Fig. S8 and S9,† while the rock salt phase is not Raman active at all (see Fig. S10a†). The small shift in the frequency of the calculated and experimental spectra of the sphalerite MgS phase may be due to electrolyte effects and the nanocrystalline nature of the discharged cathode. In a recent report, Nakayama et al. also observed the formation of a metastable zinc blende MgS phase during the discharge of a sulfur cathode in a Mg/S battery with sulfone-based magnesium electrolyte.23 This, together with our experimental and computational findings, makes us conclude that the final discharge product is indeed MgS in the zinc blende structure type. For further comparison, we determined the theoretical average discharge voltage for the conversion of hcp-Mg and rhombic sulfur into the zinc blende MgS from our DFT calculations. It amounts to ∼1.42 V, which is in good agreement with the experimentally determined average voltage (∼1.4 V) from the discharge plateau of the Mg/S cell. The formation of polysulfide species observed from the operando Raman spectra during the charging process (Fig. 4) corresponds to the reverse process of the one observed during discharge. The bands of the short chain polysulfide species (like S42− and S22−) have almost disappeared at around 1.7 V and after that the S8n− polysulfide species and S8 start to form. The intensity of the S8 bands reaches its maximum at ∼2.2 V, which confirms the complete re-conversion of the lower order polysulfide chains to S8 rings. According to the above spectroscopic experiments, the main electrochemical reactions in the whole redox process can be summarized as follows: (i) reduction of elemental bulk (S8) sulfur to long-chain polysulfides (S8n−), (ii) reduction of long-chain polysulfides (S8n−) to short-chain polysulfides (e.g. S42−/S22−), and (iii) solid-state transformation of short-chain polysulfides to MgS. Fig. 5(e) and (f) show the SEM images of the pristine cathode and the cathode after 50 cycles. These images prove that the S/NC sulfur cathode is structurally stable with no major crack formation during repeated magnesiation/de-magnesiation. To further understand the adsorption capability of Mg polysulfides towards polar and non-polar carbon host matrices, pristine carbon and nitrogen doped carbons (NCs) were kept in a polysulfide solution for 2 days, which resulted in a color change of the solution from dark red to light yellow (see Fig. S12†). The color change of the solution from red to yellow was clearly more significant for the NC carbon matrix as compared to the pristine carbon matrix, which indicates the stronger adsorption of sulfur species on the NC carbon matrix as compared to the pristine one. The electrochemical process at the cathode of Mg/sulfur batteries can be described through a solid–liquid–solid transformation.21 In that case, the interfacial electrochemical kinetics is dominated by two main factors. First, the binding affinity of the initial S8 to the host matrix surface and the adsorption of Sxn− (x = 1–8) on the latter are of importance to provide sufficient surface coverage. Second, efficient charge transfer at the cathode/electrolyte boundary requires the fast transport of electrons through the host matrix. A non-polar conductive host matrix like pristine carbon is too inert to bind polar sulfur species, resulting in low adsorption of Sxn− (x = 1–8) on the support. A polar conductive host matrix like the nitrogen doped G-MWCNT hybrid structure on the other hand can improve the adsorption of polar polysulfides and, moreover, support an efficient charge transport.
3m). They show a broad peak in the range of ∼180–350 cm−1, which nearly matches the calculated Raman band of the zinc blende phase at about 320 cm−1 as shown in Fig. S8 and S9,† while the rock salt phase is not Raman active at all (see Fig. S10a†). The small shift in the frequency of the calculated and experimental spectra of the sphalerite MgS phase may be due to electrolyte effects and the nanocrystalline nature of the discharged cathode. In a recent report, Nakayama et al. also observed the formation of a metastable zinc blende MgS phase during the discharge of a sulfur cathode in a Mg/S battery with sulfone-based magnesium electrolyte.23 This, together with our experimental and computational findings, makes us conclude that the final discharge product is indeed MgS in the zinc blende structure type. For further comparison, we determined the theoretical average discharge voltage for the conversion of hcp-Mg and rhombic sulfur into the zinc blende MgS from our DFT calculations. It amounts to ∼1.42 V, which is in good agreement with the experimentally determined average voltage (∼1.4 V) from the discharge plateau of the Mg/S cell. The formation of polysulfide species observed from the operando Raman spectra during the charging process (Fig. 4) corresponds to the reverse process of the one observed during discharge. The bands of the short chain polysulfide species (like S42− and S22−) have almost disappeared at around 1.7 V and after that the S8n− polysulfide species and S8 start to form. The intensity of the S8 bands reaches its maximum at ∼2.2 V, which confirms the complete re-conversion of the lower order polysulfide chains to S8 rings. According to the above spectroscopic experiments, the main electrochemical reactions in the whole redox process can be summarized as follows: (i) reduction of elemental bulk (S8) sulfur to long-chain polysulfides (S8n−), (ii) reduction of long-chain polysulfides (S8n−) to short-chain polysulfides (e.g. S42−/S22−), and (iii) solid-state transformation of short-chain polysulfides to MgS. Fig. 5(e) and (f) show the SEM images of the pristine cathode and the cathode after 50 cycles. These images prove that the S/NC sulfur cathode is structurally stable with no major crack formation during repeated magnesiation/de-magnesiation. To further understand the adsorption capability of Mg polysulfides towards polar and non-polar carbon host matrices, pristine carbon and nitrogen doped carbons (NCs) were kept in a polysulfide solution for 2 days, which resulted in a color change of the solution from dark red to light yellow (see Fig. S12†). The color change of the solution from red to yellow was clearly more significant for the NC carbon matrix as compared to the pristine carbon matrix, which indicates the stronger adsorption of sulfur species on the NC carbon matrix as compared to the pristine one. The electrochemical process at the cathode of Mg/sulfur batteries can be described through a solid–liquid–solid transformation.21 In that case, the interfacial electrochemical kinetics is dominated by two main factors. First, the binding affinity of the initial S8 to the host matrix surface and the adsorption of Sxn− (x = 1–8) on the latter are of importance to provide sufficient surface coverage. Second, efficient charge transfer at the cathode/electrolyte boundary requires the fast transport of electrons through the host matrix. A non-polar conductive host matrix like pristine carbon is too inert to bind polar sulfur species, resulting in low adsorption of Sxn− (x = 1–8) on the support. A polar conductive host matrix like the nitrogen doped G-MWCNT hybrid structure on the other hand can improve the adsorption of polar polysulfides and, moreover, support an efficient charge transport.
3.4. Electrochemical processes in the Mg/S cell under static conditions
The changes in the open circuit potential (OCV) of the Mg/S cell have been investigated by monitoring separately the cathode and anode potentials over a resting period of 30 h as shown in Fig. 6(a) and (b). The electrochemical measurements were carried out using a three-electrode cell with an Mg ring as the reference electrode (MgRE), Mg metal as the counter electrode (MgCE), and S/NC (3 mgsulfur cm−2) as the working electrode (WE). During the resting time at OCV, the cathode potential decreased from 1.73 V to 1.66 V after 30 h (Fig. 6(a)).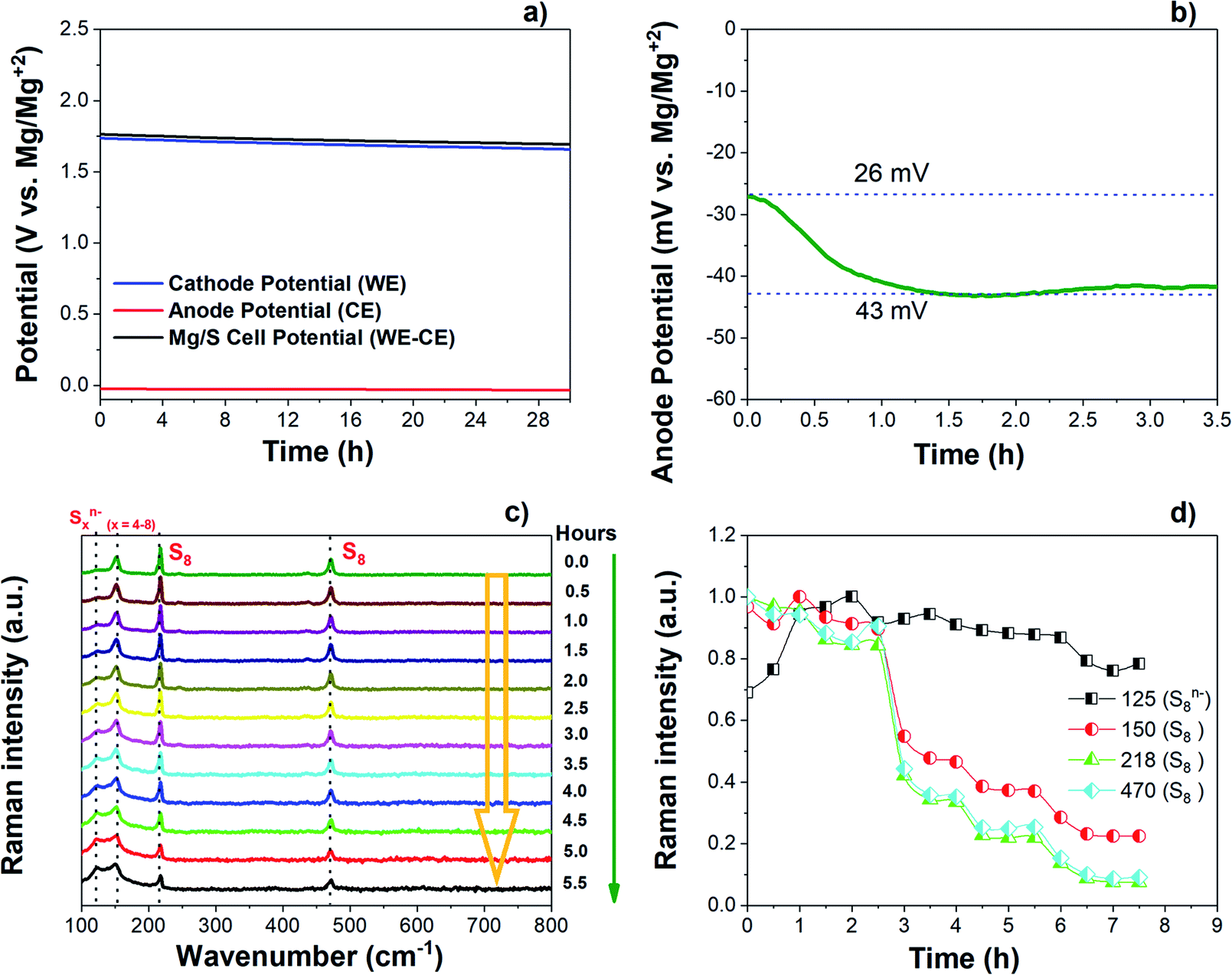 | ||
| Fig. 6 (a) Voltage profiles of the Mg/S cell kept at open circuit potential (OCV) for 30 h. (b) Increase of the anode potential (vs. MgRE) under OCV for the first 3.5 h. (c) Operando Raman spectra of the S/NC cathode in Mg/S cell over a resting period under OCV. (d) Time evolution of the intensity of Raman peaks associated with elemental sulfur and polysulfides. | ||
On the anode side, initially the potential was 26 mV with respect to MgREF. It then further increased to 43 mV after a resting period of 2 hours and finally stabilized at this potential as depicted in Fig. 6(b). In accordance with the initial increase of the anode overpotential, the anode impedance also increased during the first 2 hours from 759 Ω cm−2 to 1292 Ω cm−2, and stabilized after that (Fig. S13(a) and (b)†). This could be associated with the time required for the formation of a passivation layer on the Mg anode surface.44 During the resting period, some electrochemically inactive species may adsorb on the Mg metal surface, thus causing the high interfacial impedance and weakened ion transport properties at the interface.45–48 Meanwhile, the cathode impedance increased from the initial 190 Ω cm−2 to 265 Ω cm−2 over a resting period of 8 h as shown in Fig. S13(c and d).† The impedance study proves that the total Mg/S cell impedance is dominated by the anode side impedance, which could be due to the insulating passivation layer formation at the anode/electrolyte interface. To understand the reason for the decrease of the cathode potential during the resting time under OCV, the Raman spectra of the sulfur cathode (∼3 mgsulfur cm−2) were collected during the resting period of an Mg/S cell. Fig. 6(c) and (d) illustrate the corresponding spectra and the time evolution of the Raman intensities during the resting period. The operando Raman spectra show a decrease in the intensity of the bulk sulfur peaks and an increase in the intensity of the peaks related to long-chain polysulfides. This indicates that extended contact of the sulfur cathode with the electrolyte during the resting period could promote the reduction of bulk sulfur leading to the formation of higher order polysulfide species, which then results in a reduction of the cathode potential.49
3.5. Impedance study under dynamic conditions
To determine the evolution of the cell impedance under dynamic conditions, a three-electrode cell was used. During this study, the cell was kept for an equilibration time of 30 minutes at each potential. From the impedance study under static conditions, we have already observed that the total cell impedance is dominated by the anode side impedance. Hence in this study, we mainly monitored the variation of Mg anode side impedance with respect to the cathode potential. Fig. 7(a)–(d) show the Nyquist plots obtained for the Mg anode side impedances and total cell impedances with respect to the cathode discharge potentials from 2.1 V to 0.2 V. To get a better understanding of the processes occurring during discharge/charge cycling, the Nyquist plots were analyzed using a circuit model as shown in Fig. S15.†14,50 In the equivalent circuit of the Mg anode (see Fig. S15†), R1 includes the electrolyte and current collector resistance, and R2//Q2 and R3//Q3 represent the resistances and constant phase elements (non-ideal capacitance with regard to the suppressed semicircles) of the layers formed at the interface of the Mg anode. In the Nyquist plots, the depressed semicircle in the high frequency (500 kHz to 100 Hz) region (Fig. 7(a) and (c)) can contribute to a non-blocking interface layer with comparatively less resistance, as represented by R2//Q2 in the fitting circuit. As we go to the low frequency region (500 kHz to 0.7 Hz), the Nyquist plots (Fig. 7(b) and (d)) become a depressed semicircular capacitive loop with high charge transfer resistance, corresponding to R3//Q3 in the fitting circuit.45 The circuit fitted values of R1, R2, Q2, R3 and Q3 are summarized in Table S1 (ESI†). As the cathode potential decreased from 2.1 to 0.2 V (discharge of the Mg/S cell), the anode side impedance also decreased. From Fig. 7, it is very clear that the charge transfer resistances of the depressed semicircles in the high frequency and low frequency regions decrease when applying an oxidation potential to the Mg anode. On the other hand, when a reduction potential was applied to the Mg anode (charging of the Mg/S cell), an increase of the charge transfer resistances of the depressed semicircles was observed (Fig. S14†). This decrease/increase of the Mg anode interfacial impedances indicates that the chemical species contained in the Mg anode passivation layer (especially polysulfide species) could become electrochemically active or inactive according to the application of oxidation/reduction electrode potentials.5,45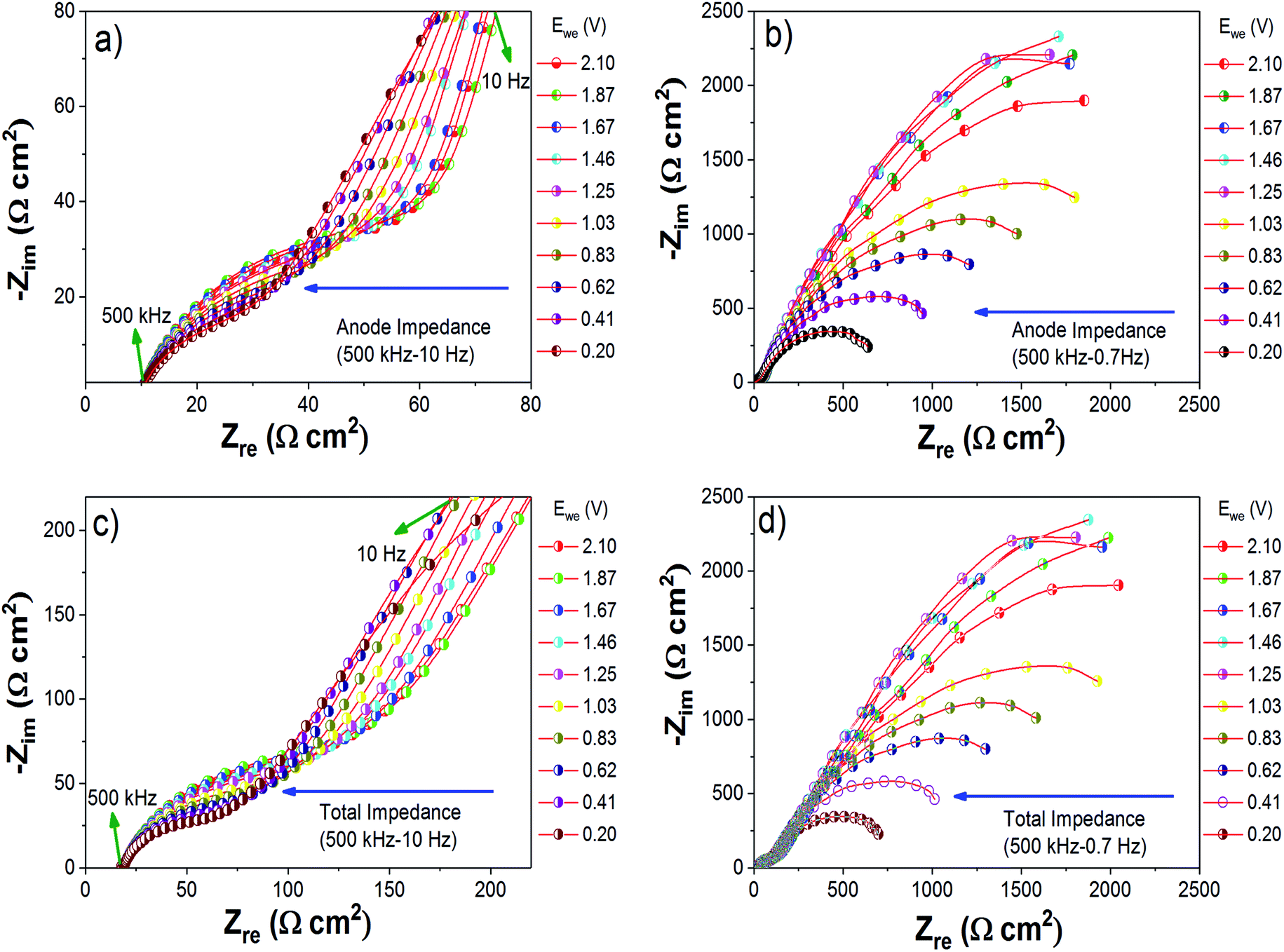 | ||
| Fig. 7 Changes in the impedance spectra of the Mg anode of a Mg/S cell under different cathode potentials over the frequency range of (a) 500 kHz to 10 Hz, and (b) 500 kHz to 0.7 Hz. Variations of the total Mg/S cell impedance under different cathode potentials over the frequency range of (c) 500 kHz to 10 Hz, and (d) 500 kHz to 0.7 Hz. The spectra were collected during the discharge of the Mg/S cell. | ||
4. Conclusion
In summary, we have developed a new cathode for Mg/S batteries using nitrogen doped graphene and MWCNT hybrid nanostructures and investigated its electrochemical performances with low and high sulfur loadings (0.5–3 mgsulfur cm−2). Operando Raman spectroscopy of the Mg/S cell showed the formation of MgS at the end of discharge from bulk sulfur (S8) through a series of long and short chain polysulfides such as S8n−/S4n−/S2n−/MgS. As the final discharge product, a nano-crystalline phase, most likely corresponding to the zinc blende phase of MgS was obtained and compared to the mechano-chemically synthesized rock salt phase of MgS. During charging, MgS converted back to S8, showing the high reversibility of the process. Separate impedance studies on the Mg anode and sulfur cathode sides under static conditions prove that the total cell impedance is mainly dominated by the Mg anode impedance due to the formation of a passivation layer. During the cycling of the Mg/S cell, the amount of polysulfide species lost to the anode side via dissolution significantly contributes to the increase of the anode overpotential and resistances. Impedance studies under dynamic conditions prove that the oxidation/reduction potentials applied to the electrodes have a major influence on the evolution of the anode impedance. This could be due to the oxidation/reduction of chemical species present at the anode interphase and further interphase studies are needed in order to fully understand the process. Effective ways to improve the Mg/S cell performance include (i) the design of a sulfur cathode with strong adsorption of polysulfides, and (ii) the protection of the Mg anode from detrimental species by introducing an electrochemically stable and Mg ion conductive interphase between the anode and electrolyte.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge funding from Bundesministerium für Bildung und Forschung (BMBF) of Germany via the “MagSiMal” project (03XP0208). This project has received partial funding from the European Union's Horizon 2020 research and innovation programme under grant agreement No. 824066. This work contributes to the research performed at CELEST (Center for Electrochemical Energy Storage Ulm-Karlsruhe) and was funded by the German Research Foundation (DFG) under Project ID 390874152 (POLiS Cluster of Excellence).References
- P. Saha, M. K. Datta, O. I. Velikokhatnyi, A. Manivannan, D. Alman and P. N. Kumta, Prog. Mater. Sci., 2014, 66, 1–86 CrossRef CAS.
- R. Mohtadi and F. Mizuno, Beilstein J. Nanotechnol., 2014, 5, 1291–1311 CrossRef PubMed.
- H. D. Yoo, I. Shterenberg, Y. Gofer, G. Gershinsky, N. Pour and D. Aurbach, Energy Environ. Sci., 2013, 6, 2265–2279 RSC.
- H. S. Kim, T. S. Arthur, G. D. Allred, J. Zajicek, J. G. Newman, A. E. Rodnyansky, A. G. Oliver, W. C. Boggess and J. Muldoon, Nat. Commun., 2011, 2, 427 CrossRef PubMed.
- Z. Zhao-Karger, R. Liu, W. Dai, Z. Li, T. Diemant, B. P. Vinayan, C. Bonatto Minella, X. Yu, A. Manthiram, R. J. Behm, M. Ruben and M. Fichtner, ACS Energy Lett., 2018, 3, 2005–2013 CrossRef CAS.
- P. Bonnick, E. Nagai and J. Muldoon, J. Electrochem. Soc., 2018, 165, A6005–A6007 CrossRef CAS.
- L. Medenbach and P. Adelhelm, Cell Concepts of Metal–Sulfur Batteries (Metal = Li, Na, K, Mg), Strategies for Using Sulfur in Energy Storage Applications, 2017 Search PubMed.
- T. Gao, X. Li, X. Wang, J. Hu, F. Han, X. Fan, L. Suo, A. J. Pearse, S. B. Lee, G. W. Rubloff, K. J. Gaskell, M. Noked and C. Wang, Angew. Chem., Int. Ed., 2016, 55, 9898–9901 CrossRef CAS PubMed.
- N. A. C. N. Wagner, D. Wittmaier and K. A. Friedrich, State of the Art of Batteries of the 4th Generation, 2015 Search PubMed.
- B. P. Vinayan, Z. Zhao-Karger, T. Diemant, V. S. K. Chakravadhanula, N. I. Schwarzburger, M. A. Cambaz, R. J. Behm, C. Kübel and M. Fichtner, Nanoscale, 2016, 8, 3296–3306 RSC.
- Z. Zhao-Karger, J. E. Mueller, X. Zhao, O. Fuhr, T. Jacob and M. Fichtner, RSC Adv., 2014, 4, 26924–26927 RSC.
- Z. Zhao-Karger, M. E. Gil Bardaji, O. Fuhr and M. Fichtner, J. Mater. Chem. A, 2017, 5, 10815–10820 RSC.
- Z. Zhang, Z. Cui, L. Qiao, J. Guan, H. Xu, X. Wang, P. Hu, H. Du, S. Li, X. Zhou, S. Dong, Z. Liu, G. Cui and L. Chen, Adv. Energy Mater., 2017, 7, 1602055 CrossRef.
- A. Du, Z. Zhang, H. Qu, Z. Cui, L. Qiao, L. Wang, J. Chai, T. Lu, S. Dong, T. Dong, H. Xu, X. Zhou and G. Cui, Energy Environ. Sci., 2017, 10, 2616–2625 RSC.
- Y. Zhang, J. Xie, Y. Han and C. Li, Adv. Funct. Mater., 2015, 25, 7300–7308 CrossRef CAS.
- Z. Zhang, S. Dong, Z. Cui, A. Du, G. Li and G. Cui, Small Methods, 2018, 2, 1800020 CrossRef.
- M. Salama, R. Attias, B. Hirsch, R. Yemini, Y. Gofer, M. Noked and D. Aurbach, ACS Appl. Mater. Interfaces, 2018, 10, 36910–36917 CrossRef CAS PubMed.
- X. Zhou, J. Tian, J. Hu and C. Li, Adv. Mater., 2018, 30, 1704166 CrossRef PubMed.
- X. Yu and A. Manthiram, Small Methods, 2017, 1, 1700217 CrossRef.
- A. Robba, A. Vizintin, J. Bitenc, G. Mali, I. Arčon, M. Kavčič, M. Žitnik, K. Bučar, G. Aquilanti, C. Martineau-Corcos, A. Randon-Vitanova and R. Dominko, Chem. Mater., 2017, 29, 9555–9564 CrossRef CAS.
- T. Gao, X. Ji, S. Hou, X. Fan, X. Li, C. Yang, F. Han, F. Wang, J. Jiang, K. Xu and C. Wang, Adv. Mater., 2018, 30, 1704313 CrossRef PubMed.
- G. Bieker, J. Wellmann, M. Kolek, K. Jalkanen, M. Winter and P. Bieker, Phys. Chem. Chem. Phys., 2017, 19, 11152–11162 RSC.
- Y. Nakayama, R. Matsumoto, K. Kumagae, D. Mori, Y. Mizuno, S. Hosoi, K. Kamiguchi, N. Koshitani, Y. Inaba, Y. Kudo, H. Kawasaki, E. C. Miller, J. N. Weker and M. F. Toney, Chem. Mater., 2018, 30, 6318–6324 CrossRef CAS.
- W. Hummers and R. Ofeman, J. Am. Chem. Soc., 1958, 80, 1339 CrossRef CAS.
- B. P. Vinayan and S. Ramaprabhu, Nanoscale, 2013, 5, 5109–5118 RSC.
- B. P. Vinayan, K. Sethupathi and S. Ramaprabhu, J. Nanosci. Nanotechnol., 2012, 12, 6608–6614 CrossRef CAS PubMed.
- B. P. Vinayan, T. Diemant, X.-M. Lin, M. A. Cambaz, U. Golla-Schindler, U. Kaiser, R. Jürgen Behm and M. Fichtner, Adv. Mater. Interfaces, 2016, 3, 1600372 CrossRef.
- B. P. Vinayan and S. Ramaprabhu, J. Mater. Chem. A, 2013, 1, 3865–3871 RSC.
- B. P. Vinayan, R. Nagar and S. Ramaprabhu, Langmuir, 2012, 28, 7826–7833 CrossRef PubMed.
- W. J. Lee, U. N. Maiti, J. M. Lee, J. Lim, T. H. Han and S. O. Kim, Chem. Commun., 2014, 50, 6818–6830 RSC.
- P. Wang and M. R. Buchmeiser, Adv. Funct. Mater., 2019, 0, 1905248 CrossRef.
- H. Kuwata, M. Matsui and N. Imanishi, J. Electrochem. Soc., 2017, 164, A3229–A3236 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- J. Hannauer, J. Scheers, J. Fullenwarth, B. Fraisse, L. Stievano and P. Johansson, ChemPhysChem, 2015, 16, 2755–2759 CrossRef CAS PubMed.
- Y. Gorlin, A. Siebel, M. Piana, T. Huthwelker, H. Jha, G. Monsch, F. Kraus, H. A. Gasteiger and M. Tromp, J. Electrochem. Soc., 2015, 162, A1146–A1155 CrossRef CAS.
- K. Yen Peng, L. Chen and A. F. Yee, Nanotechnology, 2011, 22, 295709–295717 CrossRef PubMed.
- N. A. Cañas, D. N. Fronczek, N. Wagner, A. Latz and K. A. Friedrich, J. Phys. Chem. C, 2014, 118, 12106–12114 CrossRef.
- G. J. Janz, J. R. Downey, E. Roduner, G. J. Wasilczyk, J. W. Coutts and A. Eluard, Inorg. Chem., 1976, 15, 1759–1763 CrossRef CAS.
- O. El Jaroudi, E. Picquenard, A. Demortier, J.-P. Lelieur and J. Corset, Inorg. Chem., 2000, 39, 2593–2603 CrossRef CAS PubMed.
- B. P. Vinayan, T. Diemant, X.-M. Lin, M. A. Cambaz, U. Golla-Schindler, U. Kaiser, R. Jürgen Behm and M. Fichtner, Adv. Mater. Interfaces, 2016, 3, 1600372 CrossRef.
- Y. V. Mikhaylik and J. R. Akridge, J. Electrochem. Soc., 2004, 151, A1969–A1976 CrossRef CAS.
- O. Tutusaus, R. Mohtadi, N. Singh, T. S. Arthur and F. Mizuno, ACS Energy Lett., 2017, 2, 224–229 CrossRef CAS.
- X. Qu, Y. Zhang, N. N. Rajput, A. Jain, E. Maginn and K. A. Persson, J. Phys. Chem. C, 2017, 121, 16126–16136 CrossRef CAS.
- P. Canepa, G. S. Gautam, R. Malik, S. Jayaraman, Z. Rong, K. R. Zavadil, K. Persson and G. Ceder, Chem. Mater., 2015, 27, 3317–3325 CrossRef CAS.
- R. E. Doe, R. Han, J. Hwang, A. J. Gmitter, I. Shterenberg, H. D. Yoo, N. Pour and D. Aurbach, Chem. Commun., 2014, 50, 243–245 RSC.
- M. Marinescu, T. Zhang and G. J. Offer, Phys. Chem. Chem. Phys., 2016, 18, 584–593 RSC.
- J. Genescá, L. Betancourt and C. Rodríguez, Corrosion, 1996, 52, 502–507 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ta09155f |
| This journal is © The Royal Society of Chemistry 2019 |