
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInfluence of bromide content on iodide migration in inverted MAPb(I1−xBrx)3 perovskite solar cells†
Rodrigo
García-Rodríguez
 *a,
Dominic
Ferdani
a,
Samuel
Pering
a,
Peter J.
Baker
b and
Petra J.
Cameron
*a
*a,
Dominic
Ferdani
a,
Samuel
Pering
a,
Peter J.
Baker
b and
Petra J.
Cameron
*a
aDepartment of Chemistry, University of Bath, Bath BA27AY, UK. E-mail: rgr25@bath.ac.uk; p.j.cameron@bath.ac.uk
bISIS Pulsed Neutron and Muon Source, Rutherford Appleton Laboratory, Harwell Oxford, Didcot OX110QX, UK
First published on 20th September 2019
Abstract
The effect of a systematic increase in the bromide content on mixed anion methyl ammonium lead halide, MAPb(I1−xBrx)3, perovskite solar cells is investigated. We show that at a critical bromide concentration (7.5%) we supress the slow impedance response from the cells. We link the changing impedance spectrum to a large increase in the activation energy for iodide motion. These results are corroborated by muon spin relaxation measurements, where we show that at the concentration of bromide typically used in high performance perovskite solar cells (17%) there is no sign of iodide motion in powders. Finally, we show JV curve hysteresis as a function of bromide content. The scan rate at which the maximum hysteresis index is observed does not change as the % Br is increased, leading us to conclude that the low frequency impedance response and the JV curve hysteresis are not caused by the same mobile ions.
Introduction
Research into perovskite solar cells (PSC) has been a focus of the photovoltaic community over the last 10 years, mainly as no other photovoltaic technology has observed a more pronounced rise in the efficiency of devices. PSC have increased from an efficiency of 3.8% in 2009 (ref. 1) to a certified efficiency of 20.9% for a 1 cm2 cell2,3 and recently to 24.2% for an aperture area of 0.096 cm2.4 The steep rise in efficiency is partly a consequence of lessons learned during the development of other emerging photovoltaics technologies such as thin film solar cells,5,6 organic solar cells7 and dye-sensitized solar cells.8,9 Due to the ionic nature of the components, perovskite materials are fairly easy to synthesize via solution-based methods10–13 and at the same time, the performance of PSC has proven to be very resilient to defects.14,15 It is also possible to change the chemical composition of the lead halide perovskites to effectively and systematically tune their bandgap,16–18 making them particularly attractive for a wide variety of applications including light emitting diodes16,19,20 and tandem solar cells.21,22 However, in spite of these promising features, the development of perovskite solar cells still presents important challenges: low stability, particularly under high humidity conditions,23–25 and a hysteretic behavior that appears to be intrinsic to lead halide perovskite devices.12,13,26–28 Stability and hysteresis have been closely linked to the ionic nature of the perovskites. Several studies have shown that a very small activation energy has to be overcome for the iodide anion (0.2–0.7 eV) and methyl ammonium cation (MA+) (0.5–0.8 eV) to move through the perovskite structure.11–13,29–33 The consensus is that the activation barrier to iodide and methyl ammonium motion is low enough to contribute to the low stability of the perovskite devices.11–15,28,29,31,32 Hysteretic behavior has been related to ion migration and the redistribution of charge inside the perovskite film following the application of an electrical potential.11,12 Experimental evidence of ion migration has been found by employing photo-thermal induced resonance microscopy,34 by polarizing the perovskite structure (also known as poling) and then employing complementary techniques such as X-ray diffraction (XRD) to analyze the chemical composition of the perovskite after poling,28,30,35 and via temperature-dependent impedance spectroscopy31,33 and current–potential curves.11,13,32 The activation energies obtained from these characterization techniques are in the range of the values predicted theoretically.It has been proposed that ion motion can be altered by changing the chemical composition of the perovskite. In a theoretical study, Lin et al.36 found that the presence of bromide in methylammonium lead iodide increased ion diffusion barriers and hindered the movement of ions in the perovskite due to a more compact crystal structure. By substituting iodide ions with the smaller bromide ions, Noh et al.37 and Ruess et al.38 managed to increase the stability of the perovskite under high humidity conditions. The increase in stability was also attributed to a more compact structure. High bromide concentrations (>20%) are difficult to study as phase separation into iodide and bromide rich regions occurs under illumination.38–41 Yoon et al.39 proposed that anion segregation is highly influenced by the concentration of defect sites, and found that a stoichiometric excess of the halides in the perovskite composition may help to reduce phase separation. Hysteresis can also be modified by changing the configuration of the devices.28,42–44 Although changes to the perovskite structure have been shown to strongly influence material stability and I–V curve hysteresis, a systematic experimental study on the influence of anion mixing on iodide migration in perovskite cells has not been reported.
Here we study the influence of anion ratios on methyl ammonium lead halide devices by gradually increasing the bromide concentration in inverted MAPbI3 perovskite solar cells. We use impedance spectroscopy measurements at different temperatures to relate the bromide concentration to the activation energy for iodide migration within the material. We show that iodide motion is strongly suppressed at low concentrations of bromide. Our impedance results are corroborated by muon spin relaxation measurements which show that for the mixed halide perovskite MAPb(I0.83Br0.17)3, no iodide diffusion is observed. By measuring current–voltage curves at different scan rates, we analyze the influence of bromide content on the hysteretic behavior and determine the scan rate at which the hysteresis is highest. We then compare the information on ion motion obtained from hysteresis and impedance measurements, highlighting fundamental differences between both characterization techniques.
Experimental
All chemicals were purchased from Sigma-Aldrich unless otherwise noted. [6,6]-Phenyl-C61-butyric acid methyl ester (PCBM) was purchased from Ossila (Sheffield, United Kingdom).Perovskite solar cells with inverted configuration
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v mixture of dimethyl formamide (DMF)/dimethyl sulfoxide (DMSO) was prepared. The solution was stirred until the chemicals were completely dissolved. For the cells with different bromide content, a 1.25 M solution of lead bromide (PbBr2) and methyl ammonium bromide (MABr) in a 4:1 v/v mixture of DMF/DMSO was prepared. Depending on the bromide concentration required, the corresponding ratios of MAPbI3 and the MAPbBr3 were mixed. The solution was stirred for 30 minutes to homogenize and was filtered through a PTFE syringe filter (45 μm).
:1 v/v mixture of dimethyl formamide (DMF)/dimethyl sulfoxide (DMSO) was prepared. The solution was stirred until the chemicals were completely dissolved. For the cells with different bromide content, a 1.25 M solution of lead bromide (PbBr2) and methyl ammonium bromide (MABr) in a 4:1 v/v mixture of DMF/DMSO was prepared. Depending on the bromide concentration required, the corresponding ratios of MAPbI3 and the MAPbBr3 were mixed. The solution was stirred for 30 minutes to homogenize and was filtered through a PTFE syringe filter (45 μm).
Optical characterization
UV-Vis transmittance and reflectance measurements were measured in a PerkinElmer Lambda 750 S UV/Vis Spectrometer, from 350 nm to 900 nm and with a step size of 1 nm. To account for internal reflections, the absorption of the material was calculated from the reflectance and transmission measurements according with A ≈ T/(1 − R)2.45 To calculate the bandgap of the material we employed Tauc plots for direct allowed transitions. To determine the chemical structure of the perovskite material, X-ray diffraction measurements were made with a STOE Transmission Diffractometer System STADI P, employing Cu-Kα radiation, scanning from 10° to 70° (2θ), with a 0.01 step size and an integration time of 20 seconds per point.Current–voltage measurements
Current voltage curves of the solar cells were measured with a Keithley 2601A potentiostat under 1 sun illumination intensity provided by a single source, Class AAA solar simulator (T S Space Systems LTS, Bucks, United Kingdom). The illumination intensity was calibrated with a certified WPVS monocrystalline silicon reference cell with a KG3 filter (Fraunhofer, ISE). 1.2 V was applied to each cell prior each measurement under illumination. A linear potential scan was applied from 1.2 V to 0 V and immediately from 0 V to 1.2 V at a scan rate of 100 mV s−1 with a step size of 10 mV. A mask of (0.25 × 0.25) cm2 was employed to avoid additional contributions of scattered light in the glass. Each substrate accommodated 6 small solar cells.Impedance spectroscopy measurements
Impedance spectroscopy and hysteresis measurements were performed with a ModuLab XM PhotoEchem system (Solartron Analytical). Before each measurement, the cell was under a 470 nm LED (Thorlabs M470L3) illumination (42.5 cm−2) for 15 minutes to obtain a stable open circuit potential. The impedance measurements were made at open circuit potential under LED illumination and employing a small amplitude perturbation of 10 mV in the 100 kHz to 100 mHz frequency range. The electrochemical impedance spectrum was analyzed using Z-View software. The samples were mounted in a desiccator filled cell holder to prevent degradation. The temperature was controlled with a Peltier element and a Precision Peltier Temperature Controller TEC-1091 (Meerstetter Engineering, Rubigen, Switzerland).Hysteresis measurements
A black mask with an aperture of (2.5 × 2.5) mm2 was used to avoid additional contributions of scattered light to the sample. The cell was illuminated with a blue LED (Thorlabs 470 nm) at open circuit for 15 minutes or until the open circuit potential did not change over time. The illumination intensity employed was 4.4 mW cm−2. 1.2 V was applied for 5 seconds prior to each scan. The cell was measured from open circuit conditions (Voc) to short circuit conditions (Jsc) and immediately after from Jsc to Voc. After each scan the cell was illuminated at open circuit potential for 2 minutes. The scan rates employed are presented in Table 1. The cell was measured from fast to slow scan rates.| # | Scan rate (mV s−1) | # | Scan rate (mV s−1) | # | Scan rate (mV s−1) |
|---|---|---|---|---|---|
| 1 | 7000 | 8 | 300 | 15 | 10 |
| 2 | 5000 | 9 | 200 | 16 | 7 |
| 3 | 3000 | 10 | 100 | 17 | 5 |
| 4 | 2000 | 11 | 70 | 18 | 3 |
| 5 | 1000 | 12 | 50 | 19 | 2 |
| 6 | 700 | 13 | 30 | 20 | 1 |
| 7 | 500 | 14 | 20 | 21 | 0.5 |
Muon spectroscopy
Perovskite powders were prepared by a hot-casting method. To make a MAPbBr3, a 1.9 M solution of MA and PbBr2 in anhydrous DMF was prepared by stirring the precursors at 60 °C for one hour. The solution was subsequently cast onto a clean glass Petri dish at 110 °C and left for an hour to form a thick orange film. The film was scratched from the Petri dish to produce orange MAPBr3 powder. The structure of the MAPBr3 powder was confirmed by pXRD analysis. The synthesis procedure for the mixed halide perovskites was the same with the ratio of PbI2 and PbBr2 adjusted to give the desired stoichiometry.Muon spin relaxation experiments were performed at the ISIS pulsed muon facility using the HIFI instrument. Approximately 1 g of sample was pressed into a metal sample holder forming a disc of approximately 1 mm thickness. The temperature was controlled between 40 and 410 K using a closed cycle refrigerator and hot stage. Measurements were taken at 4 different longitudinal fields at each temperature. Initial calibration of the instrument was performed by applying a 100 G transverse field. X-Ray diffraction patterns were obtained using a Bruker axs D8 advance powder X-ray diffractometer with Cu-Kα source and Ge monochromator.
Results and discussion
MAPb(I1−xBrx)3 films were prepared, where x = 0, 2.5%, 5%, 7.5%, 25% and 100%. The X-ray diffractograms for the films (Fig. 1) show the expected peaks associated with the (110) and (220) planes of a 3D perovskite structure.38 As the amount of bromide increases, the peaks are displaced towards larger values of 2θ (the peak positions have been normalized to the peak of the underlying FTO substrate, see Fig. S1†). This displacement has been attributed to changes in the crystal structure due to the smaller size of the bromide ion compared to the iodine ion, and the fact that there is a gradual shift from the tetragonal phase of pure MAPbI3 to the cubic phase of pure MAPbBr3.17,41,46 Previous reports have shown a reduction in the lattice parameters of 2–3 pm when changing between pure MAPbI3 and 10% Br perovskites.17,41,46,47 The tetragonal to cubic phase transition has been reported to occur between 13–20% bromide,17 or at even higher bromide concentrations (>30%).47 It has also been reported that as the amount of bromide is increased, phase segregation can occur, particularly when the film is under illumination.16,40,41 Phase separation causes the 28.8° peak to split into two peaks, associated with iodide and bromide rich regions.40,41 No phase separation was observed in these samples, although these films were not exposed to light before the XRD spectrum was measured. Peak splitting was observed in pure MAPbI3, which is attributed to the (004) (28.11°) and (220) (28.36°) reflections of the tetragonal phase of the perovskite;17 the peak splitting disappears with bromide addition. In order to avoid the complication of large scale phase segregation in our measurements, only bromide substitution ratios ≤7.5% were investigated. | ||
| Fig. 1 XRD measurements for perovskite films with different bromide content, (a) 2θ from 10° to 65° and (b) from 25° to 35°. In (b) the main peaks for the tetragonal (004) (28.11°) (purple dot), (220) (28.36°) (green dot) and cubic phases (200) (red dot) are highlighted. | ||
In the next step, inverted perovskite solar cells containing different amounts of bromide were prepared and characterised. The highest reported efficiency for an inverted perovskite cell with NiO and PCBM contacts is 20.5%.48 This efficiency was achieved with a doped hole transport layer, a modified aluminium cathode and a perovskite with a chemical composition of MAPbI3−xClx. In our lab we prepare simple inverted NiO cells with top efficiencies close to 15% (Fig. S2†), using a combination of anti-solvent treatments and solvent annealing. In order to make a direct comparison between cells with different bromide ratios, the cells used in this study have slightly lower efficiencies. It was much more important that direct comparisons could be made between devices with different compositions. We did not use solvent annealing, as we were concerned that it would lead to differences in crystal size between films with different bromide content. In this study cells were also prepared with an emphasis on stability and reproducibility. The cells were able to maintain a stable open circuit voltage over several hours to allow us to carry out reliable temperature dependent impedance measurements.
The influence of bromide content on perovskite solar cell performance is summarized in Fig. S3.† In Fig. S3a† the increase in Voc as a function of bromide content is related with the change in the bandgap,16–18,36,49 as shown in Fig. S4 and Table S1.† The change in current density (Fig. S3b†) is related to the absorption of the perovskite material.17 There is a slight decrease in fill factor with increasing bromide content. All the cells showed very low hysteresis under illumination at 1 sun (Fig. S5†).
It should be noted that the performance of the devices was higher when measured from Jsc to Voc (Fig. S5,† so called ‘inverted hysteresis’). Inverted hysteresis has previously been reported for cells measured using negative preconditioning potentials.50,51 Existing literature on inverted hysteresis refers to n–i–p perovskite cells and underlines the importance of preconditioning on the IV curve measured.50,51 Almora et al. also proposed that inverted hysteresis is more likely to be observed in mixed-perovskite structures.51 In our p–i–n cells, the preconditioning potential was always above the Voc of the perovskite cell and inverted hysteresis appeared not only in mixed-halide perovskites, but also for pure MAPbI3 cells (Fig. S5†). Inverted hysteresis disappears for pure MAPbBr3 devices, although a direct comparison is difficult due to the lower efficiency of MAPbBr3 solar cells (Fig. S3†). Our results show that inverted hysteresis is not only dependent on the preconditioning potential and the composition of the perovskite, as reported previously.50,51
Impedance spectroscopy (IS) was used to evaluate the influence of bromide content on ion movement. All IS was carried out at open circuit under illumination. IS is widely used to characterize PSC and the spectra vary a little depending on cell configuration and the measuring conditions used.31,52–60 The Nyquist plots for PSC typically show 2–3 clear semi-circles, with a fourth feature that extends below the real axis at very low frequencies. In our results on both standard31 and inverted33 MAPbI3 cells, the Nyquist plots show three semicircles when cells are measured at 25 °C under illumination (Fig. 2).We will refer to the three features as the high frequency (hf), mid-frequency (mf) and low frequency (lf) response of the device. There is still considerable debate about the interpretation of impedance spectra for PSC in the literature. There is consensus that the hf response is due to the geometric capacitance, Cgeo, coupled to a resistance, Rhf.31,53–55Rhf is related to the recombination resistance, although as Ebadi et al. recently pointed out it could also contain a contribution from charge injection.61 It is sometimes suggested that a transport resistance, caused by percolation of charges through the perovskite or the mesoporous oxide contact, also needs to be included.61 However, as transfer of charge through the perovskite film is very rapid,62 the transport resistance should be negligible in efficient cells.
 | ||
| Fig. 2 Nyquist plots for perovskite solar cells as a function of the bromide content. The measurements were made at 25 °C. The high frequency (hf), mid-frequency (mf) and low frequency (lf) regions are displayed. Frequency values are highlighted in each plot: 1 = 100 kHz, 2 = 10 kHz, 3 = 1 kHz, 4 = 100 Hz, 5 = 10 Hz and 6 = 1 Hz. | ||
In the paper by Pockett et al. we suggested that the mf and lf response from the cell were due to a coupled ionic-electronic response.31 The ionic distribution inside the device is modulated by the applied voltage, this in turn modifies electron injection and recombination currents and hence causes the mf and lf impedance response. The, mf, semicircle has been associated with charge recombination,31,55,63 although the nature of the ions causing the response in this frequency region it is still a matter of debate. The lf semicircle is associated with iodide ion modulated recombination/injection processes.10,31,63,64 Moia et al. have developed a promising equivalent circuit which uses transistors to model the effect of ion modulation on electronic recombination across the interface.65 The model can reproduce both the low frequency ‘capacitive’ and ‘inductive’ behavior inside PSC. Jacobs et al. use a drift diffusion model to simulate the impedance response for PSC.66 They show that ‘giant photo induced capacitances’, ‘induction loops’ and apparent ‘negative capacitances’ at low frequency can all be explained by the coupling of ion migration and electronic current. The only downside of models based on transistors and semi-conductor drift diffusion models is that complex numerical solutions are required to simulate the impedance response. In our own work we also do not use an equivalent circuit to fit our data. We are taking an experimental approach – linking systematic temperature dependent IS experiments to physical processes occurring in the cell. The temperature dependent time-constant for the low frequency process can be used to extract an activation energy31,33,64 which agrees with the majority of experimental and computational activation energies for iodide migration (0.3–0.6 eV).11–13,29–33 We recently reported that cation substitution into MAPbI3 moves the lf process to such low frequencies that it can no longer be seen in the impedance spectrum. This corresponds to a large increase in the activation energy for iodide motion inside the perovskite. Here we clearly show that the lf semi-circle can also be strongly affected by anion substitution.
The Nyquist plot for pure MAPbI3 is shown in Fig. 2a. Three clear semicircles are observed, with the third semicircle appearing at frequencies lower than 100 Hz. As the amount of bromide is increased, the diameter of the low frequency semicircle decreases. For 5.0% bromide content, the semicircle is still visible although noticeably smaller than for pure MAPbI3. When the bromide concentration reaches 7.5%, the low frequency semicircle has disappeared completely. The Nyquist plot for pure MAPbBr3 (Fig. S6†), also shows only two clear semicircles with no trace of a third low frequency semicircle observed.
Computational studies have reported that the substitution of iodide with bromide inside the perovskite structure increases the activation energy for the migration of iodide ions.36 The substitution of the smaller bromide ions makes the perovskite crystal more compact,36 Ruess et al. suggested that this increases the interaction between the anions and MA+ cations.38 Svane et al. also looked at the cation–halide interaction energies in MAPbI3 and MAPbBr3 in detail.67 They concluded that the hydrogen bond energy is very similar in both compounds, and that the total electrostatic energy of interaction is greater in MAPbI3 than in MAPbBr3. Other studies have found that bromide ions form a stronger bond with Pb2+ than iodide ions.39,68
The increase in activation energy for iodide motion in mixed halide perovskites is likely to be due to local lattice distortions. Substitution of iodide with the smaller bromide ion distorts the crystal lattice, changing ion diffusion pathways and hence increasing the activation energy for iodide motion. We recently showed that just 5% cation substitution creates enough local distortion to substantially increase the activation barrier for iodide motion.33
To investigate further, impedance spectra were collected at a range of different temperatures (10–40 °C). Fig. 3 shows Cole (imaginary impedance as a function of frequency) and Nyquist plots at selected temperatures for cells with 5%, 6.3% and 7.5% Br substitution. Cole plots are shown as the hf, mf and lf processes can be observed as clear “peaks” in the impedance and shifts in frequency with temperature can be more clearly observed. For perovskites with 5.0% bromide content, the low frequency peak (associated with the third lf semicircle in the Nyquist plot) is observed at all the temperatures measured; the peak of the lf process is shifted towards higher frequencies as the temperature is increased. This is expected for a temperature activated process; the iodide motion is faster at higher temperatures. However, when the bromide concentration is increased to 7.5%, the third semicircle completely disappears, even at temperatures as high as 40 °C the lf semicircle is not visible, indicating the process that causes it has been suppressed.
 | ||
| Fig. 3 (a) Cole plot of perovskite solar cells as a function of temperature and bromide concentration: 5.0%, 6.3% and 7.5% bromide content. The main peaks at different frequencies are highlighted. (b) Nyquist plots of the perovskite with 6.3% bromide concentration as a function of temperature. Frequency values are highlighted: 1 = 100 kHz, 2 = 10 kHz, 3 = 1 kHz, 4 = 100 Hz, 5 = 10 Hz and 6 = 1 Hz. | ||
An intermediate bromide concentration of 6.3% was chosen to analyze the evolution of this low frequency process. Interestingly, cells with 6.3% Br only show suppression of iodide motion at low temperature. At 10 °C only two peaks are observed with no trace of the low frequency peak. As the temperature is increased to 17 °C and beyond, the low frequency process appears. The process becomes faster as the temperature is increased and the peak in the Cole plot is observed at higher frequencies. This response shows that at 6.3% bromide concentration the iodide motion is not fast enough to affect the response at low temperatures. As the temperature is increased the amount of iodide motion increases, bringing the low frequency response into the measurement window. This effect can be fully appreciated in Fig. 3b, where the evolution of the low frequency semicircle with temperature shows the appearance of the low frequency response when going from 10 °C to 25 °C. These results suggest that there is a critical concentration of bromide over which iodide migration will be suppressed.
We used the time constants from the temperature dependent impedance measurements to obtain activation energies for the low and mid-frequency processes.31 The results are summarized in Fig. 4. The increase in the lf activation energy with bromide concentration (red dots in Fig. 4) support the argument that iodide motion is suppressed as the bromide content is increased. This trend is consistent with the results presented in Fig. 2 and 3. Unfortunately impedance spectroscopy gives no information on the location of the ions that are causing the lf response i.e. whether we are measuring the influence of iodide ions moving in the bulk, along grain boundaries or at the interfaces with the hole and electron transporting contacts.
 | ||
| Fig. 4 Activation energy for the middle and low frequency semicircles as a function of bromide content. Dashed lines have been included as a guide for the eye. The activation energy for the low frequency process for the 6.3% bromide is highlighted as it was obtained with only three temperatures. | ||
We cannot conclusively say whether the ions are modulating bulk recombination or interfacial recombination/injection. IS only provides the time scale related with the process, it does not give any information on the pathway of iodide migration/diffusion.
It is worth noting that while the high frequency process is largely independent of temperature, the mid-frequency peak shifts to higher frequencies with temperature, suggesting that this is another temperature activated process (Fig. S7†). As outlined above, the nature of the mid-frequency process is not completely understood. Our results show that it is a thermally activated process that is not strongly influenced by the bromide concentration. The mf activation energy is lowest for the pure MAPbI3 (0.3 eV) and it increases to values between 0.4–0.6 eV for mixed bromide devices where it fluctuates but does not increase sharply with Br content. The mf response has previously been shown to be particularly sensitive to illumination,31 and has been associated with charge recombination.31,55,63
The mid-frequency process has an associated time constant between 0.1 and 10 ms, it is 2–3 orders of magnitude faster than the lf process. In contrast the activation energies for the mf and lf processes are very similar in MAPbI3 cells. Here we measured 0.30 ± 0.08 eV for the lf and 0.37 ± 0.10 eV for the mf process in pure MAPbI3 cells. It has been suggested that ion migration via grain boundaries should occur on faster time scales than migration through the bulk of the perovskite.15 This leads us to think that the mf semicircle may be related to iodide migration along grain boundaries or at the interfaces with the selective contacts, whereas the lf process is caused by iodide migration in the bulk of the crystals. If this is correct, anion and cation33 substitution suppresses bulk iodide diffusion, but does not strongly influence the motion of ions at the crystal surface. Temperature-dependent impedance measurements controlling the grain size of the perovskite may shed some light into this phenomenon but is outside the scope of this work. Equally, it would be useful to measure the temperature dependent impedance of cells where the crystallite surfaces have been passivated.69
It should be noted that at very low frequencies the impedance response in the Nyquist plots drops below the real axis (Fig. S8†). This response has been associated with transient injection currents,61 however no trend in the very low frequency response of our cells was obtained as a function of bromide concentration.
Muon spin relaxation (μSR) was used as a complementary technique to further investigate iodide diffusion in MAPb(I1−xBrx)3 powders. The muons used in this study stop inside the bulk of the crystals and can act as a direct probe for bulk iodide motion.33 μSR has become a standard technique for analysis of ion migration in lithium and sodium battery materials.70–73 Recently, we used it to study the properties of ion migration in methyl ammonium lead iodide samples as well as mixed cation samples.33 μSR involves implanting a 100% spin polarized positive muon into a sample. The muons decay inside the sample, emitting a positron which is detected. Fluctuating electronic fields in the target material impact upon the spin of the muon, changing the decay angle of the positron and these changes can be used to gain information on the environment the muon experiences prior to decay. One possible way that the local environment of the muon can be altered is by the motion of nearby ions that have a nuclear magnetic moment. Bromide has a similar spin (3/2) and nuclear magnetic moment (+2.12μN for 79Br and +2.27μN for 81Br) as Li+ (3/2 and +3.26μN), Na+ (3/2 and +2.22μN) and I− (5/2 and +2.81μN) making it feasible to detect its migration using μSR. We recently showed that it is possible to detect iodide diffusion in MAPbI3 above 350 K.33
Here we carried out μSR on pure MAPbBr3 and on MAPb(I0.83Br0.17)3. Powder samples of MAPbBr3 and MAPb(I0.83Br0.17)3 were prepared and characterized with pXRD (Fig. S9†). For the study of MAPBr3, measurements were taken at regular temperature intervals between 50 and 400 K with four different longitudinal field measurements (0, 5, 10 and 20 Gauss) taken at each temperature. The resulting data was fit using a dynamic Gaussian Kubo Toyabe function multiplied by an exponential relaxation (eqn (1)):72
| A0PLF = AKTGDGKT(Δ, ν, t, HLF)exp(−λt) + ABG | (1) |
The terms A0, ABG, AKT refer to the initial, background and sample asymmetry, respectively. PLF is the polarization in a longitudinal field, GDGKT refers to a dynamic Gaussian Kubo Toyabe (KT) distribution function, Δ is the local field formed by nuclear magnetic moments at the muon implantation site, ν is the dynamic fluctuation rate of the local field, t is time and HLF is the applied longitudinal magnetic field. Finally, exp(−λt) is used to model the depolarization of muons caused by electronic moments, the rate of fluctuation is so rapid that it can be modelled by a simple exponential function with λ as the rate of relaxation. A small non-zero value of λ (less than 0.02 MHz) was found to be necessary to consistently describe the data across the full temperature range and can be attributed to a slight difference between the field distribution in the samples and the Gaussian approximation of the GDGKT function. An example of raw muon data for MAPbBr3 at 100 K is shown in Fig. S10.†
The dynamic fluctuation rate, ν, can be linked to ions moving in the perovskite powders. By fitting the asymmetry data across the full temperature range, it is possible to obtain values for ν with respect to temperature (Fig. 5). We have presented the data for MAPbI3 previously, but it is reproduced here for clarity. In MAPbI3ν first increases between about 100 K and 150 K. This increase in fluctuation rate at low temperatures is caused by the muons interacting with the MA cations which can change orientation inside the lattice (they are not diffusing at these temperatures).74 Above 150 K, cation tumbling becomes too fast to be measured by μSR. At temperatures greater than 250 K in MAPbI3 there is a second increase in ν which is due to the motion of iodide ions. ν increases steeply after 350 K as iodide diffusion strongly influences the measured response. By fitting the increase in ν as a function of temperature, an activation energy for iodide motion can be obtained.
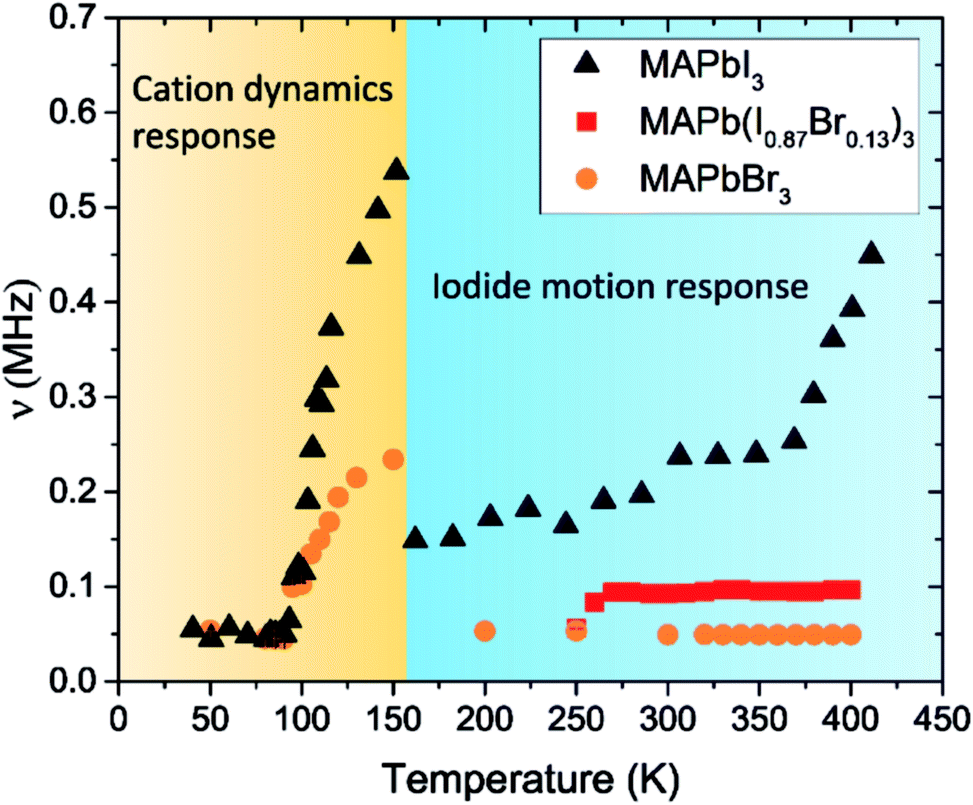 | ||
| Fig. 5 Temperature dependence of ν for MAPbI3, MAPbBr3 and MAPb(I0.83Br0.17)3. The regions where the cation and anion response are expected are highlighted. The results of MAPbI3 have been adapted with permission.33 | ||
The ν data for MAPbBr3 shows only one increase in fluctuation between 100 K and 150 K, again this is where the cations would be expected to have enough energy to reorient inside the lattice.
After 150 K the fluctuation rate decreases back down to 0.05 MHz where it remains flat for the rest of the temperature range. The results of the MAPBr3 study show that cation dynamics are still prevalent at low temperature and the process has an Ea of 0.026 eV (Fig. S11†). This compared to an activation energy of 0.072 eV for the cations in MAPbI3.33 The complete absence of any high temperature increase in fluctuation rate shows that it was not possible to detect bromide ions moving in the same way that we were able to detect iodide motion in pure MAPbI3.33
The mixed halide perovskite MAPb(I0.83Br0.17)3 was studied between 250 and 400 K as this is the temperature range where iodide motion is observed in pure MAPbI3 (Fig. 5).33 The data was fit using the same parameterization as the MAPbBr3 study and the resulting temperature dependence of ν is shown in Fig. 5. The fluctuation rate was not affected by the increase in temperature. This indicates that bulk iodide diffusion was not detectable in the mixed halide sample. This agrees with the impedance results which show bromide inhibiting iodide diffusion.
In the final part of this study, the effect of anion mixing on J–V curve hysteresis is investigated. Hysteresis in perovskite cells has been associated with ion migration and charge redistribution in the perovskite crystal, and it has also been used as an indirect way to study ion migration in perovskite solar cells.12,13,26
Several groups have investigated the dependence of hysteresis on scan rate and related the response to the timescale of the redistribution of charge as the applied external potential changes.11,26,75 By measuring hysteresis across a broad range of scan rates the timescale at which maximum hysteresis is observed can be reported.13,75,76
Our impedance and μSR results suggest that bromide content strongly influences iodide motion in the perovskite structure. We therefore expected bromide content to influence the scan rate at which peak hysteresis is observed. We measured J–V curves with different scan rates (Fig. S12–S16†) and obtained the degree of hysteresis as a function of scan rate and bromide concentration according to the procedure presented in the ESI (eqn S1†). The results are summarized in Fig. 6. The PSC showed inverted hysteresis at the majority of scan rates investigated. It should be noted that for these measurements the cells were measured at fixed temperature under blue LED illumination. The degree of hysteresis was much higher under blue light than under 1 sun illumination (Fig. S5†). The difference in the degree of hysteresis may be related with differences in the penetration depth of the light, and as a consequence the generation profile of carriers inside the perovskite, as was proposed by Contreras-Bernal et al.52 This could be related to the experimental finding that hysteresis is closely related to the nature of the selective contacts.28,42–44
 | ||
| Fig. 6 Degree of hysteresis as a function of scan rate and bromide content in perovskite solar cells. The scan rate at which the peak of hysteresis is observed for each concentration is presented. The blue shade represents the region where normal hysteresis is present. | ||
Fig. 6 shows that, surprisingly, the scan rate at which the maximum hysteresis is observed does not change noticeably with small additions of bromide. However, there are clear changes for pure bromide devices which show no hysteresis up to 1000 mV s−1.
It has already been reported that “hysteresis-free” devices, such as inverted perovskite cells are simply devices where the timescale of the process responsible of the hysteretic behavior is not observable under the measurement conditions presented (e.g. hysteresis is low at 100–200 mV s−1, but not at other scan rates).13,77 Although the inverted perovskite cells measured here show very low hysteresis under 1 sun illumination and a scan rate of 100 mV s−1 (Fig. S5†), our results show that under blue light the hysteresis increases at faster scan rates (Fig. 6).
The peak hysteresis for our inverted cell is remarkably similar to peak hysteresis reported by Levine et al. for pure MAPbI3.13 They found that peak hysteresis for inverted cells appeared at higher scan rates than for traditional n–i–p cells (between 300–1500 mV s−1), the shift was attributed to differences between the selective contacts. Levine et al.13 and Bryant et al.77 also found that lowering the temperature, and hence slowing ion motion, in inverted perovskite solar cells increases the degree of hysteresis.
The results presented in Fig. 6 highlight the differences between hysteresis and impedance measurements (Fig. 2 and 3). A direct relationship between low frequency impedance response and hysteresis has been recently proposed by Ebadi et al.61 Their results showed that devices with a negative capacitive response at low frequencies were more likely to show hysteretic behavior with a stronger dependence on scan rate. However, our results show that changes in the impedance response at low frequencies are not necessarily observed in hysteresis measurements, and vice versa. Although both, low-frequency impedance10,31,33,63,64 and hysteresis12,13,26 measurements, are usually related with ion migration, these measurements are fundamentally different.
For the impedance measurements we apply a small potential perturbation (10 mV) to ensure linearity on the system;31,78 for hysteresis measurements the changes in potential are larger (from Voc to Jsc and vice versa) and we are no longer under steady state conditions. Care needs to be taken when comparing steady state and dynamic measurements. However, it is interesting to roughly compare ‘timescales’. The impedance results for the low frequency semicircle appear at frequencies below 100 Hz. If the amplitude of the small perturbation is 10 mV, it means that at 1 Hz there will be a change of 20 mV, e.g., when the measurement frequency is 1 Hz, we have a potential change of 20 mV s−1. At 100 Hz the change in potential will be 2000 mV s−1, which means that the low frequency semicircle is related with changes in potential of approximately 2000 mV s−1 and lower, which will include most of the scan rates employed for the hysteresis measurements in this study. However, with impedance measurements we observe a clear change with the inclusion of bromide, while for hysteresis measurements there are only small differences. In some of the hysteresis measurements it is possible to observe two different peaks in the degree of hysteresis, possibly related with two processes with different time constants, as previously reported by Bryant et al.77
All our evidence supports the argument that the low frequency process in impedance is related to ion motion, as previously reported in the literature.10,31,63,64 We go a step further and relate this process with iodide migration, a result which is supported by muon spectroscopy. Bromide substitution strongly affects both the impedance measurements and the muon spectroscopy; it does not affect hysteresis. There is considerable evidence to suggest that ion migration causes hysteresis. However, the nature of the hysteresis process is clearly more complicated.
There are several possible reasons for the differences between IS and hysteresis measurements. As mentioned above, we believe that faster ion migration at grain boundaries/interfaces can still occur in all the devices and the easy motion of surface ions in response to the applied voltage could explain IV hysteresis. IV curve hysteresis has also been more closely linked to the nature of the hole and electron selective contacts used in the cells, and it is possible that it is the interaction of surface ions with the contact that is causing hysteresis.
Conclusions
Our results show that by incorporating small amounts of bromide into the perovskite structure, the low frequency impedance response is strongly affected. We attribute this change to a suppression of bulk iodide motion, probably due to local distortions/contractions in the crystal structure with the introduction of small Br ions as increasing Br substitution increases the activation energy for iodide motion. We show there is a critical bromide concentration after which the iodide migration can no longer be observed. Muon spin relaxation measurements support the conclusion that the incorporation of bromide in the perovskite inhibits bulk iodide motion. In contrast, hysteresis measurements show that the scan rate at which the hysteresis is largest is not related to the amount of bromide inside the perovskite. Although both are influenced by ion motion, hysteresis and impedance measurements appear to provide different information about the perovskite device. By increasing understanding of two of the most common techniques used to measure perovskite solar cells, IV curves measurements and impedance spectroscopy, we are moving one step closer to understanding the fundamental processes in perovskite devices.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Rodrigo García-Rodríguez gratefully acknowledges CONACYT for support through a postdoctoral scholarship. Sam Pering was supported by the University of Bath, Dominic Ferdani was supported by the Centre for Doctoral Training in Sustainable Chemical Technologies (studentship under grant EP/L016354/1). We thank the STFC for the allocated muon beamtime. Muon data is available electronically at the following site https://doi.org/10.5286/ISIS.E.RB1810384. Additional data that support the findings of this study are available from the corresponding author, PJC, upon reasonable requestNotes and references
- A. Kojima, K. Teshima, Y. Shirai and T. Miyasaka, J. Am. Chem. Soc., 2009, 131, 6050–6051 CrossRef CAS.
- M. A. Green, Y. Hishikawa, E. D. Dunlop, D. H. Levi, H.-E. Jochen, Y. Masahiro and A. W. Y. Ho-Baillie, Prog. Photovolt. Res. Appl., 2019, 27, 3–12 CrossRef.
- W. S. Yang, J. H. Noh, J. J. Nam, Y. C. Kim, S. Ryu, J. Seo and S. Il Seok, Science, 2015, 348, 1234–1237 CrossRef CAS.
- M. A. Green, E. D. Dunlop, D. H. Levi, J. Hohl-Ebinger, M. Yoshita and A. W. Y. Ho-Baillie, Prog. Photovolt. Res. Appl., 2019, 27, 565–575 CrossRef.
- J. Ramanujam and U. P. Singh, Energy Environ. Sci., 2017, 10, 1306–1319 RSC.
- T. D. Lee and A. U. Ebong, Renewable Sustainable Energy Rev., 2017, 70, 1286–1297 CrossRef CAS.
- L. Lu, T. Zheng, Q. Wu, A. M. Schneider, D. Zhao and L. Yu, Chem. Rev., 2015, 115, 12666–12731 CrossRef CAS PubMed.
- A. Hagfeldt, G. Boschloo, L. Sun, L. Kloo and H. Pettersson, Chem. Rev., 2010, 110, 6595–6663 CrossRef CAS PubMed.
- B. E. Hardin, J. Snaith and M. D. Mcgehee, Nat. Photonics, 2012, 6, 162–169 CrossRef CAS.
- L. Contreras, J. Idígoras, A. Todinova, M. Salado, S. Kazim, S. Ahmad and J. A. Anta, Phys. Chem. Chem. Phys., 2016, 18, 31033–31042 RSC.
- C. Eames, J. M. Frost, P. R. F. Barnes, B. C. O'Regan, A. Walsh and M. S. Islam, Nat. Commun., 2015, 6, 1–8 Search PubMed.
- M. L. Petrus, J. Schlipf, C. Li, T. P. Gujar, N. Giesbrecht, P. Müller-Buschbaum, M. Thelakkat, T. Bein, S. Hüttner and P. Docampo, Adv. Energy Mater., 2017, 7, 1700264 CrossRef.
- I. Levine, P. K. Nayak, J. T. W. Wang, N. Sakai, S. Van Reenen, T. M. Brenner, S. Mukhopadhyay, H. J. Snaith, G. Hodes and D. Cahen, J. Phys. Chem. C, 2016, 120, 16399–16411 CrossRef CAS.
- E. Mosconi, D. Meggiolaro, H. J. Snaith, S. D. Stranks and F. De Angelis, Energy Environ. Sci., 2016, 9, 3180–3187 RSC.
- Y. Yuan and J. Huang, Acc. Chem. Res., 2016, 49, 286–293 CrossRef CAS PubMed.
- A. F. Gualdrón-Reyes, S. J. Yoon, E. M. Barea, S. Agouram, V. Muñoz-Sanjosé, Á. M. Meléndez, M. E. Niño-Gómez and I. Mora-Seró, ACS Energy Lett., 2018, 54–62 Search PubMed.
- J. H. Noh, S. H. Im, J. H. Heo, T. N. Mandal and S. Il Seok, Nano Lett., 2013, 13, 1764–1769 CrossRef CAS PubMed.
- N. Pellet, J. Teuscher, J. Maier and M. Grätzel, Chem. Mater., 2015, 27, 2181–2188 CrossRef CAS.
- O. A. Jaramillo-Quintero, R. S. Sánchez, M. Rincón and I. Mora-Sero, J. Phys. Chem. Lett., 2015, 6, 1883–1890 CrossRef CAS PubMed.
- B. Zhao, S. Bai, V. Kim, R. Lamboll, R. Shivanna, F. Auras, J. M. Richter, L. Yang, L. Dai, M. Alsari, X. J. She, L. Liang, J. Zhang, S. Lilliu, P. Gao, H. J. Snaith, J. Wang, N. C. Greenham, R. H. Friend and D. Di, Nat. Photonics, 2018, 12, 783–789 CrossRef CAS.
- K. A. Bush, A. F. Palmstrom, Z. J. Yu, M. Boccard, R. Cheacharoen, J. P. Mailoa, D. P. McMeekin, R. L. Z. Hoye, C. D. Bailie, T. Leijtens, I. M. Peters, M. C. Minichetti, N. Rolston, R. Prasanna, S. Sofia, D. Harwood, W. Ma, F. Moghadam, H. J. Snaith, T. Buonassisi, Z. C. Holman, S. F. Bent and M. D. McGehee, Nat. Energy, 2017, 2, 1–7 Search PubMed.
- E. E. Giles, L. Tomas, B. A. Kevin, P. Rohit, G. Thomas, W. Jacob Tse-Wei, M. P. David, V. George, M. L. Rebecca, M. Richard, P. Axel, S. J. Daniel, B. A. Rebecca, P. B. Jay, P. S. Elizabeth, S. J. Rebecca, M. Wen, M. Farhad, C. Bert, B. Aslihan, B. Hans-Gerd, B. Stacey, G. Feliciano, H. M. Laura, J. B. Michael, M. D. Michael and S. J. Henry, Science, 2016, 354, 861 CrossRef PubMed.
- H. S. Kim, J. Y. Seo and N. G. Park, ChemSusChem, 2016, 9, 2528–2540 CrossRef CAS PubMed.
- S. Yang, W. Fu, Z. Zhang, H. Chen and C. Z. Li, J. Mater. Chem. A, 2017, 5, 11462–11482 RSC.
- J. Berry, T. Buonassisi, D. A. Egger, G. Hodes, L. Kronik, Y. L. Loo, I. Lubomirsky, S. R. Marder, Y. Mastai, J. S. Miller, D. B. Mitzi, Y. Paz, A. M. Rappe, I. Riess, B. Rybtchinski, O. Stafsudd, V. Stevanovic, M. F. Toney, D. Zitoun, A. Kahn, D. Ginley and D. Cahen, Adv. Mater., 2015, 27, 5102–5112 CrossRef CAS PubMed.
- W. Tress, N. Marinova, T. Moehl, S. M. Zakeeruddin, M. K. Nazeeruddin and M. Grätzel, Energy Environ. Sci., 2015, 8, 995–1004 RSC.
- C. Eames, J. M. Frost, P. R. F. Barnes, B. C. O'Regan, A. Walsh and M. S. Islam, Nat. Commun., 2015, 6, 1–8 Search PubMed.
- M. De Bastiani, G. Dell'Erba, M. Gandini, V. D'Innocenzo, S. Neutzner, A. R. S. Kandada, G. Grancini, M. Binda, M. Prato, J. M. Ball, M. Caironi and A. Petrozza, Adv. Energy Mater., 2016, 6, 1–9 Search PubMed.
- J. M. Azpiroz, E. Mosconi, J. Bisquert and F. De Angelis, Energy Environ. Sci., 2015, 8, 2118–2127 RSC.
- Y. Yuan, Q. Wang, Y. Shao, H. Lu, T. Li, A. Gruverman and J. Huang, Adv. Energy Mater., 2016, 6, 1501803 CrossRef.
- A. Pockett, G. Eperon, N. Sakai, H. Snaith, L. M. Peter and P. J. Cameron, Phys. Chem. Chem. Phys., 2017, 19, 5959–5970 RSC.
- S. Meloni, T. Moehl, W. Tress, M. Franckevicius, M. Saliba, Y. H. Lee, P. Gao, M. K. Nazeeruddin, S. M. Zakeeruddin, U. Rothlisberger and M. Graetzel, Nat. Commun., 2016, 7, 10334 CrossRef CAS PubMed.
- D. Ferdani, S. Pering, D. Ghosh, P. Kubiak, A. Walker, S. E. Lewis, A. L. Johnson, P. J. Baker, S. Islam and P. J. Cameron, Energy Environ. Sci., 2019, 12, 2264–2272 RSC.
- Y. Yuan, J. Chae, Y. Shao, Q. Wang, Z. Xiao, A. Centrone and J. Huang, Adv. Energy Mater., 2015, 5, 1500615 CrossRef.
- Z. Xiao, Y. Yuan, Y. Shao, Q. Wang, Q. Dong, C. Bi, P. Sharma, A. Gruverman and J. Huang, Nat. Mater., 2015, 14, 193–198 CrossRef CAS PubMed.
- C. Lin, S. Li, W. Zhang, C. Shao and Z. Yang, ACS Appl. Energy Mater., 2018, 1, 1374–1380 CrossRef CAS.
- J. H. Noh, S. H. Im, J. H. Heo, T. N. Mandal and S. Il Seok, Nano Lett., 2013, 13, 1764–1769 CrossRef CAS PubMed.
- R. Ruess, F. Benfer, F. Böcher, M. Stumpp and D. Schlettwein, ChemPhysChem, 2016, 17, 1505–1511 CrossRef CAS PubMed.
- S. J. Yoon, M. Kuno and P. V. Kamat, ACS Energy Lett., 2017, 2, 1507–1514 CrossRef CAS.
- M. Hu, C. Bi, Y. Yuan, Y. Bai and J. Huang, Adv. Sci., 2015, 3, 6–11 Search PubMed.
- E. T. Hoke, D. J. Slotcavage, E. R. Dohner, A. R. Bowring, H. I. Karunadasa and M. D. McGehee, Chem. Sci., 2015, 6, 613–617 RSC.
- H.-S. Kim, I.-H. Jang, N. Ahn, M. Choi, A. Guerrero, J. Bisquert and N.-G. Park, J. Phys. Chem. Lett., 2015, 6, 4633–4639 CrossRef CAS PubMed.
- J. Xu, A. Buin, A. H. Ip, W. Li, O. Voznyy, R. Comin, M. Yuan, S. Jeon, Z. Ning, J. J. McDowell, P. Kanjanaboos, J. P. Sun, X. Lan, L. N. Quan, D. H. Kim, I. G. Hill, P. Maksymovych and E. H. Sargent, Nat. Commun., 2015, 6, 1–8 RSC.
- Y. Shao, Z. Xiao, C. Bi, Y. Yuan and J. Huang, Nat. Commun., 2014, 5, 5784 CrossRef CAS PubMed.
- J. I. Pankove, Optical Processes in Semiconductors, Dover, New Jersey, 1971 Search PubMed.
- R. G. Niemann, A. G. Kontos, D. Palles, E. I. Kamitsos, A. Kaltzoglou, F. Brivio, P. Falaras and P. J. Cameron, J. Phys. Chem. C, 2016, 120, 2509–2519 CrossRef CAS.
- B. W. Park, B. Philippe, S. M. Jain, X. Zhang, T. Edvinsson, H. Rensmo, B. Zietz and G. Boschloo, J. Mater. Chem. A, 2015, 3, 21760–21771 RSC.
- S. Yue, K. Liu, R. Xu, M. Li, M. Azam, K. Ren, J. Liu, Y. Sun, Z. Wang, D. Cao, X. Yan, S. Qu, Y. Lei and Z. Wang, Energy Environ. Sci., 2017, 10, 2570–2578 RSC.
- R. G. Niemann, A. G. Kontos, D. Palles, E. I. Kamitsos, A. Kaltzoglou, F. Brivio, P. Falaras and P. J. Cameron, J. Phys. Chem. C, 2016, 120, 2509–2519 CrossRef CAS.
- G. A. Nemnes, C. Besleaga, V. Stancu, D. E. Dogaru, L. N. Leonat, L. Pintilie, K. Torfason, M. Ilkov, A. Manolescu and I. Pintilie, J. Phys. Chem. C, 2017, 121, 11207–11214 CrossRef CAS.
- O. Almora, P. Lopez-Varo, K. T. Cho, S. Aghazada, W. Meng, Y. Hou, C. Echeverría-Arrondo, I. Zimmermann, G. J. Matt, J. A. Jiménez-Tejada, C. J. Brabec, M. K. Nazeeruddin and G. Garcia-Belmonte, Sol. Energy Mater. Sol. Cells, 2019, 195, 291–298 CrossRef CAS.
- L. Contreras-Bernal, M. Salado, A. Todinova, L. Calio, S. Ahmad, J. Idígoras and J. A. Anta, J. Phys. Chem. C, 2017, 121, 9705–9713 CrossRef CAS.
- E. Guillén, F. J. Ramos, J. A. Anta and S. Ahmad, J. Phys. Chem. C, 2014, 118, 22913–22922 CrossRef.
- L. Contreras-Bernal, C. Aranda, M. Valles-Pelarda, T. T. Ngo, S. Ramos-Terrón, J. J. Gallardo, J. Navas, A. Guerrero, I. Mora-Sero, J. Idigoras and J. A. Anta, J. Phys. Chem. C, 2018, 122, 5341–5348 CrossRef CAS.
- A. Pockett, G. E. Eperon, T. Peltola, H. J. Snaith, A. Walker, L. M. Peter and P. J. Cameron, J. Phys. Chem. C, 2015, 119, 3456–3465 CrossRef CAS.
- B. Suarez, V. Gonzalez-Pedro, T. S. Ripolles, R. S. Sanchez, L. Otero and I. Mora-Sero, J. Phys. Chem. Lett., 2014, 5, 1628–1635 CrossRef CAS PubMed.
- K. Cao, H. Li, S. S. Liu, J. Cui, Y. Shen and M. Wang, Nanoscale, 2016, 8, 8839–8846 RSC.
- I. Zarazua, G. Han, P. P. Boix, S. Mhaisalkar, F. Fabregat-Santiago, I. Mora-Seró, J. Bisquert and G. Garcia-Belmonte, J. Phys. Chem. Lett., 2016, 7, 5105–5113 CrossRef CAS PubMed.
- I. Zarazúa, S. Sidhik, T. Lopéz-Luke, D. Esparza, E. De La Rosa, J. Reyes-Gomez, I. Mora-Seró and G. Garcia-Belmonte, J. Phys. Chem. Lett., 2017, 8, 6073–6079 CrossRef PubMed.
- A. R. Pascoe, N. W. Duffy, A. D. Scully, F. Huang and Y. B. Cheng, J. Phys. Chem. C, 2015, 119, 4444–4453 CrossRef CAS.
- F. Ebadi, N. Taghavinia, R. Mohammadpour, A. Hagfeldt and W. Tress, Nat. Commun., 2019, 10, 1574 CrossRef PubMed.
- S. D. Stranks, G. E. Eperon, G. Grancini, C. Menelaou, M. J. P. Alcocer, T. Leijtens, L. M. Herz, A. Petrozza and H. J. Snaith, Science, 2013, 342, 341–344 CrossRef CAS PubMed.
- A. Guerrero, G. Garcia-Belmonte, I. Mora-Sero, J. Bisquert, Y. S. Kang, T. J. Jacobsson, J. P. Correa-Baena and A. Hagfeldt, J. Phys. Chem. C, 2016, 120, 8023–8032 CrossRef CAS.
- M. Bag, L. A. Renna, R. Y. Adhikari, S. Karak, F. Liu, P. M. Lahti, T. P. Russell, M. T. Tuominen and D. Venkataraman, J. Am. Chem. Soc., 2015, 137, 13130–13137 CrossRef CAS PubMed.
- D. Moia, I. Gelmetti, P. Calado, W. Fisher, M. Stringer, O. Game, Y. Hu, P. Docampo, D. Lidzey, E. Palomares, J. Nelson and P. R. F. F. Barnes, Energy Environ. Sci., 2019, 12, 1296–1308 RSC.
- D. A. Jacobs, H. Shen, F. Pfeffer, J. Peng, T. P. White, F. J. Beck and K. R. Catchpole, J. Appl. Phys., 2018, 124, 225702 CrossRef.
- K. L. Svane, A. C. Forse, C. P. Grey, G. Kieslich, A. K. Cheetham, A. Walsh and K. T. Butler, J. Phys. Chem. Lett., 2017, 8, 6154–6159 CrossRef CAS PubMed.
- S. J. Yoon, K. G. Stamplecoskie and P. V. Kamat, J. Phys. Chem. Lett., 2016, 7, 1368–1373 CrossRef CAS PubMed.
- M. Abdi-Jalebi, Z. Andaji-Garmaroudi, S. Cacovich, C. Stavrakas, B. Philippe, J. M. Richter, M. Alsari, E. P. Booker, E. M. Hutter, A. J. Pearson, S. Lilliu, T. J. Savenije, H. Rensmo, G. Divitini, C. Ducati, R. H. Friend and S. D. Stranks, Nature, 2018, 555, 497–501 CrossRef CAS PubMed.
- J. Sugiyama, K. Mukai, H. Nozaki, M. Harada, K. Kamazawa, Yutakaikedo, M. Månsson, O. Ofer, E. J. Ansaldo, J. H. Brewer, K. H. Chow, Isaowatanabe, Y. Miyake and T. Ohzuku, Phys. Procedia, 2012, 30, 105–108 CrossRef CAS.
- M. Månsson and J. Sugiyama, Phys. Scr., 2013, 88, 068509 CrossRef.
- J. Sugiyama, K. Mukai, Y. Ikedo, H. Nozaki, M. Månsson and I. Watanabe, Phys. Rev. Lett., 2009, 103, 147601 CrossRef PubMed.
- T. E. Ashton, J. V. Laveda, D. A. MacLaren, P. J. Baker, A. Porch, M. O. Jones and S. A. Corr, J. Mater. Chem. A, 2014, 2, 6238 RSC.
- D. Ghosh, P. Walsh Atkins, M. S. Islam, A. B. Walker and C. Eames, ACS Energy Lett., 2017, 2, 2424–2429 CrossRef CAS.
- A. Pockett and M. J. Carnie, ACS Energy Lett., 2017, 2, 1683–1689 CrossRef CAS.
- S. Van Reenen, M. Kemerink and H. J. Snaith, J. Phys. Chem. Lett., 2015, 6, 3808–3814 CrossRef CAS PubMed.
- D. Bryant, S. Wheeler, B. C. O'Regan, T. Watson, P. R. F. Barnes, D. Worsley and J. Durrant, J. Phys. Chem. Lett., 2015, 6, 3190–3194 CrossRef CAS.
- D. Pitarch-Tena, T. T. Ngo, M. Vallés-Pelarda, T. Pauporté and I. Mora-Seró, ACS Energy Lett., 2018, 3, 1044–1048 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ta08848b |
| This journal is © The Royal Society of Chemistry 2019 |