
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Selective acid leaching: a simple way to engineer cobalt oxide nanostructures for the electrochemical oxygen evolution reaction†
Mingquan
Yu
a,
Kendra Solveig
Belthle
a,
Cengiz
Tüysüz
b and
Harun
Tüysüz
 *a
*a
aMax-Planck-Institut für Kohlenforschung, Kaiser-Wilhelm-Platz 1, Mülheim an der Ruhr, D-45470, Germany. E-mail: tueysuez@kofo.mpg.de
bFaculty of Education, Uşak University, Turkey
First published on 18th September 2019
Abstract
Developing a simple and cost-effective strategy to construct earth-abundant catalysts is in high demand for diverse applications. Herein, a general and facile strategy is developed to engineer cobalt oxide nanostructures via selective acid leaching for the electrochemical oxygen evolution reaction (OER). A leaching process is implemented to selectively remove CoMoO4 by treating mixed Co–Mo oxides in diluted hydrochloric acid solution, resulting in the formation of sub-5 nm particles and a threefold increase in the specific surface area (up to 150 m2 g−1). The leached oxides exhibit superior OER activity to pristine oxides as a result of (i) a larger surface area, (ii) phase purification to expose more active Co3O4 species to the reactant, and (iii) faster charge transfer kinetics for the OER. This strategy can be also applied to a broader range of earth-abundant metals, where a second metal (Li, Ca, and Mg) is selectively leached out, which results in a material with a larger surface area and enhanced catalytic performance for the OER. Moreover, various metal oxides with a high surface area, such as NiO and Fe2O3, can be prepared via this simple synthetic method. This work will pave a new practical way for the production of high surface area catalysts for diverse applications.
1. Introduction
A high demand for renewable energy has arisen due to limited fossil fuel resources, which have been used as the main energy source and causing environmental issues. As a clean energy carrier that has the capacity to satisfy the global energy demand, hydrogen can be generated by electrochemical water splitting by utilizing solar energy without CO2 emission.1–4 The bottleneck of this technique for its commercialization lies on the anode side where oxygen is produced, namely the oxygen evolution reaction (OER).5,6 As OER possesses sluggish kinetics as a result of the formation of oxygen–oxygen bonds and four-electron transfer, an efficient catalyst is required to lower the overpotential and energy loss for the OER.6–9 The most efficient OER catalysts have been identified as IrO2 and RuO2, which are fabricated into thin films and employed in industrial proton exchange membrane water electrolyzers.7,10,11 However, these are not ideal catalysts as they suffer from lack of stability and durability under a high potential.8,12,13 Moreover, the scarcity and high cost of the noble metals (Ir and Ru) make them impractical for large scale water electrolysis. Therefore, an urgent need for economic hydrogen production is to develop alternative catalysts, which are composed of earth abundant-elements and catalyze the OER with high efficiency and long-term stability.Over the past years, research has focused on studying first-row (3d) transition metal-based catalysts to minimize the energy input for the OER.6,10,14,15 Accordingly, various categories of OER catalysts have been developed, including 3d metal-based oxides,7,16,17 (oxy)hydroxides,17–21 phosphides,22–25 borates,26,27 and perovskites.28,29 Among these, Co3O4 has drawn particular interest not only due to its abundance and good catalytic performance,30–33 but also because it can be easily constructed into desirable morphologies and shapes.34,35 There are diverse approaches to fabricate Co3O4 into nanostructures with a high specific surface area (SSA), which is a key parameter for escalating the reaction rate of the OER.30,34 Esswein et al. prepared Co3O4 nanoparticles with average sizes of 5.9 nm, 21.1 nm, and 46.9 nm through the hydrothermal method where the highest SSA and OER activities were obtained for the smallest nanoparticles.30 Nanocasting is a very effective method to fabricate well-defined nanostructures. Our group has utilized silica-based templates to prepare ordered mesoporous Co3O4 with a crystallite size of ∼8 nm and an SSA of ∼105 m2 g−1.4,36 Recently, laser technology has been employed to synthesize ∼5 nm Co3O4 nanoparticles via ablation or fragmentation of a bulk cobalt precursor in a liquid solution.37,38 However, these methodologies are not practical since they suffer from (1) high production cost, (2) low yield, and (3) complicated procedures. To overcome these drawbacks, our group has designed a facile route using bio-waste templates to produce Co3O4 nanoparticles with large scalability. However, the bio-waste templated Co3O4 possessed an unsatisfactory SSA of ∼50 m2 g−1 and lower OER activity compared with ordered mesoporous Co3O4.39 In this regard, it is desirable to develop an approach to engineer the Co3O4 nanostructure to enhance the SSA which can provide a larger number of catalytically active centers.
In this study, we reported a simple selective acid leaching strategy to engineer the Co3O4 nanostructures for electrochemical water oxidation. Through a coffee-waste (CW) templating method by following a simple impregnation-calcination process,39 mesoporous Co–Mo oxides were prepared with a mixed crystal phase of Co3O4 and CoMoO4. With post-treatment, the CoMoO4 phase was selectively removed by using diluted HCl solution, which leads to the formation of sub-5 nm Co3O4 nanoparticles and more than threefold enhancement of the SSA, up to 150 m2 g−1. The leached oxides exhibited enhanced OER activity due to a larger SSA and more active Co3O4 species which were exposed to the reactant. Additionally, we demonstrated that this leaching strategy can be also applied to selectively remove other earth-abundant metals from mixed Co-M oxides (M = Li, Ca, and Mg). The developed method is versatile and can be also applied to prepare nanoscale Fe2O3 and NiO with high surface areas.
2. Experimental section
2.1 Synthesis of coffee-waste templated metal oxides
Coffee-waste templated metal oxides were prepared through a simple impregnation-calcination procedure.39 Co(NO3)2·6H2O (>98%, Honeywell Fluka) and H3(P(Mo3O10)4)·H2O (ACS reagent, Aldrich) were used as metal precursors in this study, with a designed Co/Mo ratio (2/1, 4/1, 8/1 and 16/1) well mixed for the synthesis of Co–Mo oxides. Coffee waste (TIZIO Premium Caffè) was collected from the tray of a coffee machine and washed with hot water until it became colorless, and afterwards was used as the template. The weight ratio of the coffee waste template to metal precursors was maintained at 2/1. For a small batch, 4 g of pre-washed coffee waste was added to the metal precursor solution for impregnation, whereas 20 g of pre-washed coffee waste was used as the template for large batch synthesis of Co4Mo oxides. After drying, the composite materials were calcined at 400 °C for 4 h with a ramping rate of 2 °C min under air.To prepare Co4M oxides (M = Li, Mg, and Ca), LiNO3 (>95%, Riedel-de Haën), Mg(NO3)2·6H2O (>99%, Honeywell Fluka), and Ca(NO3)2·6H2O (>99%, Alfa Aesar) were used as precursors and mixed with Co(NO3)2·6H2O with a Co/M ratio of 4/1, followed by impregnation into 4 g of pre-washed coffee and the same calcination step.
For the synthesis of Ni4Ca oxides and Fe4Ca oxides, Ni(NO3)2·6H2O (>98%, Sigma-Aldrich) and Fe(NO3)3·9H2O (>98%, Sigma-Aldrich) were used as precursors and mixed with Ca(NO3)2·6H2O with a M/Ca ratio of 4/1, followed by the same impregnation and calcination steps.
2.2 Leaching process
Concentrated HCl solution (37–38%, J. T. Baker) was diluted with distilled water to prepare leaching solutions of various concentrations (0.01 M, 0.05 M, 0.1 M and 0.2 M). The powder samples were then added to the diluted acid solution and kept shaking at a speed of 480 rpm for a certain period of time (5 min, 30 min, 2 h and 6 h). Afterwards, the leached oxides were collected by centrifugation and washed with water, followed by drying in air at 80 °C overnight.2.3 Characterization
Powder X-ray diffraction (XRD) patterns were collected at room temperature on a Stoe theta/theta diffractometer in Bragg–Brentano geometry using Cu K Kα1/2 radiation. Transmission electron microscopy (TEM) images of the samples were captured at 100 kV using an H-7100 electron microscope from Hitachi. High resolution TEM (HR-TEM) and scanning TEM (STEM) images were taken on HF-2000 and Hitachi S-5500 microscopes, respectively. SEM-EDX measurements were performed using a Hitachi S-3500N electron microscope. N2-sorption isotherms were measured using a 3Flex Micrometrics at 77 K. Prior to the measurements, the samples were degassed under vacuum at a temperature of 150 °C for 10 h. Brunauer–Emmett–Teller (BET) surface areas were determined in the relative pressure range between 0.06 and 0.2. X-ray photoelectron spectroscopy (XPS) measurements were carried out with a SPECS GmbH spectrometer with a hemispherical analyzer (PHOIBOS 150 1D-DLD). The monochromatic Al Kα X-ray source (E = 1486.6 eV) was operated at 100 W. An analyzer pass energy of 20 eV was applied for the narrow scans. The medium area mode was used as the lens mode. The base pressure during the experiment in the analysis chamber was 5 × 10−10 mbar. The binding energy scale was corrected for surface charging by using the C 1s peak of contaminant carbon as the reference at 284.5 eV.2.4 Electrochemical measurements
Electrochemical measurements were carried out in a typical three-electrode configuration using a rotating disc electrode (Model: AFMSRCE, PINE Research Instrumentation), and a hydrogen reference electrode (HydroFlex, Gaskatel) and Pt wire were used as the reference electrode and counter electrode, respectively. 1 M KOH (85%, VWR Chemicals) solution was filled in a Teflon cell as the electrolyte. Argon was continuously purged through the cell to remove oxygen during the measurement. The temperature of the electrolyte was kept at 25 °C using a water circulation system. Working electrodes were fabricated by depositing catalysts on the center circle of glass carbon (GC) electrodes (PINE, 5 mm diameter, 0.196 cm2 area). Before depositing materials, the surface of each GC electrode was polished with an Al2O3 suspension (1.0 and 0.05 μm, Allied High Tech Products, Inc.) and thoroughly cleaned with distilled water by sonication for 5 min. To prepare the catalyst ink, 4.8 mg of the catalyst powder was dispersed in a mixed solution containing 0.75 mL of H2O, 0.25 mL of 2-propanol and 50 μL of Nafion (5% in a mixture of water and alcohol). Then the solution mixture was put in a sonication bath for 30 min to disperse the solid catalyst in the solution. After that, 5.25 μL of the catalyst ink was dropped onto the GC electrode (catalyst loading, 0.12 mg cm−2) and dried under light irradiation for 10 min. The GC electrode was then connected to the rotating disc electrode for OER measurements.Linear sweep voltammetry (LSV) curves were measured by sweeping the potential from 0.7 V to 1.7 V vs. RHE with a scan rate of 10 mV s−1. Cyclic voltammetry (CV) measurements were carried out in a potential range between 0.7 and 1.6 vs. RHE with a scan rate of 50 mV s−1. Impedance spectra were measured at an anodic potential of 1.6 V vs. RHE, and the spectra were collected from 105 Hz to 0.1 Hz with an amplitude of 5 mV. The electrochemical active surface area (ECSA) was estimated by measuring the capacitive current in a non-faradaic region. Briefly, CV with increasing scan rates ranging from 20 to 180 mV s−1 was performed in the potential window from 1.2 to 1.3 V vs. RHE. By plotting the capacitive current against the scan rate, the double layer capacitance (Cdl) is obtained as half of the slope and the ECSA can be derived from the equation ECSA = Cdl/Cs, where Cs is the specific capacitance and its value is chosen as 0.04 mF cm−2 in alkaline solution.10 To perform the long-term stability test, an electrode was fabricated by drop-casting the catalyst ink onto the surface of carbon fiber paper (0.5 × 1 cm2) as well, where the catalyst loading was kept at 0.5 mg cm−2. A chronopotentiometric test was performed by using controlled current electrolysis at a constant current density of 10 mA cm−2. In all measurements, the IR drop was compensated at 85%, and a rotating disc electrode configuration was maintained at a rotation speed of 2000 rpm for the measurements of GC electrodes.
3. Results and discussion
As illustrated in the scheme (Fig. 1a), the process of fabricating ultra-small Co3O4 nanoparticles can be divided into two steps. The first step is to synthesize mesoporous Co–Mo oxides through a hard-templating method by using coffee-waste grounds as a sustainable template.39 Briefly, CW templates were impregnated with an aqueous solution containing the desired ratio of metal precursors, followed by calcination/combustion to remove the carbon-based template. Before the combustion of the carbon based template, metal precursors are converted to crystallize the mixed oxides, i.e. Co3O4 and CoMoO4. | ||
| Fig. 1 (a) Schematic illustration of the acid-leaching process to engineer the structure of CW-templated Co–Mo oxides for the electrocatalytic OER. Wide-angle XRD patterns of CW-templated Co–Mo oxides and Co3O4 before (b) and after (c) leaching in 0.1 M HCl solution for 2 h. | ||
Transition-metal molybdates can possess three different monoclinic structures (α, β, and γ) depending on the synthetic conditions. The γ-CoMoO4-type structure appears at high pressure (above 60 kbar) and crystallizes into a relatively dense structure. On the other hand, experimentally observed CoMoO4 generally has α- and β-phases, which have a more open structure on the basis of defective cubic close-packing of oxygen atoms.40,41 β to α phase transition is observed at elevated temperatures (above 700 °C).40,41 The low-temperature form, i.e. the β-CoMoO4 phase, has a crystal structure where Co2+ and Mo6+ cations are located in distorted octahedral and tetrahedral sites, respectively, which makes it unstable under harsh conditions.41,42 Thus, an acid-driven leaching process can be applied to remove the cobalt molybdenum oxide phase selectively. For this, a leaching process was carried out by immersing the cobalt molybdenum oxide powders in diluted HCl solution at room temperature. The CoMoO4 phase, which should be a β-phase after calcination at 400 °C, was selectively leached out in an acid environment. We attribute the leaching to the open and disordered structure of β-CoMoO4 that is not stable in the presence of acid, where the defective oxygen sites are easily coupled with protons leading to the dissolution of the CoMoO4 phase, whereas Co3O4 with a compact spinel structure is more resistant to acid corrosion.43
X-ray diffraction (XRD) analysis was first conducted to study the leaching effect on the composition of cobalt molybdenum mixed oxides with Co/Mo ratios of 16, 8, 4, and 2. As shown in the XRD patterns of pristine oxides (Fig. 1b), distinct reflections at 2θ = 19.0°, 31.3°, 36.8°, and 44.8° are exhibited on CW-templated Co3O4, representing the (111), (220), (311), and (400) facets of the Co3O4 spinel structure. Due to the much larger radius of the Mo cation compared to the Co cation, the incorporation of Mo into the Co3O4 spinel via substituting tetrahedral Co2+ or octahedral Co3+ is unlikely. Thus, a second crystal phase of CoMoO4 appeared with the incorporation of Mo into the mixed oxides. The emerged diffraction peaks at 2θ = 23.3° and 26.5° corresponded to the (021) and (002) planes of β-CoMoO4, respectively.41 It is worth noting that the diffraction peaks of the CoMoO4 phase are sharper than those of the Co3O4 phase, suggesting that CoMoO4 crystalized into larger crystallites. This should be related to a higher decomposition temperature of the Mo precursor (phosphomolybdic acid) compared to that of the Co precursor (cobalt nitrate). It has been reported that the decomposition of phosphomolybdic acid occurs at a temperature above 400 °C,44,45 while cobalt nitrate could be completely decomposed into Co3O4 at 210 °C.46 As a result, phosphomolybdic acid was decomposed at high temperature and crystallized with Co3O4 into larger CoMoO4 crystals. In the case of the mixed oxides with the lowest Mo ratio (Co/Mo 16/1), the XRD patterns displayed reflections at the same angles as those of pure Co3O4, where no additional reflections were observed belonging to CoMoO4. We attribute this to a small amount of CoMoO4 that was not detected by XRD. Upon a leaching process in 0.1 M HCl solution for 2 h, all the mixed oxides exhibited a pure Co3O4 spinel phase (Fig. 1c), which endorses selective leaching of the CoMoO4 phase.
To further confirm the selective leaching of Mo species, elemental compositions of all the oxides were determined by energy-dispersive X-ray spectroscopy (EDX) before and after the leaching process, and the results are summarized in Fig. S1–S3 and Tables S1, S2.† The actual ratio of Co/Mo in the mixed oxides was relatively well-matched with the stoichiometry of Co and Mo precursors, as shown in Fig. S2 and Table S1.† For the leached mixed oxides, dramatic loss of Mo was determined after acid-leaching, leading to a significantly higher ratio of Co/Mo. This confirms that Mo species were selectively removed under the acidic conditions (Fig. S3 and Table S2†), which is in line with the XRD measurements. Moreover, this acid leaching process was able to wash out most impurities from the coffee waste grounds (Tables S1 and S2†). This is crucial for determining the catalytic performance of the oxides as it helps minimize the effect from impurities.
After confirming the selective leaching of Mo by structural and elemental analyses, its effect on the textural parameters was evaluated by N2 physisorption measurements. The specific surface areas (SSAs) were calculated from the adsorption isotherms via the Brunauer–Emmett–Teller (BET) method. As shown in Fig. S4,† a typical type IV isotherm for mesoporous materials was observed for all the Co–Mo oxides before and after the acid leaching process. Fig. 2 summarizes the calculated BET surface areas of oxides before and after the leaching process.
 | ||
| Fig. 2 Summarized values of the BET surface area (left axis) and incremental factors on the BET surface area (right axis) of CW-templated Co–Mo oxides and Co3O4 before and after leaching in 0.1 M HCl solution for 2 h. | ||
The pristine oxides with various Mo contents showed a similar SSA in the range of 36–55 m2 g−1 (Table S1†). A slight decrease of SSAs was observed with increasing Mo amount where the lowest SSA (36 m2 g−1) was obtained for Co2Mo oxides. This can be ascribed to the larger particle size of CoMoO4 as observed from the XRD patterns. After acid leaching and selective removal of CoMoO4, SSAs of leached oxides increased significantly. In addition to a higher SSA, selective removal of CoMoO4 also creates more porosity within the framework of particles, which can be seen from the increased pore volume of the mixed oxides after leaching (Tables S1 and S2†). Comparing the increase of SSAs between leached and unleached oxides, the higher designed ratio of Mo leads to a more pronounced increase of SSAs (up to 3.3 fold) as a result of acid leaching (Fig. 2). Among them, acid-leached Co4Mo oxides showed the highest SSA of 150 m2 g−1, significantly higher than that of ordered mesoporous Co3O4 (∼105 m2 g−1) which is replicated from ordered mesoporous silica.4
Electron microscopy measurements were further conducted to characterize the morphology and structure of the samples. As shown in the transmission electron microscopy (TEM) images (Fig. S5†), a typical structure consisting of nano-sized crystals was observed for both pristine and acid-leached Co3O4. For the selected Co4Mo oxide sample, the average particle size was around 6 nm, and after acid leaching, this was reduced to below 5 nm (Fig. 3a and e). A closer examination of these particles before acid leaching displays clear crystal planes with a spacing of 0.23 and 0.21 nm (Fig. 3b), corresponding to the (311) and (400) lattices of the Co3O4 spinel, respectively.
 | ||
| Fig. 3 High resolution TEM images (a and b), dark field HR-SEM image (c), HR-SEM and the corresponding elemental mapping images (d) of the pristine Co4Mo oxide. High resolution TEM images (e and f), dark field HR-SEM image (g), HR-SEM and the corresponding elemental mapping images (h) of Co4Mo oxides after the leaching process in 0.1 M HCl solution for 2 h. The marked circles (orange color) in (b) and (c) represent the CoMoO4 particles embedded in the matrix of Co3O4. Insets in (a) and (e) show the corresponding particle size distribution. Insets in (b) and (f) are the corresponding close-up of the marked rectangles (white color). | ||
Additionally, much larger particles were also found in this pristine Co4Mo oxide (yellow circles in Fig. 3b). The larger particles isolated from small Co3O4 particles (Fig. S6†) and the lattices fringes with a spacing of 0.34 nm can be indexed to the (002) planes of CoMoO4. The formation of both small Co3O4 and large CoMoO4 particles was observed in the scanning electron microscopy (SEM) image of the pristine Co4Mo oxide (Fig. 3c), where large CoMoO4 particles were integrated with the matrix of small Co3O4 particles. In Fig. 3d, the SEM-energy dispersive X-ray spectroscopy (EDX) mapping images show a uniform distribution of Co and O elements, while the Mo element was concentrated on the large particles of CoMoO4. Furthermore, EDX analysis was conducted on representative locations to examine the elemental composition. As seen in Fig. S7,† less than 2 at% Mo was detected in the area mostly consisting of Co3O4 particles, whereas a much higher amount of Mo was detected in the area where larger CoMoO4 particles are present. These results illustrate a consolidation of smaller Co3O4 and larger CoMoO4 particles within the pristine mixed oxide, which is well consistent with the XRD analysis.
Upon the removal of CoMoO4 by acid leaching, two changes were observed from the high-resolution TEM and SEM images of Co4Mo oxides (Fig. 3e–g). The large CoMoO4 particles were not observed throughout the sample; instead, a uniform structure with interconnected ultra-small nanoparticles was obtained. Characteristic lattice planes of the Co3O4 spinel were also seen for these nanoparticles (inset of Fig. 3f). This observation is supported by the absence of the CoMoO4 phase in the XRD patterns. The SEM-EDX mapping revealed that Co, Mo, and O elements were not localized, but homogeneously dispersed over the entire region. The residual Mo species in leached oxides were possibly trapped within the Co3O4 framework, which protected them from the etching solution. An overview image and EDX mapping of leached samples showed that the leached sample contained only around 2 atomic percent Mo (Fig. S8†), which is not detectable by XRD. The second change is the decrease in the particle size of the leached Co4Mo oxide, where the average particle size of Co3O4 particles decreased from 6.2 nm to 4.8 nm after leaching in HCl solution. Both interparticle voids resulting from the removal of CoMoO4 and the formation of sub-5 nm Co3O4 particles contribute to a three-fold enhancement of the SSA of acid-leached Co4Mo oxide to 150 m2 g−1.
X-ray photoelectron spectroscopy (XPS) was further conducted to study the changes in the surface electronic structure of Co4Mo oxides after hydrochloride acid leaching. In the high-resolution Co 2p XPS spectra (Fig. S9a and b†), characteristic peaks of Co2+ and Co3+ were fitted to the Co 2p3/2 and Co 2p1/2 peaks of the Co4Mo oxide before and after acid leaching.16,38 A significant difference is that the acid-leached oxide possessed an increased ratio of Co3+/Co2+ on the surface, as the peaks ascribed to Co3+ became dominant in the Co 2p3/2 and Co 2p1/2 spectra compared with those of the un-leached sample. This supports the fact that the leaching treatment selectively removed the CoMoO4 phase, leading to a higher ratio of Co3+/Co2+ on the surface. For the Mo 3d region (Fig. S9c and d†), the spectra comprise two regions assigned to Mo 3d3/2 and Mo 3d5/2 spin–orbit levels of Mo6+ species, suggesting that the Mo element exists in its highest valence state on the surface of the Co4Mo oxide.47
In order to study the effect of various leaching conditions to optimize the leaching process, additional experiments were carried out by varying the leaching parameters, e.g. acid concentration and leaching time. A large batch of CW-templated Co4Mo oxides was first prepared and employed as a starting material for the acid leaching process. A mixed-phase of Co3O4 and CoMoO4 was detected from XRD patterns where a higher ratio of the CoMoO4 phase was obtained in comparison with the previous small batch synthesis (Fig. S10a†). As discussed above, the incorporation of CoMoO4 with larger particle size decreases the SSA of mixed oxides, and this is supported by a smaller BET surface area of 27 m2 g−1 (Fig. S10b†). The concentration of the acid leaching solution was first optimized with the same leaching time (2 h). As shown in Fig. S11,† 2 h of leaching with 0.01 M HCl solution is not capable of completely removing the CoMoO4 phase. A significant amount of Mo as well as trace impurities remained within the samples (Table S3†). Upon increasing the acid concentration from 0.05 to 0.2 M, the leached oxides showed a pure phase of Co3O4 (Fig. S13†). The EDX results also confirm the effective removal of Mo species and trace impurities (Fig. S11†). Accordingly, a threefold enhancement of SSAs (∼100 m2 g−1) was achieved with a high concentration of HCl solution, much higher than that of the samples after leaching with 0.01 M HCl (59 m2 g−1, Fig. S14 and Table S3†). It should be mentioned that 0.2 M HCl solution was able to etch out the Co3O4 phase as well since more than 60% of the weight was lost during the leaching process.
To optimize the leaching time, the concentration of the HCl solution was fixed at 0.05 M, and the leaching time was varied. Five minutes leaching time was already sufficient to leach out the CoMoO4 phase and results in a threefold increase of the SSA of the oxide (Fig. S12–S14†). With prolonged leaching time from 30 min to 6 h, a slight increase of the SSA was obtained (Table S4†). For large practical applications, it is essential to operate the process under mild conditions in terms of safety and production cost. Hereby, we optimized the leaching process by utilizing 0.05 M HCl aqueous solution for only 30 min of leaching. This simple two-step process shows great potential for practical production of high surface area cobalt oxides.
After a detailed structural analysis, the electrocatalytic properties towards the OER were evaluated in 1 M KOH by loading the catalysts on the glassy carbon electrode, following the protocol proposed by the Jaramillo group.10Fig. 4a and b depict the linear scanning voltammetry (LSV) curves of Co3O4 and Co–Mo oxides before and after the acid leaching process (0.1 M HCl solution for 2 h), respectively. In comparison with pristine Co3O4, the mixed oxides were more efficient in a low potential region (below 1.65 VRHE), requiring a smaller overpotential to reach a current density of 10 mA cm−2 (Table S5†). With increasing applied potential, Co3O4 became more active towards the OER by delivering a higher catalytic current. This is attributed to the lower Tafel slope of pure Co3O4 than the mixed oxides (inset of Fig. 4a and Table S5†).
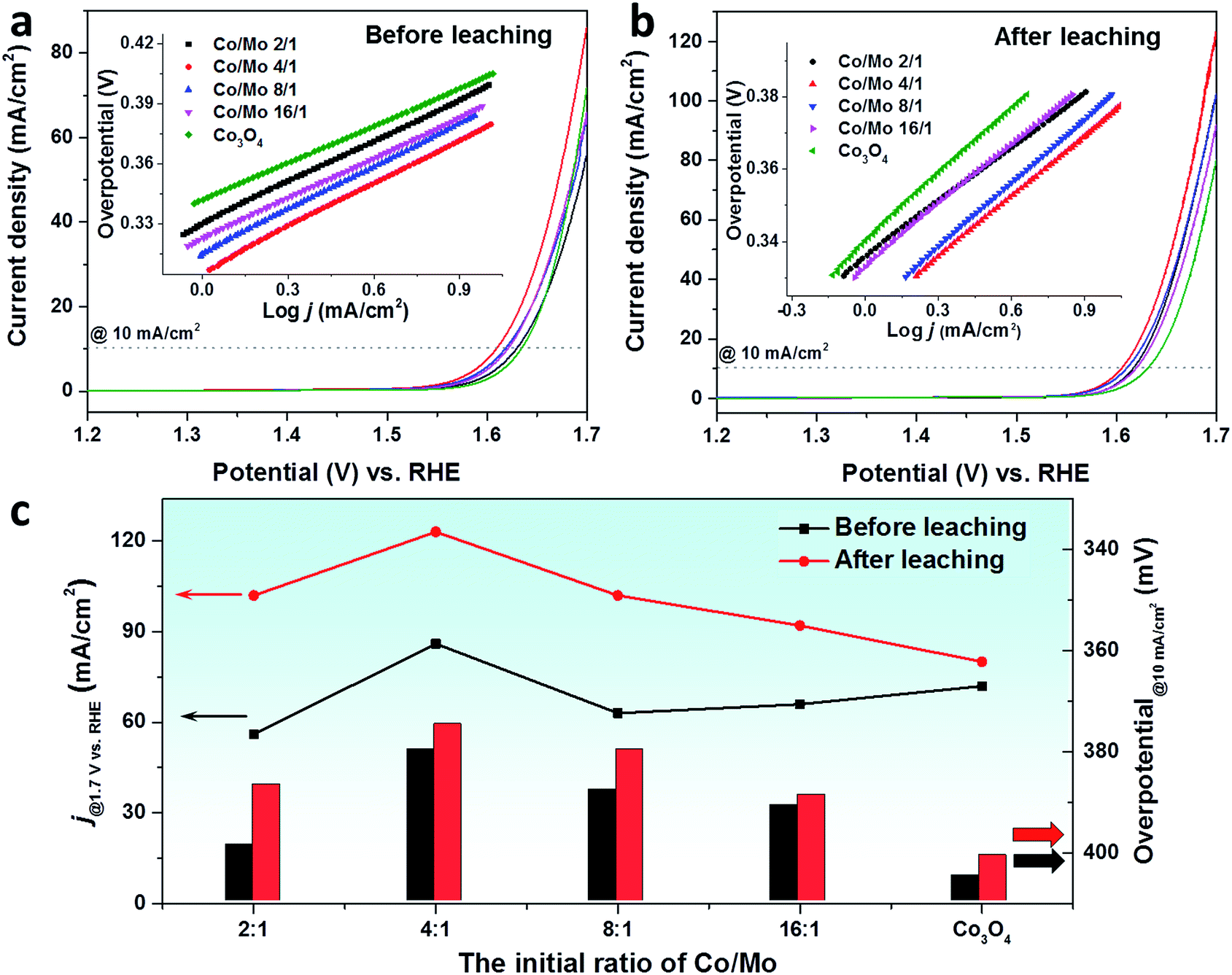 | ||
| Fig. 4 The LSV curves of CW-templated Co–Mo oxides and Co3O4 before (a) and after (b) leaching in 0.1 M HCl solution for 2 h. Insets in (a) and (b) show the corresponding Tafel plots. (c) A comparison on the current density at 1.7 VRHE (lines, left axis) and the overpotential at 10 mA cm−2 (columns, right axis) of the oxides before and after acid leaching. | ||
Upon leaching with 0.1 M HCl solution, the Co–Mo oxides exhibited superior OER activity to Co3O4 in the whole applied potential region. For all mixed oxides, the overpotential to reach 10 mA cm−2 was significantly decreased. It has been reported that the incorporation of Mo could activate the cobalt-based catalyst by modulating the electronic structure of cobalt to achieve the optimized adsorption energy for the reaction intermediates.48,49 An in situ X-ray adsorption study on the Co–Mo based electrocatalyst confirmed the changes of valence states of Co and Mo elements. When a potential of 1.4 VRHE is applied, the oxidation state of Mo decreased while Co was readily oxidized to a higher valence, similar to that of active CoOOH species.48 In addition, porous CoMoO4 has been reported to be an efficient OER catalyst.47 These could explain the catalytic performance enhancements of mixed Co–Mo oxides compared to Co3O4. As is expected, a decrease was also observed for Tafel slopes of mixed oxides upon the acid leaching process (inset of Fig. 4a and b). Among the leached oxides, the highest OER activity was achieved with the Co4Mo oxide that has the highest SSA. At an applied potential of 1.7 VRHE, the acid-leached Co4Mo oxide catalyst achieved a current density of 123 mA cm−2, outperforming the benchmark ordered mesoporous Co3O4 (∼105 mA cm−2).50
In order to compare the OER activity of pristine oxides and acid-leached oxides, the current densities at 1.7 VRHE and the overpotential to reach 10 mA cm−2 are summarized in Fig. 4c. A significant enhancement was seen for the current densities of all leached oxides, especially for the Co–Mo oxides. This escalation is mainly related to the change of the SSA where Co–Mo oxides show a more pronounced increase of the SSA compared to pure Co3O4. The other key parameter to determine OER activity, i.e. the overpotential to reach a current density of 10 mA cm−2, shows an opposite trend, where a drop was observed due to acid leaching. A much smaller overpotential was required to deliver a current density of 10 mA cm−2 for Co–Mo oxides. In addition to the remaining Mo which could act as the active species for leached Co–Mo oxides, the removal of the CoMoO4 phase could create some defects within the structure which results in the superior OER activity.33,51
Electrochemical impedance spectroscopy (EIS) test was performed to assess the charge transfer properties of Co3O4 and Co4Mo oxides. Fig. 5 depicts the Nyquist plots measured at 1.6 VRHE, from which the corresponding resistances were obtained according to the Randle model (Fig. S15†).52 The resistance in the high-frequency region is ∼6 ohm for all oxides, which is related to the solution resistance of the electrochemical setup. According to the diameter of the semicircles in the low-frequency region, pristine Co3O4 showed the highest charge transfer resistance (Rct). A much lower Rct was determined for Co4Mo oxides, indicating that the incorporation of Mo could improve the charge transfer ability of the cobalt oxide electrocatalyst. Transferring electrons faster on the pristine Co4Mo oxide contributes to a higher rate of OER activity in comparison with the pristine Co3O4 electrocatalyst. After the acid leaching process, both oxides showed reduced Rct, which suggests that such a leaching process leads to an enhancement of oxygen evolution kinetics of electrocatalysts. Additionally, we measured the double-layer capacitance (Cdl) using a simple CV method in a non-faradaic potential range to estimate the electrochemically active surface area (ECSA). As shown in Fig. S16,† the acid leached Co4Mo oxide possesses a higher Cdl (0.65 mF) compared to the unleached oxide (0.47 mF). Accordingly, a larger ECSA was exhibited on the Co4Mo oxide after leaching, suggesting that the acid-leached oxide provided more active sites exposed to the reactants in the 1 M KOH electrolyte for the OER.
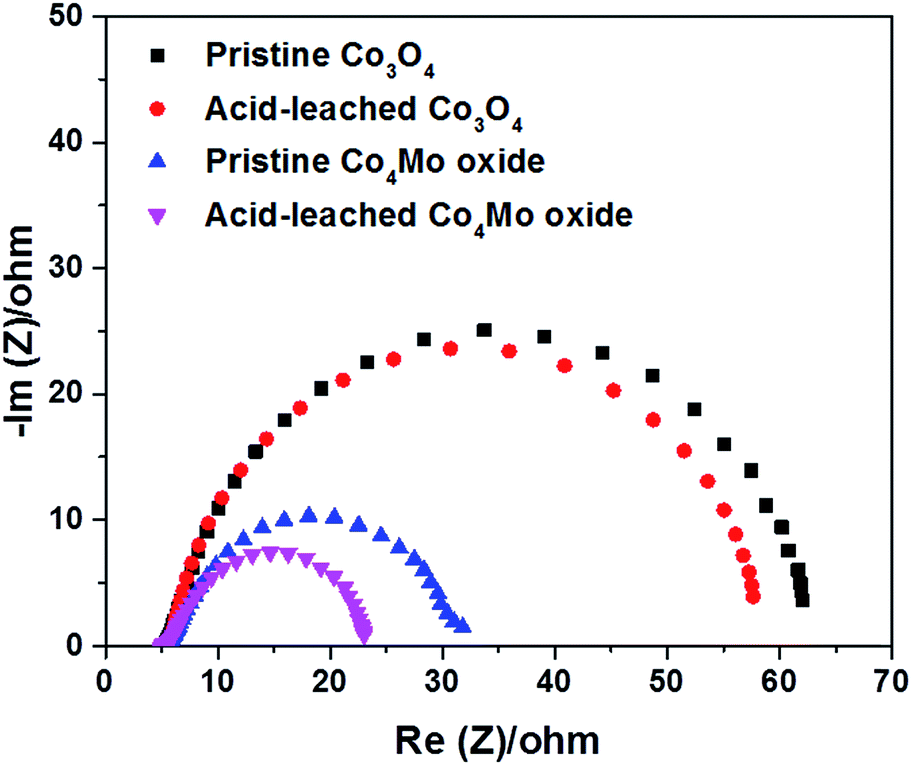 | ||
| Fig. 5 The Nyquist plots of pristine and acid-leached Co3O4 and Co4Mo oxides in 0.1 M HCl solution for 2 h. | ||
Furthermore, a stability study was carried out by a chronopotentiometric test at a constant current density of 10 mA cm−2. The long-term stability test that was conducted with the rotating disk electrode shows significant catalyst detachment after 3 h under the applied bias. However, we could overcome this detachment issue by depositing the sample on carbon fiber paper to be employed as the anode. As seen in Fig. S17,† no significant change was observed in the required potential for the acid-leached Co4Mo oxide to keep delivering the catalytic current for 12 h, proving the long-term stability of the acid-leached Co4Mo oxide.
In order to demonstrate the general applicability of this selective acid-leaching strategy, three other earth-abundant metals were selected and incorporated into the Co4M oxides (M = Li, Mg, and Ca). After the same leaching process, M-containing species were selectively washed out, as confirmed by the elemental analysis from EDX results (Fig. S18, S19 and Table S6†). The XRD results demonstrate a pure Co3O4 phase after the leaching process (Fig. S20†). Unlike the β-CoMoO4 phase with an open and defective structure, it has been shown that the well-crystalline structure of LiCoO2 and CaCO3 can be formed at milder temperatures.53,54 Thus, the crystal structure may not be the reason causing the selective leaching of these M-containing phases. Instead, their inferior stability in acidic solution could be explained by the basic tendency of these metals in the oxide form which contributes to the fast dissolution of M-containing phases even in diluted acid solution.54–56
HR-TEM images of these leached oxides were also taken, where similar nanostructures consisting of small crystalline particles were revealed (Fig. 6a–c). The enlarged images of isolated particles exhibited clear lattice fringes with a spacing of 0.24 nm, attributed to the (311) plane of the Co3O4 spinel. A distinct difference is that after acid leaching, the Co4Li oxide possessed larger particles compared to the oxides incorporated with Mg and Ca. Similarly, a significant change was exhibited in the textural parameters of these acid-leached oxides (Fig. S21†). In particular, a three-fold increase was seen in the SSA of Co4Mg and Co4Ca oxides prepared from this acid-leaching strategy (Fig. 6d), which is even higher than the SSA of metal oxides that are prepared via nanocasting by using ordered mesoporous silica as the template.16 Upon employing these oxides as OER electrocatalysts, a clear enhancement was seen in the catalytic performance of the oxides after the leaching process (Fig. S22† and 6e). It should be mentioned that Mg and Ca are among the most abundant metals and are cost-effective, which can be employed for large scale surfactant-free nanoparticle synthesis for diverse applications.
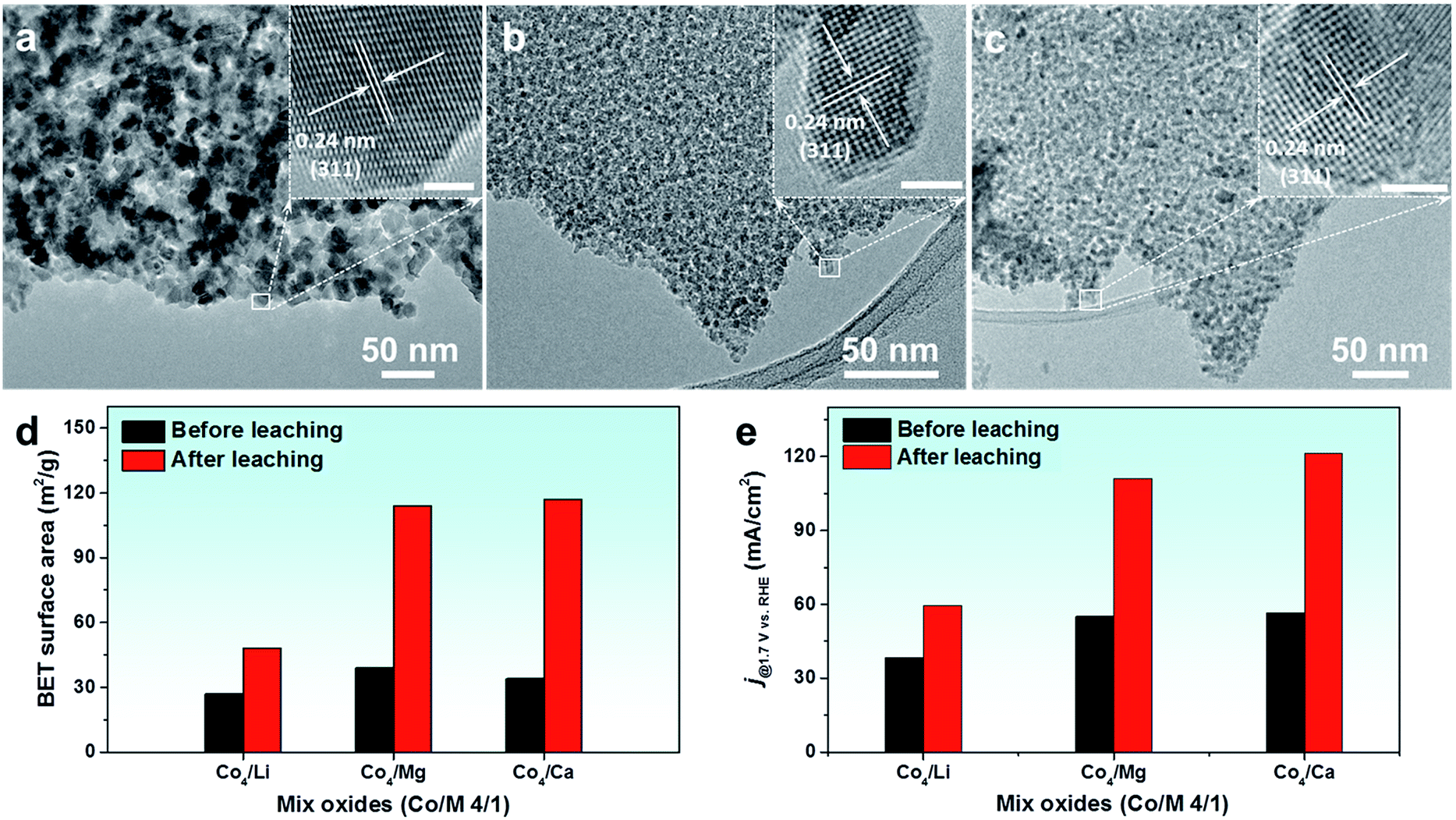 | ||
| Fig. 6 (a–c) HR-TEM images of the acid-leached Co4Li oxide, Co4Mg oxide, and Co4Ca oxide, respectively. Summarized values of (d) BET surface area and (e) current density at 1.7 VRHE of CW-templated Co4M oxides (M = Li, Mg, and Ca) before and after leaching in 0.1 M HCl solution for 2 h. | ||
In addition, this selective acid-leaching strategy can be extended to synthesis of other transition metal oxides like Fe2O3 and NiO. An acid-leaching process selectively removed Ca from Ni4Ca oxide and Fe4Ca oxide (Fig. S23, S24 and Table S7†). As a result of phase purification due to acid-leaching, a pure phase of Fe2O3 and NiO was prepared with a high specific surface area of up to 150 m2 g−1 (Fig. S25 and S26†).
4. Conclusions
In summary, we reported a general and facile acid-leaching process to engineer Co3O4 nanostructures for the electrocatalytic oxygen evolution reaction. Through a scalable coffee-waste templating method, Co–Mo oxides were prepared with mixed phases of Co3O4 and CoMoO4. The CoMoO4 phase was selectively leached out by treating the oxides with diluted HCl solution, resulting in the formation of sub-5 nm particles with a pure Co3O4 phase and a threefold increase of the specific surface area (up to 150 m2 g−1). Electrochemical measurements revealed that the acid-leached oxides exhibit superior catalytic activity to pristine oxides, which is attributed to (i) a larger surface area, (ii) phase purification to expose more active Co3O4 species to the reactant, and (iii) faster charge transfer kinetics for the OER. This strategy has widespread applicability through selectively leaching earth-abundant metals. Li, Mg and Ca species can be used for the acid-leaching process because of the resulting high surface area cobalt oxide nanocrystals. Moreover, this acid-leaching strategy can be extended to the synthesis of other metal oxides such as Fe2O3 and NiO with high surface areas of up to 150 m2 g−1. The developed process does not require harsh conditions, expensive chemicals, dedicated instruments or time-consuming operation and it can be applied to prepare nanocrystals at the industrial scale.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is financially supported by IMPRS-RECHARGE and MAXNET Energy consortium of Max Planck Society. We thank S. Palm, H. Bongard, A. Kostis, and A. Schlüter for EDX analysis and microscopy images and A. Bähr for proofreading. We would also thank J. N. Büscher and PD Dr C. Weidenthaler for XPS measurements and related discussions. The manuscript is dedicated to the memory of Prof. Dr Walter Thiel who recently passed away.Notes and references
- N. S. Lewis and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15729–15735 CrossRef CAS PubMed.
- S. Chu, Y. Cui and N. Liu, Nat. Mater., 2017, 16, 16–22 CrossRef PubMed.
- L. Schlapbach and A. Züttel, Nature, 2001, 414, 353–358 CrossRef CAS PubMed.
- X. Deng, K. Chen and H. Tüysüz, Chem. Mater., 2017, 29, 40–52 CrossRef CAS.
- M. G. Walter, E. L. Warren, J. R. McKone, S. W. Boettcher, Q. Mi, E. A. Santori and N. S. Lewis, Chem. Rev., 2010, 110, 6446–6473 CrossRef CAS PubMed.
- B. M. Hunter, H. B. Gray and A. M. Müller, Chem. Rev., 2016, 116, 14120–14136 CrossRef CAS PubMed.
- F. Song, L. Bai, A. Moysiadou, S. Lee, C. Hu, L. Liardet and X. Hu, J. Am. Chem. Soc., 2018, 140, 7748–7759 CrossRef CAS PubMed.
- N.-T. Suen, S.-F. Hung, Q. Quan, N. Zhang, Y.-J. Xu and H. M. Chen, Chem. Soc. Rev., 2017, 46, 337–365 RSC.
- F. Lyu, Y. Bai, Z. Li, W. Xu, Q. Wang, J. Mao, L. Wang, X. Zhang and Y. Yin, Adv. Funct. Mater., 2017, 27, 1702324 CrossRef.
- C. C. L. McCrory, S. Jung, I. M. Ferrer, S. M. Chatman, J. C. Peters and T. F. Jaramillo, J. Am. Chem. Soc., 2015, 137, 4347–4357 CrossRef CAS PubMed.
- G. P. Gardner, Y. B. Go, D. M. Robinson, P. F. Smith, J. Hadermann, A. Abakumov, M. Greenblatt and G. C. Dismukes, Angew. Chem., Int. Ed., 2012, 51, 1616–1619 CrossRef CAS PubMed.
- R. Kötz, H. J. Lewerenz and S. Stucki, J. Electrochem. Soc., 1983, 130, 825–829 CrossRef.
- E. Antolini, ACS Catal., 2014, 4, 1426–1440 CrossRef CAS.
- I. Roger, M. A. Shipman and M. D. Symes, Nat. Rev. Chem., 2017, 1, 0003 CrossRef CAS.
- H. S. Ahn and T. D. Tilley, Adv. Funct. Mater., 2013, 23, 227–233 CrossRef CAS.
- T. Grewe, X. Deng, C. Weidenthaler, F. Schüth and H. Tüysüz, Chem. Mater., 2013, 25, 4926–4935 CrossRef CAS.
- M. S. Burke, L. J. Enman, A. S. Batchellor, S. Zou and S. W. Boettcher, Chem. Mater., 2015, 27, 7549–7558 CrossRef CAS.
- F. Dionigi and P. Strasser, Adv. Energy Mater., 2016, 6, 1600621 CrossRef.
- M. Gong, Y. Li, H. Wang, Y. Liang, J. Z. Wu, J. Zhou, J. Wang, T. Regier, F. Wei and H. Dai, J. Am. Chem. Soc., 2013, 135, 8452–8455 CrossRef CAS PubMed.
- F. Song and X. Hu, J. Am. Chem. Soc., 2014, 136, 16481–16484 CrossRef CAS PubMed.
- G.-h. Moon, M. Yu, C. K. Chan and H. Tüysüz, Angew. Chem., Int. Ed., 2019, 58, 3491–3495 CrossRef CAS PubMed.
- Q. Fu, T. Wu, G. Fu, T. Gao, J. Han, T. Yao, Y. Zhang, W. Zhong, X. Wang and B. Song, ACS Energy Lett., 2018, 3, 1744–1752 CrossRef CAS.
- P. W. Menezes, A. Indra, I. Zaharieva, C. Walter, S. Loos, S. Hoffmann, R. Schlögl, H. Dau and M. Driess, Energy Environ. Sci., 2019, 12, 988–999 RSC.
- D. Li, H. Baydoun, B. Kulikowski and S. L. Brock, Chem. Mater., 2017, 29, 3048–3054 CrossRef CAS.
- L. Yan, L. Cao, P. Dai, X. Gu, D. Liu, L. Li, Y. Wang and X. Zhao, Adv. Funct. Mater., 2017, 27, 1703455 CrossRef.
- J. Masa, P. Weide, D. Peeters, I. Sinev, W. Xia, Z. Sun, C. Somsen, M. Muhler and W. Schuhmann, Adv. Energy Mater., 2016, 6, 1502313 CrossRef.
- P. Chen, K. Xu, T. Zhou, Y. Tong, J. Wu, H. Cheng, X. Lu, H. Ding, C. Wu and Y. Xie, Angew. Chem., Int. Ed., 2016, 55, 2488–2492 CrossRef CAS PubMed.
- J. Suntivich, K. J. May, H. A. Gasteiger, J. B. Goodenough and Y. Shao-Horn, Science, 2011, 334, 1383–1385 CrossRef CAS PubMed.
- B. Zhao, L. Zhang, D. Zhen, S. Yoo, Y. Ding, D. Chen, Y. Chen, Q. Zhang, B. Doyle, X. Xiong and M. Liu, Nat. Commun., 2017, 8, 14586 CrossRef CAS PubMed.
- A. J. Esswein, M. J. McMurdo, P. N. Ross, A. T. Bell and T. D. Tilley, J. Phys. Chem. C, 2009, 113, 15068–15072 CrossRef CAS.
- A. Bergmann, E. Martinez-Moreno, D. Teschner, P. Chernev, M. Gliech, J. F. de Araujo, T. Reier, H. Dau and P. Strasser, Nat. Commun., 2015, 6, 8625 CrossRef CAS PubMed.
- C. Zhang, M. Antonietti and T.-P. Fellinger, Adv. Funct. Mater., 2014, 24, 7655–7665 CrossRef CAS.
- J. Rosen, G. S. Hutchings and F. Jiao, J. Am. Chem. Soc., 2013, 135, 4516–4521 CrossRef CAS PubMed.
- X. Deng and H. Tüysüz, ACS Catal., 2014, 4, 3701–3714 CrossRef CAS.
- J. Wang, W. Cui, Q. Liu, Z. Xing, A. M. Asiri and X. Sun, Adv. Mater., 2016, 28, 215–230 CrossRef CAS PubMed.
- X. Deng, W. N. Schmidt and H. Tüysüz, Chem. Mater., 2014, 26, 6127–6134 CrossRef CAS.
- J. D. Blakemore, H. B. Gray, J. R. Winkler and A. M. Müller, ACS Catal., 2013, 3, 2497–2500 CrossRef CAS.
- Y. Zhou, C.-K. Dong, L. L. Han, J. Yang and X.-W. Du, ACS Catal., 2016, 6, 6699–6703 CrossRef CAS.
- M. Yu, C. K. Chan and H. Tüysüz, ChemSusChem, 2018, 11, 605–611 CrossRef CAS PubMed.
- D. Zagorac, J. C. Schön, M. Rosić, J. Zagorac, D. Jordanov, J. Luković and B. Matović, Chem. Mater., 2017, 52, 1700069 Search PubMed.
- D. Vie, E. Martínez, F. Sapiña, J.-V. Folgado, A. Beltrán, R. X. Valenzuela and V. Cortés-Corberán, Chem. Mater., 2004, 16, 1697–1703 CrossRef CAS.
- J. A. Rodriguez, S. Chaturvedi, J. C. Hanson and J. L. Brito, J. Phys. Chem. B, 1999, 103, 770–781 CrossRef CAS.
- G. Li, C. Zhang, Z. Wang, H. Huang, H. Peng and X. Li, Appl. Catal., A, 2018, 550, 67–76 CrossRef CAS.
- S. Kendell and T. Brown, React. Kinet., Mech. Catal., 2010, 99, 251–268 CAS.
- C. Rocchiccioli-Deltcheff, A. Aouissi, M. M. Bettahar, S. Launay and M. Fournier, J. Catal., 1996, 164, 16–27 CrossRef CAS.
- A. Rumplecker, F. Kleitz, E.-L. Salabas and F. Schüth, Chem. Mater., 2007, 19, 485–496 CrossRef CAS.
- M. Q. Yu, L. X. Jiang and H. G. Yang, Chem. Commun., 2015, 51, 14361–14364 RSC.
- P. Liu, S. Yang, L. Zheng, B. Zhang and H. Yang, Chem. Sci., 2017, 8, 3484–3488 RSC.
- S. Hao, L. Chen, C. Yu, B. Yang, Z. Li, Y. Hou, L. Lei and X. Zhang, ACS Energy Lett., 2019, 9, 952–959 CrossRef.
- X. Deng, S. Öztürk, C. Weidenthaler and H. Tüysüz, ACS Appl. Mater. Interfaces, 2017, 9, 21225–21233 CrossRef CAS PubMed.
- S. Wahl, S. M. El-Refaei, A. G. Buzanich, P. Amsalem, K.-S. Lee, N. Koch, M.-L. Doublet and N. Pinna, Adv. Energy Mater., 2019, 9, 1900328 CrossRef.
- C. C. L. McCrory, S. Jung, J. C. Peters and T. F. Jaramillo, J. Am. Chem. Soc., 2013, 135, 16977–16987 CrossRef CAS PubMed.
- E. Rossen, J. N. Reimers and J. R. Dahn, Solid State Ionics, 1993, 62, 53–60 CrossRef CAS.
- H. Zhu, Z. Liu, Y. Wang, D. Kong, X. Yuan and Z. Xie, Chem. Mater., 2008, 20, 1134–1139 CrossRef CAS.
- A. Auroux and A. Gervasini, J. Phys. Chem., 1990, 94, 6371–6379 CrossRef CAS.
- D. F. Shriver, Acc. Chem. Res., 1970, 3, 231–238 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ta07835e |
| This journal is © The Royal Society of Chemistry 2019 |