
Direct immobilization of an atomically dispersed Pt catalyst by suppressing heterogeneous nucleation at −40 °C†

Abstract
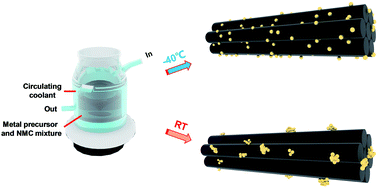
Direct deposition of isolated metal atoms onto substrates has been recognized as a simple route to obtain high performance supported atomically dispersed metals (SACs), however, the agglomeration driven by high surface energy is difficult to avoid. Herein, we demonstrate a one-pot solution synthesis to obtain atomically dispersed platinum (Pt) supported on nitrogen (N)-doped mesoporous carbon (NMC) substrates (Pt/NMC-LT) by conducting the whole synthesis at −40 °C, owing to the sluggish nucleation kinetics. We obtained the Pt/NMC-LT catalyst with superior electrochemical hydrogen evolution reaction (HER) activity and stability, in comparison with the NMC supported dominant Pt sub-nanometer cluster catalyst from solution synthesis at RT ∼ 25 °C (Pt/NMC-RT) and commercial carbon supported Pt nanoparticle catalysts (Pt/C). Lower over-potential values (only 17.0 and 49.8 mV) are needed for high HER current densities (10 and 100 mA cm−2, respectively), and no obvious degradation is observed after an accelerated durability test (ADT) for 5000 CV cycles.
 Please wait while we load your content...
Please wait while we load your content...

