
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMolten ionic oxides for CO2 capture at medium to high temperatures†
Takuya
Harada
 a,
Cameron
Halliday
a,
Aqil
Jamal
b and
T. Alan
Hatton
*a
a,
Cameron
Halliday
a,
Aqil
Jamal
b and
T. Alan
Hatton
*a
aDepartment of Chemical Engineering, Massachusetts Institute of Technology, 77 Massachusetts Avenue, Cambridge, MA 02139, USA. E-mail: tahatton@mit.edu
bResearch & Development Centre, Saudi Aramco, Dhahran 31311, Kingdom of Saudi Arabia
First published on 5th September 2019
Abstract
The development of efficient low cost CO2 capture systems is a critical challenge for mitigating climate change while meeting global energy demand. Herein, we demonstrate the first liquid absorbents for CO2 capture at medium to high temperatures (500 to 700 °C). Molten ionic oxides based on sodium borate and the mixed alkali-metal borates show remarkably fast kinetics and intrinsic regenerability, with no observable deterioration in performance over multiple absorption–desorption cycles under both temperature- and pressure-swing operations. The behavior of the molten ionic oxides is ascribed to the instantaneous formation of carbonate ions in the molten oxides without the diffusional transport restrictions imposed by solid product layers characteristic of solid adsorbents. The new liquid absorbents will enable continuous processing and thermal integration via a simple absorber–desorber arrangement, thereby overcoming the challenges previously restraining high temperature CO2 capture and opening up new opportunities in clean energy production.
Introduction
Mitigation of the substantial risks associated with changes in global climate patterns due to the accumulation of anthropogenic CO2 in the atmosphere requires the development of energy efficient carbon capture, utilization and storage (CCUS) technologies.1,2 Globally deployed CCUS technologies would enable the continued use of existing energy infrastructure in a carbon constrained world, stabilizing the energy landscape for the foreseeable future. The cost and operability of CO2 capture systems are key to determining the future pathway of global technology pervasion.3,4 Innovative breakthroughs in sorbent materials and/or processes are expected to accelerate future developments in, and implementation of, advanced low-cost CO2 capture methods.The sorbents required for CO2 capture can be classified into several groups in accordance with their operating temperature.5 For low temperature operations, various types of aqueous amines (e.g., MEA, DEA, etc.) have been considered as promising absorbents,6,7 and utilized in most of the current or planned CO2 capture systems at industrial facilities.8,9 Amine scrubbing processes, however, face several technological drawbacks due to their high energy demands, degradation of the amines, and equipment corrosion.3,10,11 As an alternative, various new methods operating at medium to high temperatures (T > 200 °C) have recently attracted attention for both pre-combustion and post-combustion processes. In pre-combustion systems, CO2 must be separated from a mixture of H2 and CO2 produced by coal gasification or steam methane reforming with water gas shift reactions, and the removal of CO2 at the temperature of the reforming or shift reactions in the range of 200 to 600 °C leads to lower energy costs for CO2 capture and an enhancement in H2 production yield.12–14 In post-combustion systems, CO2 capture from the exhaust at high temperature, in particular for next generation NGCC power plants, has advantages in both the uptake efficiency and energy economics when compared to the conventional low temperature systems.15,16 Various types of solid adsorbents, such as calcium oxide (CaO),17–19 magnesium oxide (MgO),20–22 lithium-based mixed oxides (Li2ZrO3,23–25 Li4SiO4,26–28 LiBO2,29 Li3BO3,30etc.), and layered double hydroxides (LDHs),31,32 have been proposed and studied extensively for CO2 capture at medium to high temperatures. These solid adsorbents show superior uptake performance with high capacities, thermal stability, and lower environmental impact.5,33 The CO2 capture processes utilizing solid adsorbents, however, face drawbacks with slow-uptake kinetics, low cyclic durability and complex system design due to the intrinsic physical behavior of the adsorbents as solid particles.34–36 It is recognized that the development of new advanced sorbents operating via energy efficient processes is necessary for the success of CCUS.
Herein, we identify molten ionic oxides that exist in the liquid phase at temperatures above their melting points as a new class of liquid absorbents operating at medium to high temperatures. Among the many molten ionic oxides available,37–39 we have focused on sodium and mixed alkali-metal borates. These new liquid absorbents show rapid CO2 uptake kinetics over the temperature range 500 to 700 °C with excellent cyclic regenerability. The exceptional CO2 uptake performance by the molten alkali-metal borates is discussed in detail below, as are the advantages of the potential exploitation of these molten salt absorbents in systems for CO2 capture relative to those of applications using aqueous amines and solid oxide adsorbents.
Experimental
1. Material
Lithium hydroxide (LiOH, 98%), sodium hydroxide (NaOH, 97%), potassium hydroxide (KOH, 99.9%), and boric acid (H3BO3, 99.5%) were purchased from Sigma-Aldrich. All chemicals were used as received without further purification. All water utilized in the experiments was Milli-Q (Millipore) deionised water.2. Sample preparation
A series of alkali-metal borate samples (AxB1−xOy, A: Alkali-metal), were prepared by calcination of mixed precipitants of alkali-metal hydroxide and boric acid prepared in a molar ratio, x, of 0.75. In a typical procedure for the preparation of NaxB1−xOy (x = 0.75, note that electro-neutrality dictates that y = 1.5 − x), 0.075 mol of sodium hydroxide and 0.025 mol of boric acid were dissolved in 30 ml of distilled water under vigorous stirring. The aqueous solution was dried at 120 °C for several hours to precipitate out the mixed hydroxides. The dried precipitant was ground with an agate mortar and calcined in the oven at 400 °C in air for 4 h. The obtained composite was reground into powders and calcined at 650 °C under a flow of 100% N2 for 1 h to obtain the final product.3. Sample characterization and analysis
The CO2 uptake performance by alkali-metal borates under isothermal conditions was determined by the weight variation of the borate samples under 60 ml min−1 of 100% dry CO2 (Airgas) at atmospheric pressure measured by a thermogravimetric analyzer (TGA, TGA Q50; TA Instrument). Here, samples after pre-calcination at 400 °C followed by regrinding were placed on a Pt sample pan in the TGA chamber, and underwent calcination for 1 h at a final temperature of 650 °C under a flow of 100% N2. After calcination at 650 °C, the temperature in the sample chamber was adjusted to the set temperature for the uptake analysis. Measurements of CO2 uptake were initiated by switching the gas introduced to the sample chamber from 100% N2 to 100% dry CO2. The uptake of CO2 was calculated from the change in sample weight per 1 g of the sample before the introduction of CO2 gas to the sample chamber by the molecular weight of CO2 (MCO2 = 44.0). The desorption performance at different temperatures was tracked through the decrease in weight under a flow of 100% N2 after the uptake of CO2 at 600 °C under a flow of 100% CO2 for 10 min. CO2 uptake with increasing temperature was examined at a temperature ramp rate of 5 °C min−1 from 200 °C to 800 °C under the same sample and gas flow conditions as used in the above isothermal uptake analyses. Cyclic regenerability of CO2 uptake and desorption by alkali-metal borates in a temperature-swing process was evaluated by repeated switching of the temperature in the TGA sample chamber coupled with changes in the gas flow between 100% CO2 and 100% N2. Pre-treatment of the samples was performed under the same procedure as in the isothermal CO2 uptake analysis described above. Cyclic regenerability analysis by pressure-swing was performed by switching the gas introduced to the sample chamber between 100% CO2 and 100% N2 at a constant temperature (700 °C). Pre-treatment of the samples was performed as for the isothermal CO2 uptake analysis described above. The melting points of the alkali-metal borates were identified by endothermic peaks in the heat flow with increasing temperature under a flow of 100% N2 measured by differential scanning calorimetry (DSC, SDT-Q600; TA Instrument). The phase composition and crystallographic features of the samples before and after reaction with CO2 at the reaction temperature were examined by temperature controlled in situ powder X-ray diffractometry (XRD: PANalytical X'Pert Pro Multipurpose Diffractometer with Cu-Kα X-ray (λ = 1.541 Å)), in which the samples were placed on a Pt-sheet substrate. The peaks in the XRD spectra were identified by referring to the ICDD PDF-4+2016RDB database. Fluidity of the molten alkali-metal borates at high temperature was evaluated by viscosity measurements on a rheometer equipped with an Environmental Testing Chamber (AR 2000-ETC; TA Instrument). The variation in viscosity as a function of shear rate was measured at a constant temperature (600 °C).Results and discussion
CO2 uptake by molten alkali-metal borates was examined first with sodium borate, NaxB1−xOy (x = 0.75). Fig. 1(a) shows the uptake of CO2 (Q [mmol g−1]) as the temperature increased at a rate of 5 °C min−1 under a flow of 100% CO2. It is clear that the absorption of CO2 by sodium borate at this composition increased dramatically once the temperature was raised to the onset temperature (Ton) at around 570 °C. After the sharp increase at Ton, the uptake rapidly reached full capacity (∼4.6 mmol g−1) in a few minutes and leveled off at higher temperatures. Fig. 1(b) shows the isothermal CO2 uptake by the same sample at different temperatures, which proceeded at a rapid rate for temperatures greater than 550 °C. The fastest uptake was recorded at 650 °C, where 99% of the full uptake capacity (∼5.26 mmol g−1) was attained after 1 min reaction with CO2. At 500 °C, an initial induction period of a few minutes was evident before initiation of the rapid uptake. At 450 °C, there was no substantial absorption of CO2. The sodium borate uptake characteristics at 600 °C are shown in Fig. 1(c) to be significantly different from those of previously-reported solid adsorbents CaO and Li3BO3; the results are normalized by the uptake capacities attained following 1 h reaction with CO2. With the solid adsorbents, the uptake rate decreased as the reaction proceeded, resulting in a gradual approach to the full uptake capacity with increasing reaction time. In contrast, the uptake by sodium borate jumped to the full capacity without any slowing of the uptake rate. Fig. 1(d) shows the CO2 desorption curves at different temperatures under a flow of 100% N2 for the same sodium borate sample (NaxB1−xOy (x = 0.75)) following CO2 uptake, from which it is evident that desorption of CO2 occurs at temperatures higher than 650 °C. Here, CO2 was first released rapidly and then more slowly as desorption proceeded. With higher regeneration temperatures, the desorption proceeded at a faster rate. At 800 °C, desorption of 80% pre-absorbed CO2 was completed within 10 min following the introduction of N2 gas to the test chamber. The cyclic regenerability of the sorbents and the extent of desorption are important characteristics of these materials if they are to be used in practical CO2 capture systems. From Fig. 1(e) and (f) it is clear that NaxB1−xOy (x = 0.75) exhibits excellent cyclic regenerability with no deterioration in uptake capacity nor in reaction rate during cyclic temperature- or pressure-swing operations. Although a residual amount of CO2 was retained by the molten salt at the end of the desorption process, the absorption and desorption performance in later cycles was essentially identical to that of the initial cycle. The excellent cyclic behavior was confirmed up to 100 cycles experimentally, as shown in the ESI (Fig. S1).†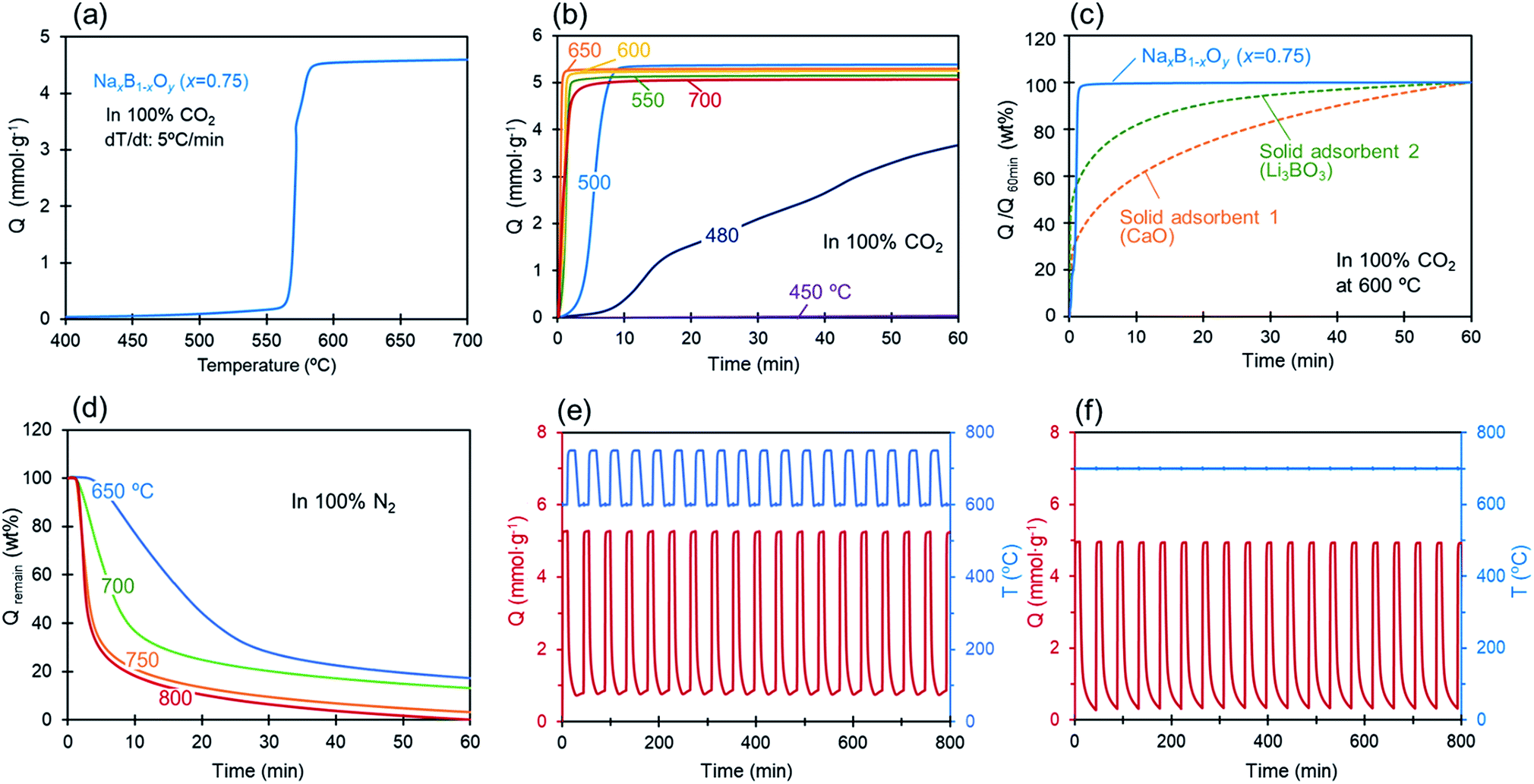 | ||
| Fig. 1 (a) CO2 uptake (Q [mmol g−1]) by NaxB1−xOy (x = 0.75) with temperature (increased at a rate of 5 °C min−1) under a flow of 100% CO2 at atmospheric pressure (1 bar), (b) isothermal CO2 uptake by NaxB1−xOy (x = 0.75) at different temperatures under a flow of 100% CO2, (c) CO2 uptake by NaxB1−xOy (x = 0.75), CaO, and Li3BO3 at 600 °C, normalized by the uptake capacity after 1 h of reaction, (d) CO2 desorption curves at different temperatures under a flow of 100% N2 for NaxB1−xOy (x = 0.75) after the uptake of CO2 under a flow of 100% CO2 at 600 °C for 10 min, (e) cyclic regenerability of CO2 uptake and the desorption by NaxB1−xOy (x = 0.75); by temperature swing operation with ΔT = 150 °C (at 600 °C under a flow of 100% CO2, and at 750 °C under a flow of 100% N2) and, (f) by pressure swing operation on repeated switching of the gas stream between 100% CO2 (pCO2 = 1 bar) and 100% N2 (pCO2 = 0 bar) at 700 °C. | ||
Fig. 2(a) shows the uptake of CO2 by alkali-metal borates with different alkali-metal species at a temperature increase rate of 5 °C min−1 under 100% CO2. Binary, A = (Li0.5Na0.5), and ternary, A = (Li0.33Na0.33K0.33), mixtures of the alkali-metals were prepared with equimolar mixtures of these alkali-metal species. The onset temperature at which the rapid uptake begins shifts to lower temperatures when the sodium is replaced by lithium (i.e., LixB1−xOy) or by a mixed species that includes lithium ((Li0.5Na0.5)xB1−xOy, (Li0.33Na0.33K0.33)xB1−xOy). With LixB1−xOy, the uptake accelerated as the temperature increased to a maximum at around 590 °C, followed by a decline at higher temperatures. With the mixed alkali-metal species the uptake rate increased even more sharply, and showed a broad maximum starting at around 570 °C. In the case of potassium borate (KxB1−xOy), however, the uptake was low over the entire temperature range studied. Cyclic regenerability with no deterioration in capacity upon repeated cycling of CO2 absorption and desorption for the most promising mixed alkali-metal species (Li0.5Na0.5)xB1−xOy is evident in Fig. 2(b). Here, the uptake was quite fast, as was the case for sodium borate, and the full uptake capacity of 7.27 mmol g−1 was attained in a few minutes under reaction with CO2.
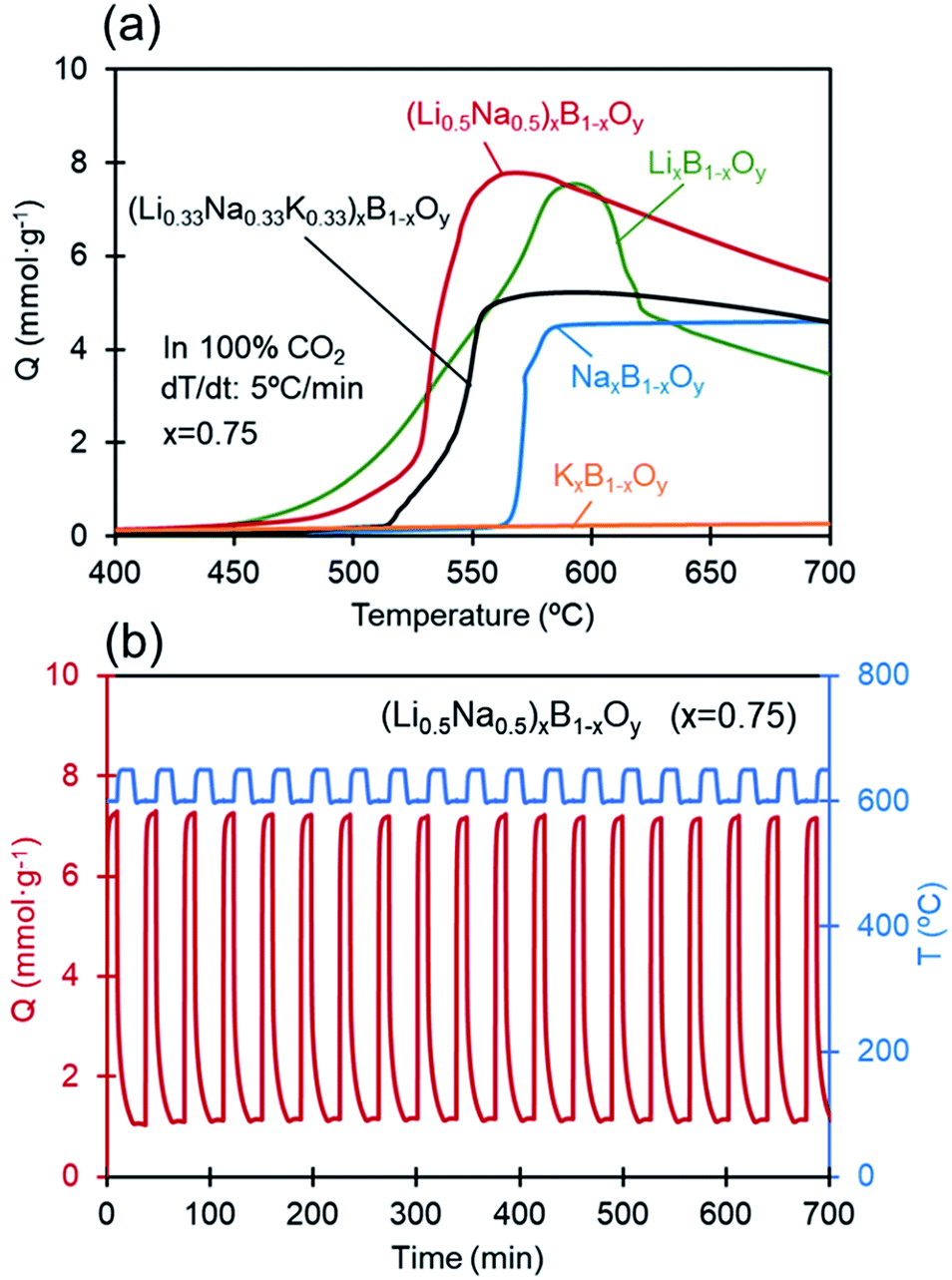 | ||
| Fig. 2 (a) CO2 uptake by alkali-metal borates (AxB1−xOy) with different alkali-metal species at x = 0.75 with temperature (increased at a rate of 5 °C min−1) under a flow of 100% CO2, (b) cyclic regenerability under repeated cycles of CO2 uptake and desorption by (Li0.5Na0.5)xB1−xOy. | ||
The results given above demonstrate that, apart from potassium borate, the alkali-metal borates perform well with high CO2 capacities and uptake rates beyond a well-defined onset temperature (Ton). A clue as to the temperature dependence of the CO2 uptake by these alkali-metal borates was provided by DSC results given in Fig. 3(a)–(c) that show changes in the physical state of the different alkali-metal borates with increasing temperature. A clear endothermic peak was observed at ∼570 °C with NaxB1−xOy (x = 0.75), whereas no peak or only a weak broad peak was apparent in the scans for LixB1−xOy (x = 0.75) and KxB1−xOy (x = 0.75). These results are an indication that the sodium borate melted to form a molten oxide at ∼570 °C, whereas the lithium borate and potassium borate did not melt or only melted partially. The fact that the onset temperature at which the steep jump in uptake capacity was observed for the sodium borate coincided with its melting point suggests that the CO2 uptake depends strongly on the physical state of the sorbent. Lithium borate remained in the solid phase up to at least 650 °C. Thus, the moderate rate at which uptake of CO2 by LixB1−xOy (x = 0.75) increases with temperature was due to a gas–solid reaction on the surfaces of solid particles, as discussed in the previous report.30 Potassium borate, which was similarly in a solid state, most likely separated into K2O and KBO2, as inferred from a database in which stable compounds of KxB1−xOy in the range of 0.5 < x < 1.0 appear not to have been confirmed.40 Here, K2O should react instantly with atmospheric CO2 and H2O to form KHCO3 which can be transformed further into K2CO3 during pre-calcination at 650 °C; this carbonate does not decompose back to K2O and CO2. The low CO2 uptake performance by KxB1−xOy (x = 0.75) can be attributed to the low CO2 uptake capacity of the mixture of KBO2 and K2CO3. The physical state and phase composition of two different sodium-containing alkali-metal borates, NaxB1−xOy (x = 0.75) and (Li0.5Na0.5)xB1−xOy (x = 0.75), at the reaction temperature for CO2 uptake were examined further by temperature controlled in situ XRD. Fig. 4(a) and (b) show the XRD spectra at 600 °C before reaction with CO2. No distinctive diffraction peaks of solid crystals were observed in the XRD spectra for either compound at 600 °C; only the peaks for the Pt-sheet used as a substrate for XRD analysis registered on the spectra. These results demonstrate that the alkali-metal borates were in a molten (liquid) state at 600 °C before their reaction with CO2. Following reaction with CO2 for 30 min, clearly defined multiple peaks appeared in the XRD spectrum at the same temperature with NaxB1−xOy (x = 0.75), as shown in Fig. 4(c), consistent with solid precipitant generation by the reaction with CO2. Identification of the peaks within the spectrum revealed that solid crystals of α-Na2CO3 and Na2B4O7 formed at 600 °C. In contrast, no clear peaks emerged for (Li0.5Na0.5)xB1−xOy (x = 0.75) even after reaction with CO2 for 30 min, as shown in Fig. 4(d), indicating that the uptake of CO2 by (Li0.5Na0.5)xB1−xOy (x = 0.75) proceeds without the generation of solid precipitants. Since the melting point of the mixed lithium–sodium carbonate is ∼500 °C at around the equimolar eutectic composition,41 it is evident that the mixed carbonates generated by the reaction with CO2 existed as ionized molten liquids. Here, the carbonate ions (CO32−) produced by the reaction of CO2 with oxygen anions (O2−) in the molten oxides could be stabilized by the coordination of double alkali-metal ions (Li+ and Na+) to the carbonate ions at the reaction temperature (600 °C).
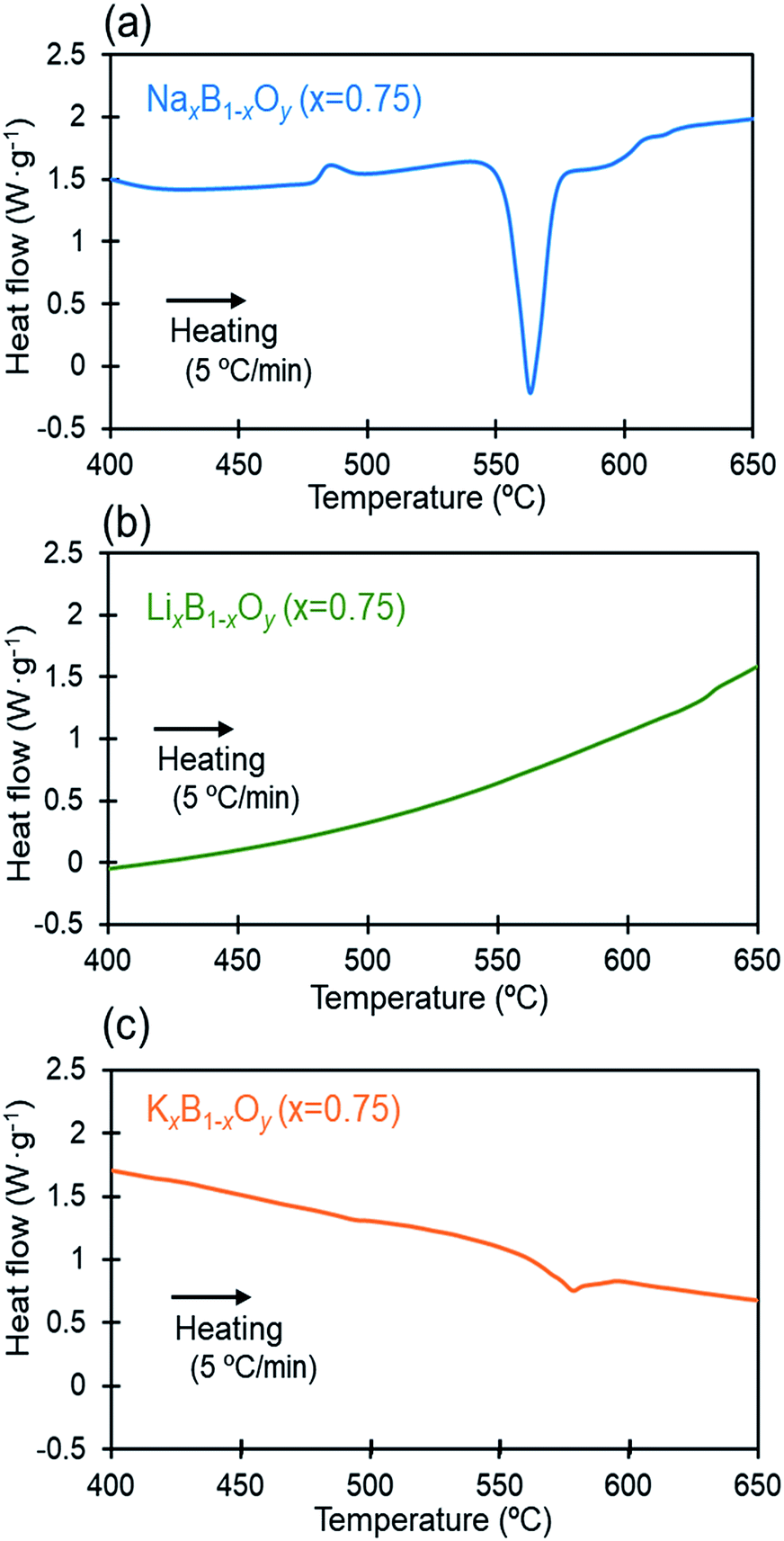 | ||
| Fig. 3 Heat flow required to increase temperature at a rate of 5 °C min−1 under 100% N2 measured by DSC; (a) on NaxB1−xOy (x = 0.75), (b) on LixB1−xOy (x = 0.75) and (c) on KxB1−xOy (x = 0.75). | ||
 | ||
| Fig. 4 In situ XRD spectra at 600 °C for NaxB1−xOy (x = 0.75) and (Li0.5Na0.5)xB1−xOy (x = 0.75), before ((a) and (b)) and after ((c) and (d)) the uptake of CO2 for 30 min. | ||
The dynamic variation in the uptake rate was examined for two of the alkali-metal borates, NaxB1−xOy (x = 0.75) and (Li0.5Na0.5)xB1−xOy (x = 0.75), in which solid precipitants were and were not generated by the reaction with CO2, respectively. Fig. 5(a) shows expanded plots of the uptake by these borates at 600 °C, focusing on the initial 2 min of absorption. The uptake rates for the two samples were significantly different. For the case of NaxB1−xOy (x = 0.75), the absorption began at a low rate initially, and then accelerated after a few seconds (t > 0.3 min) contact before leveling off after t > 0.7 min to complete the uptake reaction. In contrast, with (Li0.5Na0.5)xB1−xOy (x = 0.75), the uptake increased promptly following a quite short initial induction period, and leveled off on approach to the full uptake capacity, albeit more gradually than in the case of NaxB1−xOy (x = 0.75). The initial induction time with a low uptake rate observed for NaxB1−xOy (x = 0.75) is due to the time required for dissolution of CO2 to accumulate carbonate ions (CO32−) in the molten oxides at sufficient levels to exceed the supersaturation threshold for nucleation and growth of nuclei to form solid sodium carbonate particles (Na2CO3(s)). The accelerating increase after the initial induction period (t > 0.3 min) was described well by the Avrami–Erofeyev (Erofe'ev or Erofeev) model for nucleation and nucleus-growth,20,42–44 written as,
| −ln(1 − α) = ktn | (1) |
| [1/(1 − α)] − 1 = kt | (2) |
 | ||
| Fig. 5 (a) Variation of CO2 uptake ratio (α = Q/Q60 min) with NaxB1−xOy (x = 0.75) and (Li0.5Na0.5)xB1−xOy (x = 0.75) under a flow of 100% CO2 at 600 °C over 2 min, (b) double logarithmic plot of the Avrami–Erofeyev equation (eqn (1)) for CO2 uptake by NaxB1−xOy (x = 0.75) over the second period (0.3 min < t < 0.7 min). (c) 2D reaction control model (eqn (2)) for CO2 uptake by (Li0.5Na0.5)xB1−xOy (x = 0.75) as a function of time. | ||
The excellent cyclic regenerability realized by the alkali-metal borates can also be attributed to the liquid behavior of the molten oxides. It is well recognized that deterioration of the uptake performance in repeated uptake and desorption cycles by solid adsorbents is due to the morphological deformation of solid grains to induce pore-blockage or interparticle sintering.5,34,46 For the molten oxides, however, deterioration due to morphological deformation does not occur intrinsically due to the fluidic nature of the molten oxide. The rapid mass transfer of ionic species in the molten oxides averages out any defects or inhomogeneities in molecular distribution instantly to maintain the original uptake performance over all operation cycles. Therefore, the cyclic regenerability of the molten oxide is said to be intrinsic to the material.
Viscosity is an important property of any engineering liquid. Fig. 6 shows the viscosity of NaxB1−xOy (x = 0.75) and (Li–Na)xB1−xOy (x = 0.75) in the molten state at 600 °C. The molten ionic oxides exhibit pseudo-plastic shear-thinning behavior; the viscosities for NaxB1−xOy (x = 0.75) and (Li–Na)xB1−xOy (x = 0.75) at a shear rate of 1000 (s−1) are 790 cP and 170 cP, respectively. These fluid absorbents can be transferred through transfer lines between the absorber and desorber via simple transfer pumps, and should allow low-cost continuous CO2 capture systems operating at medium–high temperature to be established.
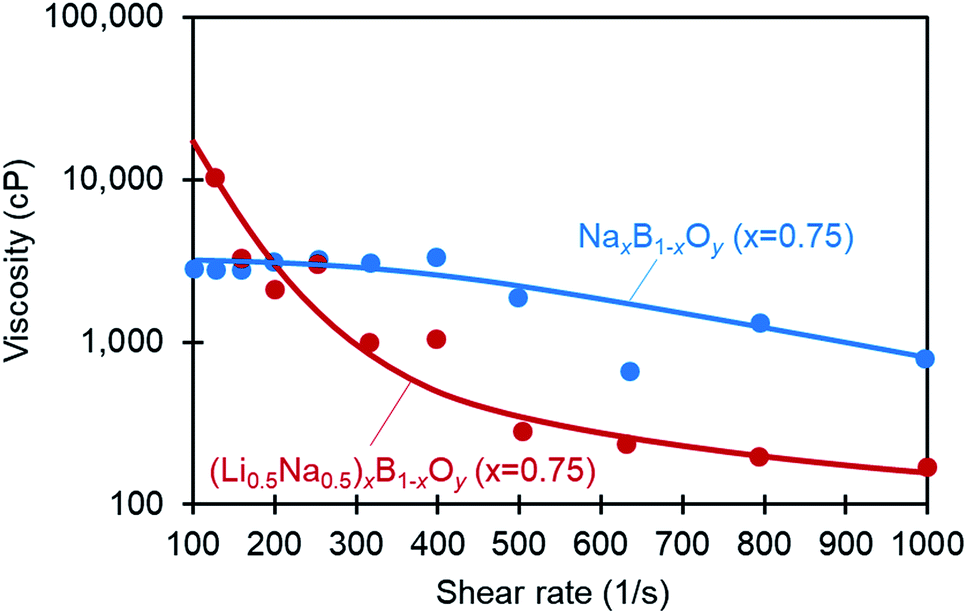 | ||
| Fig. 6 Viscosity of NaxB1−xOy (x = 0.75) and (Li0.5Na0.5)xB1−xOy (x = 0.75) in the molten state at 600 °C. | ||
Table 1 compares the characteristic features of molten ionic oxides for CO2 capture to those of two representative conventional sorbents, aqueous amines and solid oxides.6,19,25,28,36,47–52 Aqueous amines typically use a gas scrubber operating at near-ambient temperatures with simple transfer pumps to deliver the sorbent between the absorber and desorber. Solid oxides operating at medium to high temperatures are far more challenging to transfer between the vessels. Significant effort has been made to address solids handling issues, such as dual fluidized bed reactors with cyclones and conveyer-like moving beds,36,53 but solids handling and system complexity, in addition to uptake deterioration upon cycling, remain key challenges for solid oxides. Molten ionic oxides combine the benefits of the simple liquid transfer system of aqueous amines with the high uptake capacity and high operating temperature of solid sorbents. In contrast to low temperature systems, the streams leaving the process at high temperatures contain valuable heat content which could be recovered in a downstream heat recovery steam generator. Therefore, it is expected that molten ionic oxides will have a low energy penalty of capture in an integrated system. Moreover, the volumetric capacity of molten salts under operational conditions (∼12 mol CO2 L−1) is significantly greater than that of amine solutions (∼1–2 mol CO2 L−1) and fluidized particles (∼1 mol CO2 L−1 with a solids fraction of ∼2%) and thus it is anticipated that the equipment size and plant footprint could be reduced significantly. The added benefits of rapid kinetics and intrinsic regenerability are expected to make molten ionic oxides an important class of materials for future CO2 capture systems.
| Class of material | Molten ionic oxides | Amines | Solid oxides | ||
|---|---|---|---|---|---|
| Materials | NaxB1−xOy, (Li–Na)xB1−xOy | MEA, DEA, piperazine, KS-1 | CaO, MgO, Li4SiO4, Li2ZrO3, Li3BO3 | ||
| Reactor system | Liquid circulation (wet scrubbing, bubbling,…) | Liquid circulation (wet scrubbing, bubbling, EMAR,…) | Dual fluidized bed (cyclone, moving bed) | ||
| State of sorbent (active material wt%) | Molten (100 wt%) | Dissolved in solution (20–30 wt%) | Solid particles (60–100 wt%) | ||
| Process temperature (sorber) | 500–700 °C | 40–60 °C | 250–350 °C (MgO) | 500–600 °C (Li4SiO4) | 600–700 °C (CaO) |
| Process temperature (desorber) | 550–800 °C | 100–120 °C | 350–450 °C (MgO) | 600–700 °C (Li4SiO4) | 700–900 °C (CaO) |
| Enthalpy of reaction (ΔH0) | 212 ± 13 kJ mol−1 ((Li0.5Na0.5)xB1−xOy, (x = 0.75)) | 70–120 kJ mol−1 | 101 kJ·mol−1 (MgO) | 142 kJ·mol−1 (Li4SiO4) | 178 kJ·mol−1 (CaO) |
| Material capacity | ∼5 mmol·g−1 | 0.5–0.75 mmol·g−1 | ∼15 mmol·g−1 (MgO) | ∼5 mmol·g−1 (Li4SiO4) | ∼10 mmol·g−1 (CaO) |
| Working capacity in reactor | ∼12 mol CO2 per L | ∼1–2 mol CO2 per L | ∼1 mol CO2 per L (with a solids fraction of ∼2%) | ||
| Rate of uptake | Fast (<1 min to full uptake) | Moderate (few minutes to full uptake) | Slow (hours to full uptake) | ||
| Cyclic regenerability | Excellent (intrinsic to material) | Good (but material deterioration) | Poor (requires regular supply of fresh material) | ||
| Main Challenges | Gas–liquid contact | Energy penalty | System complexity, grain fragmentation, deterioration of uptake capacity | ||
Conclusions
Molten alkali-metal borates are proposed as a new class of liquid absorbents that can be exploited at medium to high temperatures, in the range of 500–700 °C, with recovery of high grade heat in secondary steam generators. These sorbents exhibit rapid kinetics for the uptake of CO2 to attain full uptake capacity of 5.2 to 7.3 mmol g−1 within 1 min under a flow of CO2 at atmospheric pressure, with intrinsic cyclic regenerability over repeated cycles of CO2 uptake and desorption under both temperature- and pressure-swing operations. The rapid CO2 uptake and superior cyclic performance are attributed to the ionic interactions in the melts and the fluidic nature of the molten oxides.Technologies based on the molten ionic oxides will be applicable to both post- and pre-combustion CO2 capture processes, in particular for next generation high-efficiency power generators, such as integrated combined cycle systems. In post-combustion, CO2 capture at high temperatures could both minimize the energy penalty by high efficiency heat recovery and reduce the size of the carbon capture system, resulting in low cost power generation without CO2 emissions. In pre-combustion, the appropriate installation of capture units for CO2 separation from mixtures of H2 and CO2 could be realized at the temperatures at which coal gasifiers and steam methane reformers with shift reactors operate and thereby improve H2 production rates through shifts in the reaction equilibria. Thus, molten ionic oxides represent a new paradigm for carbon capture, enabling energy efficient process configurations not previously feasible at high temperatures, and could be well positioned to reshape major segments of the CCUS and energy landscape in years to come.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Saudi Aramco under the MIT Energy Initiative program. The authors acknowledge the support of Dr Charles Settens at the Center of Materials Science and Engineering at MIT on the XRD analysis.References
- G. C. Institute, The Global Status of CCS: 2017, 2017 Search PubMed.
- M. Bui, C. S. Adjiman, A. Bardow, E. J. Anthony, A. Boston, S. Brown, P. S. Fennell, S. Fuss, A. Galindo, L. A. Hackett, J. P. Hallett, H. J. Herzog, G. Jackson, J. Kemper, S. Krevor, G. C. Maitland, M. Matuszewski, I. S. Metcalfe, C. Petit, G. Puxty, J. Reimer, D. M. Reiner, E. S. Rubin, S. A. Scott, N. Shah, B. Smit, J. P. M. Trusler, P. Webley, J. Wilcox and N. Mac Dowell, Energy Environ. Sci., 2018, 11, 1062–1176 RSC.
- D. Y. C. Leung, G. Caramanna and M. M. Maroto-Valer, Renewable Sustainable Energy Rev., 2014, 39, 426–443 CrossRef CAS.
- E. Blomen, C. Hendriks and F. Neele, Energy Procedia, 2009, 1, 1505–1512 CrossRef CAS.
- S. Choi, J. H. Drese and C. W. Jones, ChemSusChem, 2009, 2, 796–854 CrossRef CAS.
- G. T. Rochelle, Science, 2009, 325, 1652–1654 CrossRef CAS.
- B. Dutcher, M. Fan and A. G. Russell, ACS Appl. Mater. Interfaces, 2015, 7, 2137–2148 CrossRef CAS.
- O. Miyamoto, C. Maas, T. Tsujiuchi, M. Inui, T. Hirata, H. Tanaka, T. Yonekawa and T. Kamijo, Energy Procedia, 2017, 114, 5616–5623 CrossRef CAS.
- E. Oko, M. Wang and A. S. Joel, Int. J. Coal Sci. Technol., 2017, 4, 5–14 CrossRef CAS.
- G. T. Rochelle, Curr. Opin. Chem. Eng., 2012, 1–2, 183–190 CrossRef.
- A. Veawab, P. Tontiwachwuthikul and A. Chakma, Ind. Eng. Chem. Res., 1999, 38, 3917–3924 CrossRef CAS.
- D. Jansen, M. Gazzani, G. Manzolini, E. Van Dijk and M. Carbo, Int. J. Greenhouse Gas Control, 2015, 40, 167–187 CrossRef CAS.
- X. Zhu, Y. Shi, S. Li, N. Cai and E. J. Anthony, in Pre-combustion Carbon Dioxide Capture Materials, ed. Q. Wang, Royal Society of Chemistry, 2018, pp. 281–334 Search PubMed.
- D. P. Harrison, Ind. Eng. Chem. Res., 2008, 47, 6486–6501 CrossRef CAS.
- D. Berstad, R. Anantharaman and K. Jordal, Int. J. Greenhouse Gas Control, 2012, 11, 25–33 CrossRef CAS.
- J. Strohle, A. Lasheras, A. Galloy and B. Epple, Chem. Eng. Technol., 2009, 32, 435–442 CrossRef.
- A. M. Kierzkowska, R. Pacciani and C. R. Müller, ChemSusChem, 2013, 6, 1130–1148 CrossRef CAS.
- C. Courson and K. Gallucci, in Pre-combustion Carbon Dioxide Capture Materials, ed. Q. Wang, The Royal Society of Chemistry, 2018, pp. 144–237 Search PubMed.
- V. Manovic and E. J. Anthony, Environ. Sci. Technol., 2009, 43, 7117–7122 CrossRef CAS.
- T. Harada, F. Simeon, E. Z. Hamad and T. A. Hatton, Chem. Mater., 2015, 27, 1943–1949 CrossRef CAS.
- J. Wang, L. Huang, R. Yang, Z. Zhang, J. Wu, Y. Gao, Q. Wang, D. O'Hare and Z. Zhong, Energy Environ.Sci., 2014, 7, 3478–3518 RSC.
- W. Gao, L. Sun and Q. Wang, in Pre-combustion Carbon Dioxide Capture Materials, ed. Q. Wang, The Royal Society of Chemistry, 2018, pp. 61–143 Search PubMed.
- K. Nakagawa and T. Ohashi, J. Electrochem. Soc., 1998, 145, 1344–1346 CrossRef CAS.
- Q. Xiao, Y. Liu, Y. Zhong and W. Zhu, J. Mater. Chem., 2011, 21, 3838–3842 RSC.
- D. J. Fauth, E. A. Frommell, J. S. Hoffman, R. P. Reasbeck and H. W. Pennline, Fuel Process. Technol., 2005, 86, 1503–1521 CrossRef CAS.
- M. Kato, S. Yoshikawa and K. Nakagawa, J. Mater. Sci. Lett., 2002, 21, 485–487 CrossRef CAS.
- P. V. Subha, B. N. Nair, P. Hareesh, a. P. Mohamed, T. Yamaguchi, K. G. K. Warrier and U. S. Hareesh, J. Mater. Chem. A, 2014, 2, 12792–12798 RSC.
- M. Seggiani, M. Puccini and S. Vitolo, Int. J. Greenhouse Gas Control, 2013, 17, 25–31 CrossRef CAS.
- A. Román-Tejeda and H. Pfeiffer, J. Therm. Anal. Calorim., 2012, 110, 807–811 CrossRef.
- T. Harada and T. A. Hatton, J. Mater. Chem. A, 2017, 5, 22224–22233 RSC.
- M. K. Ram Reddy, Z. P. Xu, G. Q. Lu and J. C. D. Da Costa, Ind. Eng. Chem. Res., 2006, 45, 7504–7509 CrossRef CAS.
- Q. Wang, H. H. Tay, Z. Zhong, J. Luo and A. Borgna, Energy Environ. Sci., 2012, 5, 7526 RSC.
- J. Wang, L. Huang, R. Yang, Z. Zhang, J. Wu, Y. Gao, Q. Wang, D. O'Hare and Z. Zhong, Energy Environ. Sci., 2014, 7, 3478–3518 RSC.
- R. Barker, J. Appl. Chem. Biotechnol., 1973, 23, 733–742 CrossRef CAS.
- B. Feng, W. Liu, X. Li and H. An, Energy Fuels, 2006, 20, 2417–2420 CrossRef CAS.
- F. Fang, Z. S. Li and N. S. Cai, Ind. Eng. Chem. Res., 2009, 48, 11140–11147 CrossRef CAS.
- D. Araten, J. Appl. Chem., 1968, 18, 118–121 CrossRef CAS.
- L. Shartsis, W. Capps and S. Spinner, J. Am. Ceram. Soc., 1953, 36, 319–326 CrossRef CAS.
- O. L. G. Alderman, M. Liška, J. Macháček, C. J. Benmore, A. Lin, A. Tamalonis and J. K. R. Weber, J. Phys. Chem. C, 2016, 120, 553–560 CrossRef CAS.
- M. Kowalski, P. J. Spencer and D. Neuschütz, in Slag Atlas, ed. Verein Deutscher Eisenhuettenleute (VDEh), Verlag Stahleisen GmbH, 2nd edn, 1996, p. 51 Search PubMed.
- G. J. Janz and M. R. Lorenz, J. Chem. Eng. Data, 1961, 6, 321–323 CrossRef CAS.
- T. J. W. De Bruijn, W. A. De Jong and P. J. Van Den Berg, Thermochim. Acta, 1981, 45, 315–325 CrossRef CAS.
- A. Khawam and D. R. Flanagan, J. Phys. Chem. B, 2006, 110, 17315–17328 CrossRef CAS.
- M. Avrami, J. Chem. Phys., 1939, 7, 1103 CrossRef CAS.
- Y. S. Ho, J. Hazard. Mater., 2006, B136, 681–689 CrossRef.
- H. Lu, E. P. Reddy and P. G. Smirniotis, Ind. Eng. Chem. Res., 2006, 45, 3944–3949 CrossRef CAS.
- H. F. Svendsen, E. T. Hessen and T. Mejdell, Chem. Eng. J., 2011, 171, 718–724 CrossRef CAS.
- S. Ziaii, G. T. Rochelle and T. F. Edgar, Ind. Eng. Chem. Res., 2009, 48, 6105–6111 CrossRef CAS.
- F. Closmann, T. Nguyen and G. T. Rochelle, Energy Procedia, 2009, 1, 1351–1357 CrossRef CAS.
- I. Kim and H. F. Svendsen, Int. J. Greenhouse Gas Control, 2011, 5, 390–395 CrossRef CAS.
- N. Rodríguez, M. Alonso and J. C. Abanades, AIChE J., 2011, 57, 1356–1366 CrossRef.
- W. Liu, H. An, C. Qin, J. Yin, G. Wang, B. Feng and M. Xu, Energy Fuels, 2012, 26, 2751–2767 CrossRef CAS.
- T. Okumura, K. Yoshizawa, S. Nishibe, H. Iwasaki, M. Kazari and T. Hori, Energy Procedia, 2017, 114, 2322–2329 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ta09122j |
| This journal is © The Royal Society of Chemistry 2019 |