
Hydrogen evolution activity tuning via two-dimensional electron accumulation at buried interfaces†
Yudong
Xue
 *abcd,
Zachary S.
Fishman
ac,
Yunting
Wang
e,
Zhenhua
Pan
ac,
Xin
Shen
ac,
Rito
Yanagi
ac,
Gregory S.
Hutchings
a,
Mingzhao
Liu
f,
Shili
Zheng
b,
Yi
Zhang
b,
Eric I.
Altman
a and
Shu
Hu
*ac
*abcd,
Zachary S.
Fishman
ac,
Yunting
Wang
e,
Zhenhua
Pan
ac,
Xin
Shen
ac,
Rito
Yanagi
ac,
Gregory S.
Hutchings
a,
Mingzhao
Liu
f,
Shili
Zheng
b,
Yi
Zhang
b,
Eric I.
Altman
a and
Shu
Hu
*ac
aDepartment of Chemical and Environmental Engineering, Yale University, New Haven, Connecticut 06511, USA. E-mail: ydxueipe@hotmail.com; shu.hu@yale.edu
bNational Engineering Laboratory for Hydrometallurgical Cleaner Production Technology, CAS Key Laboratory of Green Process and Engineering, Institute of Process Engineering, Chinese Academy of Sciences, Beijing 100190, China
cEnergy Sciences Institute, Yale University, West Haven, Connecticut 06516, USA
dUniversity of Chinese Academy of Sciences, Beijing 100049, China
eSchool of Chemical and Environmental Engineering, China University of Mining and Technology (Beijing), Beijing 100083, China
fCenter for Functional Nanomaterials, Brookhaven National Laboratory, Upton, New York 11973, USA
First published on 19th August 2019
Abstract
Developing efficient earth-abundant transition metal-based electrocatalysts for the hydrogen evolution reaction (HER) is crucial for hydrogen production at scale. This paper reports that the buried electrocatalytic interfaces between Ni–Fe sulfide (NiFeS) nanosheets and TiO2 conformal coatings (about 5 nm) achieved remarkable HER activity improvement, lowering the HER overpotential from −170 mV to −107 mV at −50 mA cm−2 in a base. Non-HER active, permeable TiO2 coatings grown by atomic layer deposition (ALD) achieved continuous fine-tuning of the electronic properties at the buried TiO2/NiFeS interfaces, as a novel strategy and the main factor for electron accumulation at the interface. Core-level and valence band X-ray photoelectron spectroscopy (XPS) was used to investigate the TiO2 electronic-structure tuning effect on the charge-transfer energetics during the HER. Their alkaline HER mechanism was elucidated by supplementing characterizations of membrane permeation, Tafel slope, and synchrotron X-ray absorption spectroscopy, which verified that the buried TiO2/NiFeS interfaces are electrocatalytically active. This study offers a general strategy for improving the charge-transfer kinetics of an electrocatalytic system by confining catalysis at a permeable solid–solid interface. The broad applicability of permeable and tunable coatings potentially accelerates the optimization of earth-abundant catalysts to achieve high performance under operationally relevant conditions.
Introduction
Hydrogen has been widely regarded as one of the most promising energy carriers to fulfill our need for a clean and sustainable future.1–4 Broadly, low- and high-temperature advanced electrolysis, the solar thermochemical process, and photoelectrochemical (PEC) water splitting are considered three scalable hydrogen generation processes.5,6 Both photoelectrochemical (PEC) water splitting and photovoltaic-driven electrolysis require electrocatalysts to produce hydrogen.7,8 Among these technological pathways, development of efficient and cost-effective electrocatalysts for the hydrogen evolution reaction (HER) is critical.9–16 Moreover, while the HER in bases generally shows inferior activity than in acids because surface *OH and *H species compete for the active sites, numerous combinations of earth-abundant multi-element catalysts exist in bases, offering vast space for tunability.17 To date, platinum-group metals and oxides, such as Pt and RuO2, have been among the most efficient, but their disadvantages are high cost and low abundance.18–20 Alternatively, transition metal (Ni, Fe, Co, W, and Mo) compounds, such as metal sulfides, selenides, phosphides, nitrides, and carbides, have been proposed as efficient and low-cost alternatives to noble metals.21–26Among them, transition metal sulfides have attracted special interest as HER catalysts due to their promising efficiency in alkaline media and their ease of synthesis.27,28 Several early reports suggested that these drawbacks may be overcome by tuning surface atomic structures or confining molecular-scale intermediates; recent work has demonstrated that the HER activity of transition metal sulfides can be improved by introducing a synergistic constituent such as forming a bi-metallic sulfide.28–31 Despite this development, the performance of transition metal sulfides is still far inferior to that needed for electrolysis and photolysis.
A more recent report demonstrated tuning the interfacial electronic structures for improving electrocatalytic activity.32 In many cases, tuning of catalysts does not need dramatic changes in their surface structures or compositions, such as in the case of bi-metallic alloying. Surface structure or composition tuning will change electronic structures, a conventional means for catalytic tuning.33 Without explicitly tuning their structures or compositions, fine tuning of electronic structures for catalytic surfaces or interfaces is a new strategy. This paper is about further improvement of hydrogen evolution performance based on an optimized structure and composition of bimetallic catalysts.34 This strategy offers an orthogonal control to structural or compositional tuning and uniquely allows for fine tuning on a continuous scale beyond conventional bi-metallic alloying. A small variation in spectroscopic signatures is expected, and will be correlated with observable changes in catalytic performance. Preliminary work showed that non-active TiO2 coatings of up to 40 nm improved hydrogen evolution,29 oxygen evolution, and chlorine evolution activities.35 But their enhancement lacks clear elucidation: especially for the case of 40 nm coatings, there has been no convincing evidence to prove that there is enough reactant and product permittivity to keep up with the HER rate.
Atomic layer deposition (ALD) is a coating process that yields ultrathin overlayers with atomically precise thicknesses and exceptional conformality on high surface-area materials.34,36,37 Several recent studies have shown that a porous coating can selectively block reactants.38 Labrador et al. reported that a SiOx-coated Pt catalyst showed high selectivity for proton and H2 transport against impurity electrodeposition and catalyst poisoning.39 Vos et al. observed that MnOx thin films deposited on IrO2 model catalysts increased the selectivity of oxygen evolution over chlorine evolution, acting as a diffusion barrier that prevents Cl− transport to active sites.40 For PEC processes, impermeable, pinhole-free ALD TiO2 coatings were used to prevent photo-corrosion in the pH range between 0 and 14, while being sufficiently transparent to reach the high quantum efficiency of protected semiconductors.37,41 With a high throughput spatial technique, the ALD process is scalable for surface modification.42 While coatings were normally grown thick enough to be impermeable, a less investigated technique is using ALD ultrathin membranes to enhance the electrocatalytic activity that bulk composition tuning cannot otherwise achieve. ALD coating is also considered favorable for stabilizing catalytic surfaces and preventing intermediates from leaving the lattice under operational conditions.43
In this study, we coated conformal, non-HER active, and permeable TiO2 of 1–10 nm onto NiFe-bimetallic sulfide (NiFeS) nanosheet electrocatalysts by ALD. We exploited the effect of TiO2-coating/NiFeS-nanosheet buried hetero-junctions on their HER activity, and proposed a generic strategy for further boosting the HER activity of bi-metallic electrocatalysts. The structural and physicochemical properties, including crystalline structures, surface morphology, valence electronic structures, and interfacial band energetics, were systematically investigated. We discovered that the buried interface between TiO2 and bimetallic sulfide nanosheets can be correlated with the favorably tuned electronic structures, suggesting a host of new opportunities for enhancing H2 production at scale.
Results and discussion
The TiO2/NiFeS hetero-junction catalyst was prepared via a “bottom-up” approach consisting of a hydrothermal process and sulfuration, followed by an ALD coating process (see the ESI† for details). During ALD, TiO2 was chemically grown on the surface of NiFeS nanosheet/Ni foam using tetrakisdimethylamido-titanium (TDMAT) as the Ti precursor and H2O as the co-reactant. Ni foam was chosen as the catalyst support due to its three-dimensional internal pores with high alkali stability.44 The pulse time of H2O and TDMAT was optimized to be 0.06 and 0.25 seconds, respectively, longer than that under previously reported conditions,37 to achieve full surface coverage on NiFeS-loaded Ni foam. Fig. S1 and S2† illustrate the ALD process, which involves successive self-limiting surface reactions between the H2O and TDMAT over the NiFeS nanosheet substrate, indicating that there is enough precursor supplied to coat high surface-area materials conformally. According to the planar growth rate of 0.047 nm per cycle, the coating thicknesses were nominally 1, 2, 5, 7, and 10 nm, corresponding to 22, 43, 106, 149, and 213 ALD cycles. The surface reactions during the TDMAT exposure are expressed as eqn (1). Here, * refers to surface reactive sites.| NiFeS-Ni(Fe)OH* + Ti(N(CH3)2)4 → NiFeS-Ni(Fe)O-Ti(N(CH3)2)3* + HN(CH3)2 | (1) |
TDMAT can react with the hydroxyl groups that are formed on the NiFeS surface by a H2O pulse, and make Ti–O bonds by transferring protons (or possibly via protons attached to H2O molecules) to TDMAT's ligand complex to produce HN(CH3)2 as a product which then leaves the surface. The subsequent surface reactions during the H2O exposure are expressed as eqn (2) and (3).
| NiFeS-Ni(Fe)O-Ti(N(CH3)2)3* + H2O → NiFeS-Ni(Fe)O–Ti–OH* + 3HN(CH3)2 | (2) |
| TiOH* + Ti(N(CH3)2)4 → TiO-Ti(N(CH3)2)3* + HN(CH3)2 | (3) |
During the initial cycles, H2O will react with a surface-bound Ti(N(CH3)2)3* to yield a hydroxyl-terminated *Ti–OH surface, or H2O could bridge the two surface-(N(CH3)2)3 sites that are close by to form a *-Ti–O–Ti-* oxo bridge. The surface chemistry of NiFeS before ALD growth was carefully controlled to minimize the formation of interfacial NiOx or FeOx, and the thermodynamics of TiO2 compared to NiFeS native oxides favors the formation of NiFeS/TiO2 interfaces but not NiOx- or FeOx-rich interfaces. After the initial cycles, TiO2 was deposited on a TiO2 surface until its deposition was completed. Unreacted precursor molecules, along with volatile products, were removed in a continuous flow of inert Ar carrier gas.
X-ray diffraction (XRD) was employed for structural characterization of the as-prepared samples and those coated with varying ALD TiO2 thicknesses (1–10 nm), as shown in Fig. 1. Prior to the ALD process, the NiFeS nanosheet generated XRD patterns with diffraction peaks at 21.7°, 31.1°, 37.8°, 49.7°, and 55.4°, which were consistent with the (101), (110), (003), (113), and (300) crystal planes of hexagonal Ni3S2 (PDF no. 00-044-1418).45,46 No diffraction peaks ascribed to iron sulfides can be observed, which indicates that the iron atoms are either substitutional sites in the lattice of Ni3S2 or in an amorphous state that does not participate in X-ray scattering. In either case, the effect of iron on varying the lattice parameters is negligible. The chemical formula was NiFe1.29S1.53; calculations are documented in the ESI.† After the ALD process, no additional diffraction peaks were introduced into the NiFeS structure. Moreover, the intensity of most Ni3S2 peaks was maintained because the surface-deposited thin amorphous TiO2 barely attenuated the intensity of scattered X-rays from the underlying NiFeS crystallites.
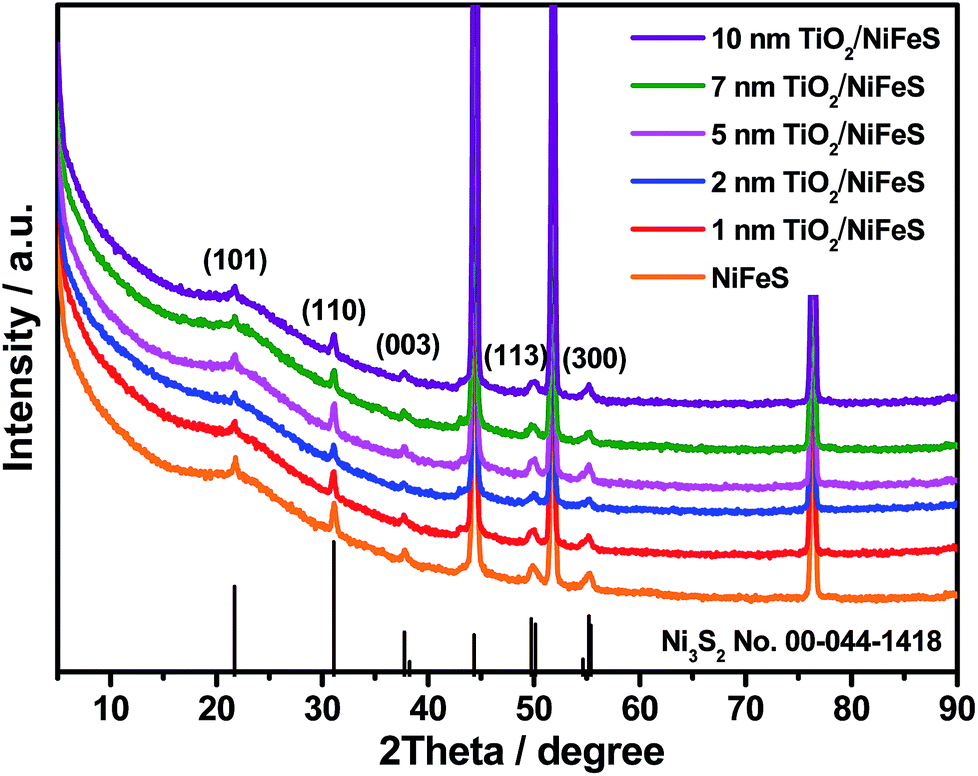 | ||
| Fig. 1 X-ray diffraction (XRD) patterns of NiFeS nanosheets: bare sample and samples with 1, 2, 5, 7, and 10 nm of atomic layer deposition (ALD) TiO2 coating. | ||
The HER performance of the NiFeS and TiO2/NiFeS electrocatalysts was compared by linear sweep voltammetry (LSV) measurements in a 1 M KOH (aq) electrolyte. As shown in Fig. 2a, the overpotentials at a current density of −50 mA cm−2 were −141, −133, −107, −132, and −185 mV for the 1, 2, 5, 7, and 10 nm ALD TiO2 modifications, respectively. The reproducibility is good based on testing at least 5 times. The overpotential measurement error was ±5 mV. Too high current densities and slow mass transport can cause current fluctuations, shown as spikes in Fig. 2a. Among these samples, the 5 nm ALD TiO2/NiFeS exhibited the best HER performance, i.e., the lowest overpotentials. To achieve a catalytic current density of −50 mA cm−2, an overpotential of only −107 mV was needed for the 5 nm TiO2/NiFeS, which is much lower than that of the as-synthesized NiFeS (−170 mV), NiFe-layered double hydroxide (NiFe-LDH, −326 mV), and Ni foam (−410 mV, Fig. S3a†). This suggests that the HER activity of the NiFeS electrode can be tuned by coating catalysts with non-catalytic ALD films grown at temperatures as low as 150 °C without substantial variations in the original surface structures and compositions. TiO2/FTO was chosen as the control to show that 5 nm TiO2 alone does not exhibit HER activity.
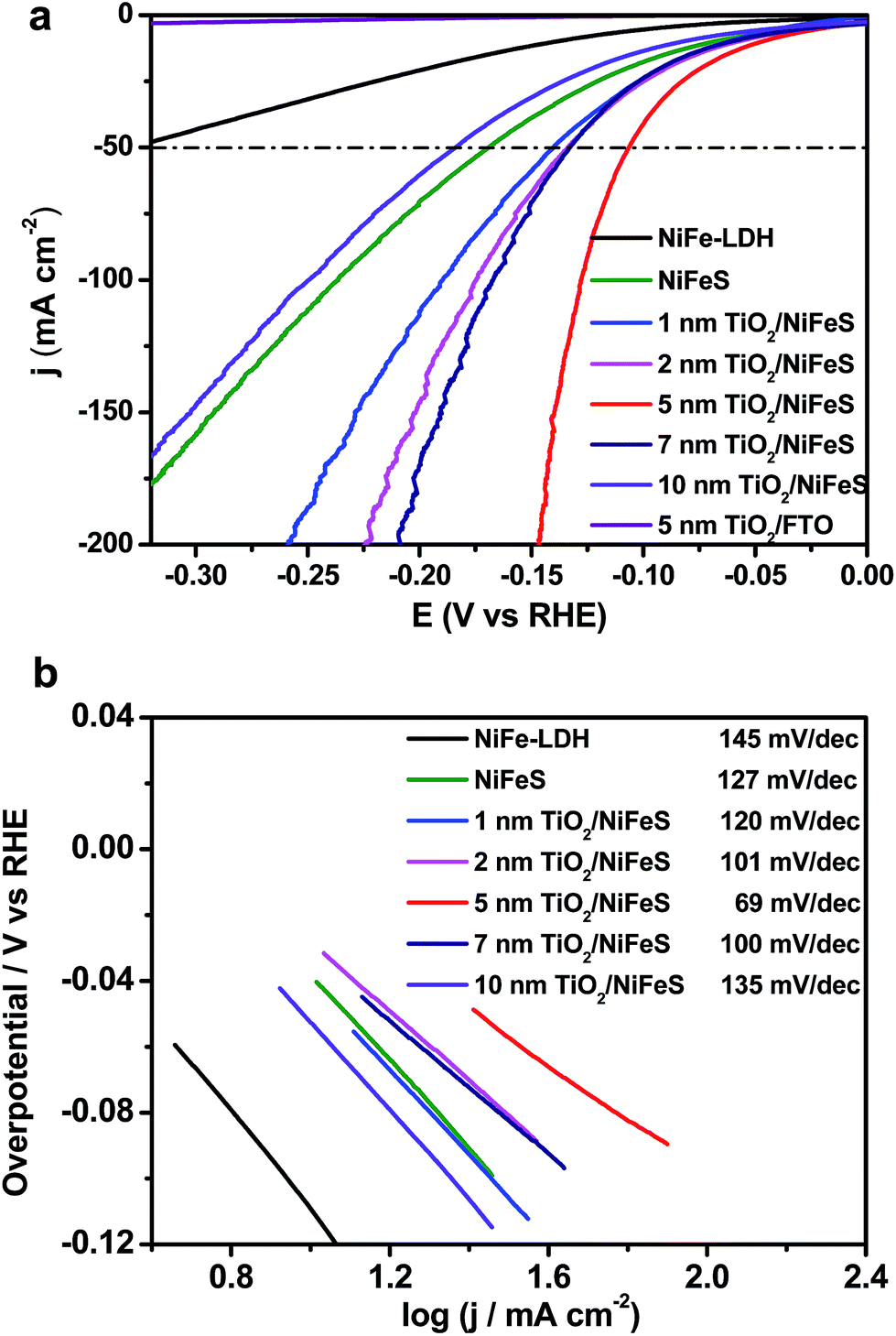 | ||
| Fig. 2 Linear sweep voltammetry (LSV) curves (a) and the respective Tafel plots (b) of the ALD TiO2/NiFeS electrodes and TiO2/FTO (fluorine-doped tin oxide) electrodes measured in a 1 M KOH (aq) solution. | ||
The electrocatalytic activity of the NiFeS electrode increases with increasing TiO2 layer thickness from 1 to 5 nm but decreases with increasing TiO2 layer thickness from 5 to 10 nm. This suggests that an optimal coating thickness is needed to achieve the most active NiFeS/TiO2/electrolyte interface. The corresponding Tafel slope of the 5 nm TiO2/NiFeS sample (69 mV dec−1) was much smaller than that of the uncoated NiFeS (127 mV dec−1), as shown in Fig. 2b. These results indicated that the 5 nm TiO2-coated sample favored faster kinetics at a reduced Tafel slope, suggesting a possible shift in the rate-determining step; we will discuss this in detail in the catalytic mechanism section.
The 5 nm TiO2/NiFeS interface was further characterized for elucidating the boosted HER activity. The overpotential of 5 nm TiO2/NiFeS at 50 mA cm−2 was comparable to that of a reported Pt/C HER catalyst.47 The turnover frequencies (TOFs)48 for 5 nm TiO2/NiFeS and uncoated NiFeS at a −100 mV overpotential were 2.72 and 0.56 s−1, respectively. The TOFs, with their calculations elaborated in the ESI and shown in Table S1,† indicated that the intrinsic HER activity of TiO2/NiFeS is about five times better than that of the uncoated NiFeS catalyst (buried TiO2/NiFeS will later be proven to be active), which is comparable with that of the state-of-the-art, earth-abundant transition metal HER catalysts. As shown in Fig. S4,† electrochemical impedance spectroscopy (EIS) analysis indicated that after 5 nm amorphous TiO2 was deposited onto the sulfide surface, the charge-transfer resistance of the TiO2/NiFeS electrode decreased slightly from 1.563 to 1.526 Ω. Moreover, the quantity of hydrogen accumulated at 10 mA cm−2 based on the TiO2/NiFeS nanocomposites matched with the calculated amount for the HER assuming a faradaic efficiency of ∼100% as shown in Fig. S5.† Their long-term HER performance testing at a fixed current density of 10 mA cm−2 for 30 hours (Fig. S6†) showed excellent stability of overpotentials, better than that of the uncoated NiFeS for 10 hours.45,49 Only the overpotential increased by 6 mV after 30 hours of operation. Besides, after HER testing, there was no change from the XRD pattern, as shown in Fig. S7.† The SEM image and XPS survey spectrum after the HER test, as shown in Fig. S8 and S9a,† indicated that TiO2 was stable in alkaline media. Trace ion analysis by inductively coupled plasma-mass spectrometry (ICP-MS) further confirmed that, compared with NiFe-LDH and NiFeS, less Ni and Fe were dissolved for the TiO2/NiFeS samples during the HER (Table S2†).50 These results also showed increased structural stability of the TiO2/NiFeS catalyst.
Fig. 3 shows the morphology and composition characterization of the as-synthesized NiFeS and 5 nm TiO2/NiFeS samples. In comparison to Fig. S10,† the nanosheet morphology of the NiFeS sample was well preserved after the sulfuration of NiFe-LDH. The original nanosheet structure morphology of TiO2-coated NiFeS (Fig. 3b and d) was well retained (Fig. 3a and c). The majority of the NiFeS surface was conformally coated by TiO2via the ALD process, i.e., ∼100% TiO2 coverage. Moreover, comparing Fig. 3c and d, thicker nanosheets were observed for the 5 nm TiO2/NiFeS sample, indicating that the nanosheet surface was uniformly covered with amorphous TiO2. The full coverage of Ti and O elements over the NiFeS nanosheet surfaces was confirmed by scanning electron microscopy-energy dispersive X-ray spectroscopy (SEM-EDX) mapping (Fig. S11†). Furthermore, as shown in Fig. 3e, the high-resolution transmission electron microscopy (HRTEM) image exhibited lattice fringes with an interplanar spacing of 0.29 nm corresponding to the spacing of the (110) crystal planes of the NiFeS nanosheets. The HRTEM structural characterization indicated that the NiFeS nanosheet basal planes had a {001}-type crystal orientation, which was reported to be catalytically active.45 Both the structural and elemental characterization experiments verified that an amorphous TiO2 layer formed a heterojunction with the underlying NiFeS nanosheets with ∼100% surface coverage on their basal planes (Fig. 3e). After ALD, the NiFeS crystal structure was maintained. As illustrated in the EDX elemental mapping images (Fig. 3f and g, and S12†), the Ni, Fe, and S elements were uniformly distributed within the NiFeS nanosheets, with the elemental intensity varying with surface morphology occasionally, while the Ti and O elements covered the NiFeS surfaces uniformly.
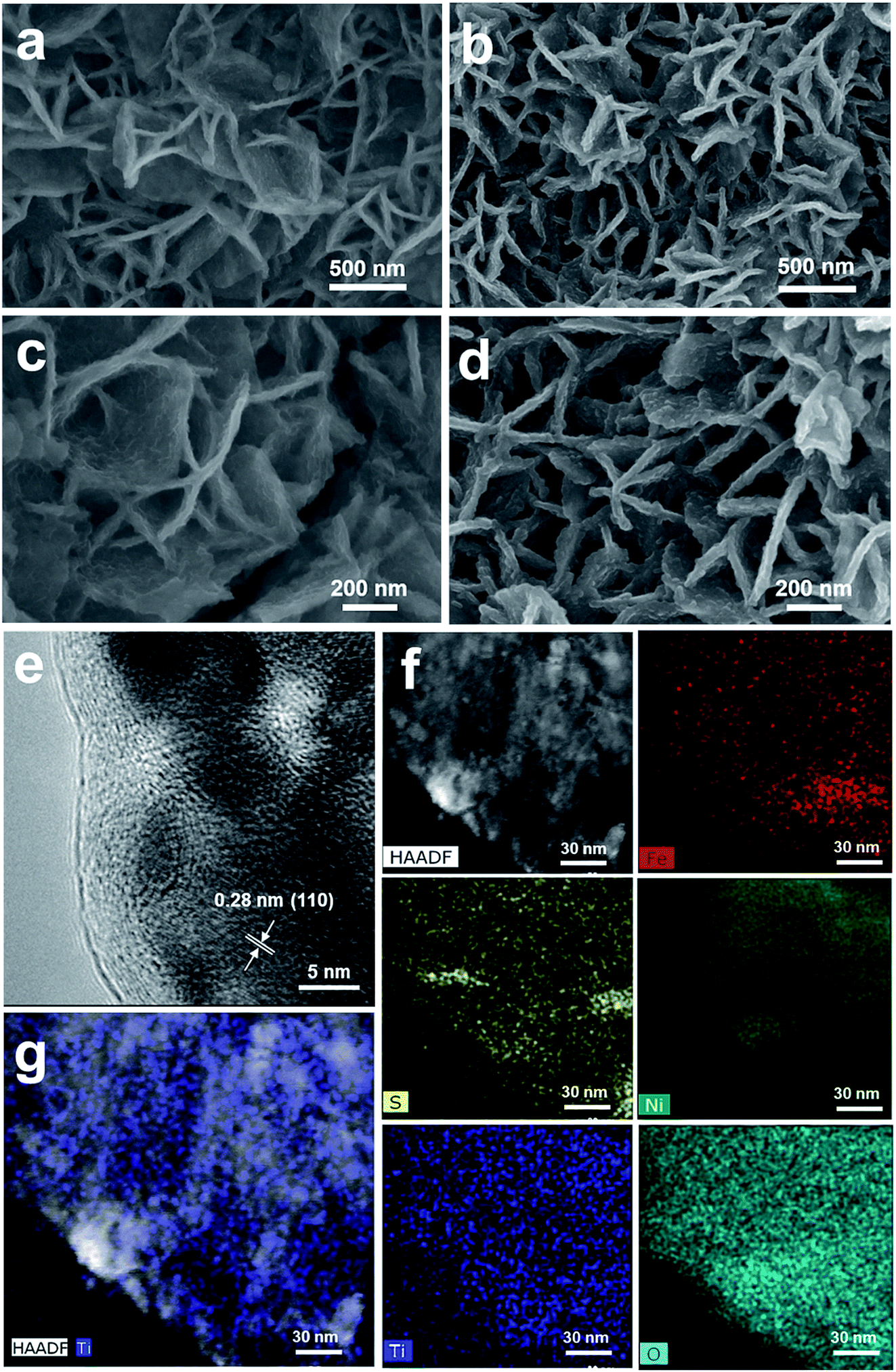 | ||
| Fig. 3 Structural and elemental characterizations of the as-synthesized NiFeS (a and c) and 5 nm ALD TiO2 grown on the NiFeS nanosheets by scanning electron microscopy (SEM) (b and d), and the high-resolution transmission electron microscopy (HRTEM) image (e), high-angle annual dark-field scanning transmission electron microscopy (HAADF-STEM) image, and energy dispersive X-ray spectroscopy (EDX) elemental mapping images of the 5 nm TiO2/NiFeS structure (f and g). | ||
The electronic structure of the buried TiO2/NiFeS heterojunction interfaces was characterized. X-ray photoelectron spectroscopy (XPS), including core-level (CL) and valence band (VB) spectra, was applied to construct the energy-band diagrams of the TiO2/NiFeS heterojunction interfaces.51 All samples were ground from their surfaces, and TiO2 was sufficiently thin for the X-ray to probe the buried TiO2/NiFeS interface. The measurement error for binding energies is below 0.03 eV. The XPS spectra of Ti 2p CL photoemission were deconvoluted as shown in Fig. 4a. In the TiO2/NiFeS sample, a small negative shift (0.2 eV) in the position of the Ti4+ 2p3/2 signal (from 458.55 to 458.35 eV) was observed, compared to TiO2 on the fluorine-doped tin oxide (FTO) substrate (Fig. 4a). Valence band (VB) XPS data showed that TiO2 stayed at the flat band after its deposition over FTO (by comparing the TiO2 valence XPS spectrum on FTO with that on TiO2 deposited on the NiFeS catalyst as shown in Fig. S9b†).37,51 Because of the comparable valence band edges for TiO2 on FTO and on NiFeS, the core-level peaks can be used to derive the band bending diagram at the NiFeS/TiO2 interface (see the ESI† for detailed explanations). The negative shift indicated that the TiO2 donated electrons to the NiFeS substrate across the buried interface. These observations indicated that NiFeS accepted electrons at the NiFeS/TiO2 interface and the very interface became a thin, two-dimensional sheet of accumulated electrons. As shown in Fig. 4b, the TiO2 O 1s CL photoemission peak-intensity shift was also negative 0.2 eV (from 529.90 to 529.70 eV), verifying the observed electron accumulation via interfacial charge transfer.52,53 Accordingly, the increase in electron density at the NiFeS nanosheet surface is evident by a positive shift in the binding energies for Ni 2p, Fe 2p, and S 2p XPS peaks, as shown in Fig. 4c, d, and e, respectively. The chemical states of Ni2+, Fe2+, and S2− were maintained after coating. The direct evidence of electron accumulation is that the band edges and core-level peaks of NiFeS further shift downwards at heterojunction interfaces. Because of the TiO2 coating, the electrons were accumulated at the buried interface, thus causing the band edges of NiFeS at the buried interface to be shifted upward by 0.5 eV relative to the band edges of the NiFeS bulk (for detailed calculations, see the ESI†).30,53 VB XPS spectra of the TiO2/NiFeS and NiFeS samples are shown in Fig. S9c† and d,† respectively. The VB XPS data of the TiO2/NiFeS (Fig. S9c†) are comparable to the reported VBM value of “leaky” TiO2 with a Ti3+-defect band inside its band gap.51 The Fermi level of both samples is located at 0 eV. The valence-band maximum (VBM) position for the TiO2 grown on NiFeS surfaces was assigned as 2.95 eV below the Fermi level of the heterojunction structure. The VB cut-off value of the NiFeS sample (Fig. S9d†) was at 0 eV, indicating that the NiFeS is either metallic or semi-metallic, which agrees with previous reports.45,46,54–56
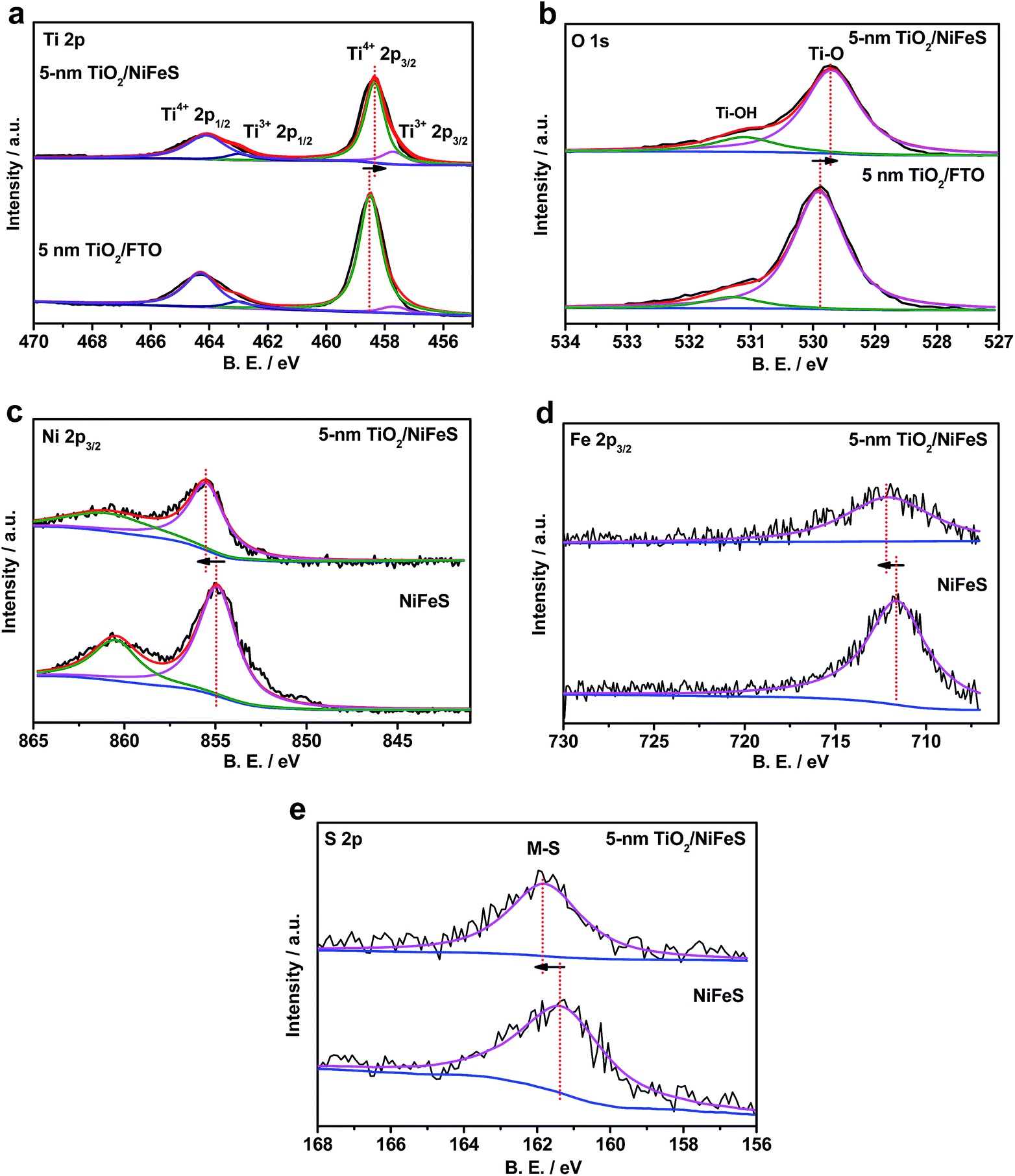 | ||
| Fig. 4 X-ray photoelectron spectroscopy (XPS) spectra of Ti 2p (a) and O 1s (b) core-level photoemissions for the 5 nm ALD TiO2 grown on the NiFeS and FTO substrates, and XPS spectra of Ni 2p (c), Fe 2p (d), and S 2p (e) core-level photoemissions comparing the 5 nm TiO2/NiFeS and bare NiFeS samples (blue represents the background. Black represents the raw data. The other colours represent fitting curves. The XPS measurement error for binding energies is below 0.03 eV). | ||
Collectively, measurements of CL and VB X-ray photoemission spectra yielded a detailed diagram for interfacial band energetics, which is shown in Fig. 5. Despite the fact that the work function of sulfides is typically considered to be lower than that of TiO2, our measurements showed that the TiO2/NiFeS interfacial chemistry by ALD resulted in band edge alignment as shown in Fig. 5a. The density of states for NiFeS charge-transport bands is not as high as that of typical metals, because the CL peak positions did shift with respect to the Fermi energy level. Therefore, the NiFeS material is considered a semi-metal rather than a conventional metal with much higher density of states.57,58 In this case, electron accumulation lowered the band edge positions of NiFeS, relative to the Fermi level of TiO2/NiFeS heterojunction structures. The semi-metallic properties of NiFeS ensure its downward band bending by 0.5 eV due to electron accumulation, while ALD TiO2 yielded an upward band bending of 0.2 eV due to electron transfer from TiO2 to NiFeS and oxygen vacancy-induced space charge. Therefore, the electrons at the buried interface were found to be delocalized and accumulated into a thin sheet and were distributed across TiO2-coated NiFeS surfaces, thus boosting reductive charge transfer. The electrons were estimated to accumulate within less than the 5 nm skin depth of NiFeS surfaces, according to the band-edge shift and density of states. The relative position of NiFeS band edges that participate in charge transfer during the HER is raised, with respect to the potential of surface *-OH and *-H states. This tuning is not possible without the formation of a NiFeS/TiO2 heterojunction interface. The band diagram analysis indicates that the present strategy is appropriate for tuning the catalytic properties of semi-metals and semiconductors.
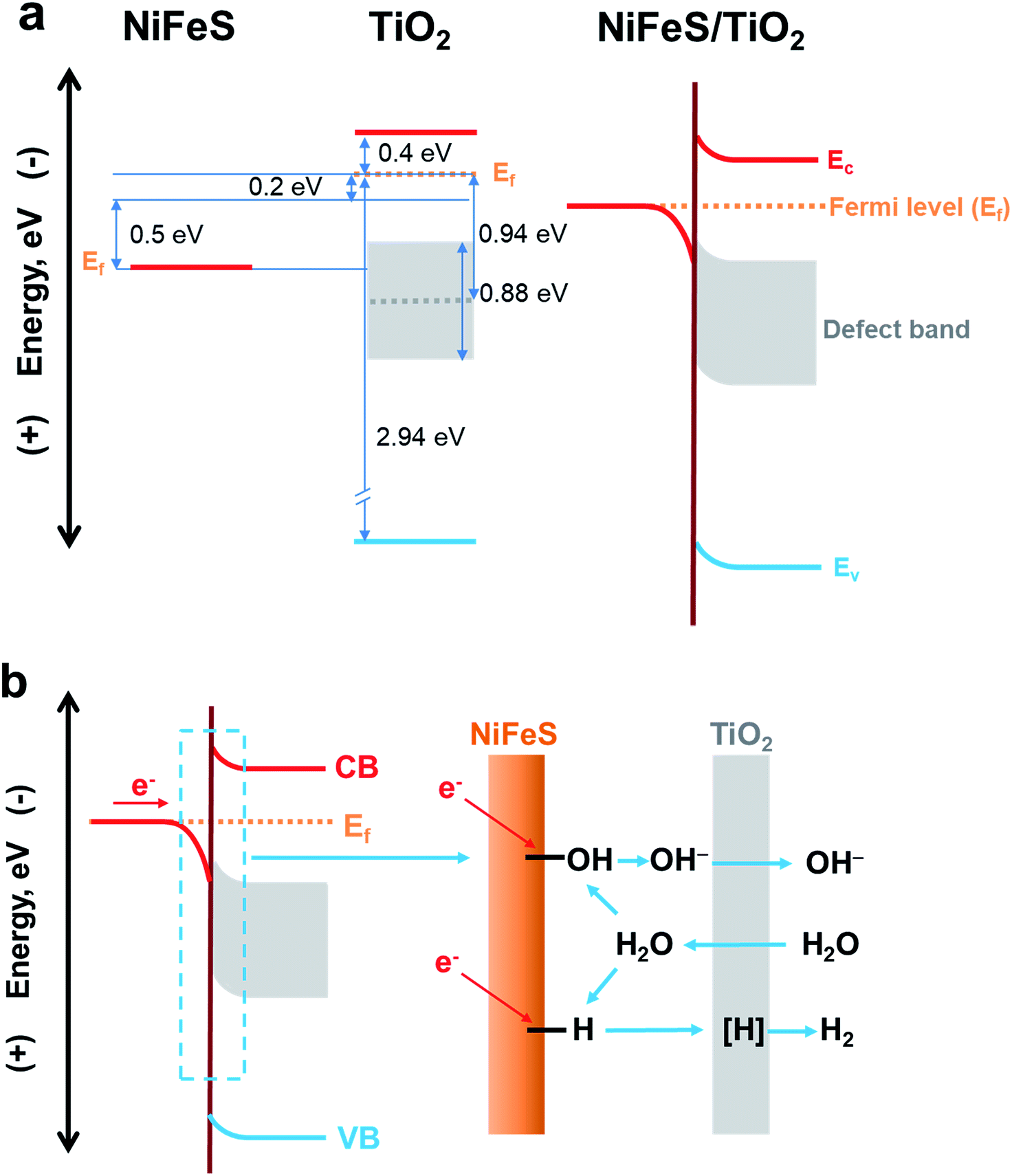 | ||
| Fig. 5 (a) Energy-band diagrams of the TiO2/NiFeS interfaces. (b) Schematic illustration of the NiFeS/TiO2 buried interface for boosting HER activity. | ||
The ALD TiO2 also functions as a sponge that efficiently transfers OH−, water or protons to the buried interface.51,59 It has been reported that metal oxides (especially TiO2) in the solution can transfer/adsorb the protons in the interfacial proton-coupled electron transfer reactions.59,60 A previous study indicated that only H-adsorbed active sites, instead of the OH bonds formed in the direct Volmer step, actively participate in the alkaline HER.61 Therefore, it is important to recognize the proton permeation properties of ALD TiO2 membranes in addition to their interfacial tuning effect. We constructed a solution-based membrane permeation test system (see Fig. S13† for the setup and Fig. S14† for the results). The original Si3N4 film covered on the window of the TEM grid blocks the OH− transfer, which ensures that the pH value of the permeation-side remains below pH 7. With a hole and without the TiO2 membrane, the concentration of OH− should increase to 0.005 mol L−1 in the original permeate compartment of deionized (DI) water by mixing with the pH 12 solution in the original compartment of the OH− source. The pH measurement without the TiO2 membrane is consistent with the above calculation, as the pH reached 11.66 after equilibration. With 5 nm TiO2 covering the window (Si3N4 dissolved in hot KOH leaving TiO2 membranes only), the pH in the permeation-side increases to 10.95 ([OH−] = 0.0009 mol L−1), indicating that the 5 nm TiO2 membrane presents considerable OH− permeation properties. After 18% OH− transfer from the original alkaline side of the membrane to the permeation side, excess amounts of K+ and OH− will accumulate on the retentate and permeate sides of the membrane, respectively. The electrochemical potential will drop across the TiO2 membrane, and the electric field formed will inhibit the OH− from further passing through the membrane. At steady-state, the permeation-side pH cannot reach 11.66 because the permittivity of K+ is less than that of OH−. Our permeation experiment also showed that TiO2 membranes can conduct protons, i.e., H+ (Fig. S14b†). It is reasonable to accept that atomic hydrogen intermediates, denoted as [H], which participated in the HER, can also be transported through the as-fabricated TiO2 semi-permeable membrane on the NiFeS surface. It is possible that the water molecules and OH− ions transported in and out of the membranes participate in the TiO2/NiFeS alkaline HER, because their sizes are in between those of H+ and SO42−. The through-membrane transport properties for H+, OH−, H2O, and the electrolyte species and the associated catalytic mechanism are a subject for further study.
The electrochemical kinetics of the TiO2/NiFeS heterostructure were compared with those of the NiFe-LDH and NiFeS nanosheets. The Tafel slope (Fig. 2b) conveys important information about the rate-determining step (RDS) of the multi-step HER process. The decrease in Tafel slope as the TiO2 coating thickness increases, in conjunction with TiO2's permittivity to a host of catalytic species, supports the activity and tuning hypothesis for the buried catalytic interfaces. Without a buried interface, the HER in alkaline media involves three main steps: the Volmer step (H2O + e− → *H + OH−), the Heyrovsky step (*H + H2O + e− → H2 + OH−), and the Tafel step (*H + *H → H2), where * indicates surface active sites.45 The corresponding Tafel slope of the Volmer, Heyrovsky, and Tafel steps as the RDS should be 120, 40, and 30 mV dec−1, respectively.17 There is an obvious shift in the mechanism with and without the TiO2 modified overlayers: the Tafel slope decreases from 127 to 69 mV dec−1. When the coating thickness increases from 5 to 10 nm, the RDS becomes limited by [H] transport as the Tafel slope increases from 69 to 135 mV dec−1.62 The mechanistic shift may be evidenced by the observed decrease in the Tafel slope from 120 to 69 mV dec−1 with increasing TiO2 coating thickness (from 1 to 5 nm). Although the Tafel slopes for bare NiFeS and NiFeS with an optimal coating thickness appear similar, the mechanism behind these measured Tafel slopes with and without TiO2 overlayers is completely different. Therefore, we have proposed one plausible mechanism though we have not yet detected the postulated molecular species at the buried catalytic interface.
| H2Osolution → H2Ointerface | (4) |
| H2O → *1-OH + *2-H (at the buried interface) | (5) |
| *2-H + e− → [H]interface | (6) |
| *1-OH + e− → OH− | (7) |
| OHinterface− → OHsolution− | (8) |
| [H]interface → [H]TiO2 surface (transport) | (9) |
| 2 [H]TiO2 surface → H2(g) | (10) |
H2O dissociation can produce surface bonded *-OH and *-H at the buried interface, followed by a competitive electron-transfer process to *-OH vs. to *-H at the Ti3+-coordinated, confined NiFeS catalytic surfaces. Then, atomic [H] will be generated after the reduction of *-H (step (6)). The [H] is considered to migrate from the interface to the catalyst/liquid interface through the 5 nm TiO2 coating. There is a correlation between the original NiFeS active sites buried by TiO2 coatings and their enhanced HER activity. It is postulated that the buried TiO2/NiFeS interfaces are the main contributor for improving HER performance. Direct observation of catalytic intermediates and active sites at buried interfaces is an ongoing challenge for in operando characterization of electrochemical processes and may be done in the future.
The TiO2/NiFeS buried interfaces also provide Ti–OH bonding sites, which may catch surface OH from NiFeS to further alleviate *-OH poisoning of the active sites and favor the Heyrovsky step. Additionally, the TiO2 after being deposited on NiFeS contains more low-coordinated Ti ions (Ti3+) than TiO2 on FTO substrates (satellite peaks next to Ti4+ peaks in Fig. 4a);63 this condition is consistent with electronic filling into a Ti3+-defect band.51 It is reasonable to consider that the catalytically active sites extended into the TiO2 phase near the TiO2/NiFeS buried interface, as Ti3+ ions were shown to present high HER activity.63 Therefore, the structural and energy-diagram characterization confirms that the tuning of NiFeS interfacial electrocatalytic properties via charge-transfer interaction and Ti3+ covalent coordination can be continuous and uniform.
At 10 mA cm−2, the stability after 30 hours of continuous operation verified the technological feasibility for photoelectrochemical water splitting. Promoting the out-diffusion of atomic hydrogen intermediates by increasing the TiO2 porosity may improve the electrochemical stability of NiFeS used in an electrolyzer. The coating is beneficial because it prevents the anticipated lattice exchange so that Ni2+ and Fe2+ cations are not liberated into the electrolyte. The Ni–H and Fe–H hydride intermediates may also favor leaving the NiFeS lattice with a relatively large equilibrium constant for their soluble species.43 One of the multiple functions of TiO2 is the physical confinement of the lattice cations or surface hydrides to prevent dissolution. This aspect was confirmed from the ICP-MS results of dissolved Ni2+ and Fe2+ (Table S2†). TiO2 coating resulted in less dissolution of the active element into the electrolyte than in the case of uncoated NiFeS catalysts. So far, we have demonstrated that the rate of the alkaline HER was improved by facilitating the Heyrovsky step on the TiO2 membrane/coating, optimizing the [H] formation and transport at the TiO2/NiFeS buried interface.
In order to better understand the atomic-scale structure of amorphous TiO2 membranes during electrocatalysis, the X-ray absorption spectra (XAS) of 5 nm TiO2/NiFeS and TiO2/fused silica (TiO2/silica) were acquired at the National Synchrotron Light Source II (NSLS II), shown in Fig. 6. Both TiO2/NiFeS and TiO2/silica gave only one peak in the pre-edge region near 4966 eV, confirming that the ALD TiO2 is amorphous on NiFeS and quartz.64 TiO2/NiFeS showed a slight decrease in the Ti absorption-edge energy, which is considered to be a consequence of the reduction of Ti cations in the TiO2 coating.65 TiO2 on NiFeS catalysts showed a Ti coordination environment similar to that in the TiO2/silica, as revealed in X-ray absorption near-edge structure (XANES) spectra. Two obvious XANES peaks at 4985.8 eV and 4999.0 eV, marked D1 and D2, were observed in both TiO2/silica and TiO2/NiFeS. In comparison, the ratio of D1/D2 of the TiO2/NiFeS (1.099) was significantly higher than that of TiO2/silica (1.044). The larger D1/D2 ratio indicates a stronger charge-transfer state contribution, which suggests more electron exchange between NiFeS and the TiO2 coating,66 thereby resulting in the accumulation of electrons at the TiO2/NiFeS interface. The XANES analysis indicated that the TiO2 layer was in a more reduced electronic state. XAS analysis is in accordance with the XPS results of the low valence (Ti3+) state in the coating. Moreover, comparing the TiO2/NiFeS catalyst before and after the 30 hour HER operation, identical Ti K-edge XANES spectra were obtained, again indicating that the electronic structure of the TiO2 coating is stable after long-term operation.
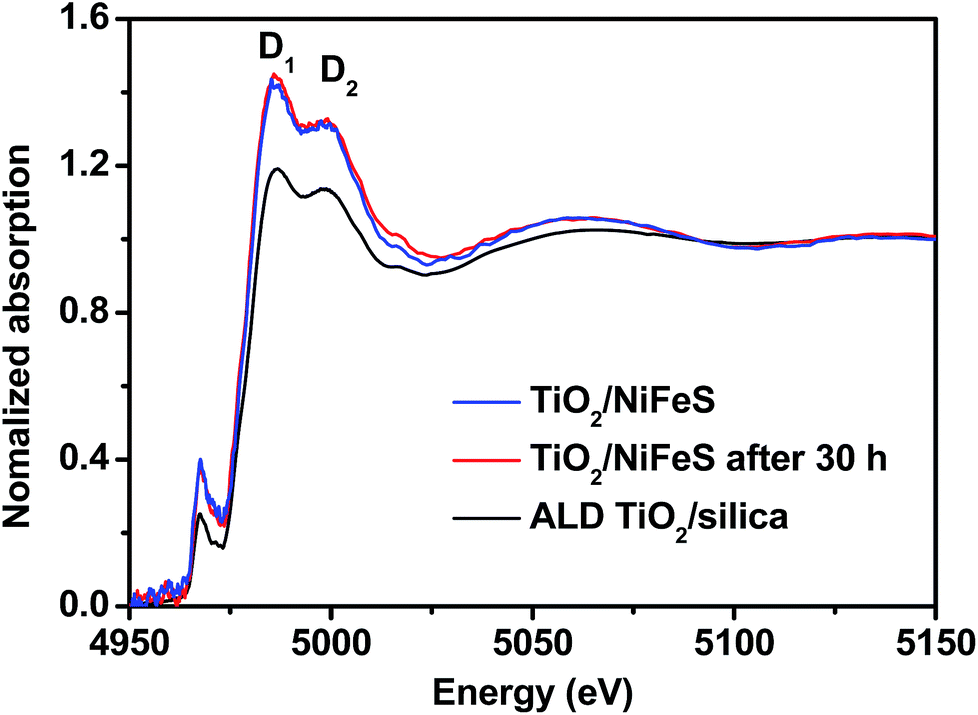 | ||
| Fig. 6 Comparison of Ti K-edge X-ray absorption near edge structure (XANES) data collected on the as-prepared TiO2/silica and 5 nm TiO2/NiFeS nanosheets attached to Ni foam, before and after 30 hours of HER operation. | ||
Conclusions
In summary, TiO2/NiFeS heterostructured HER electrocatalysts were synthesized by atomic layer deposition (ALD) coating over hydrothermally grown NiFeS nanosheets. The TiO2 layers were uniformly coated over the surface of NiFeS, resulting in fine tuning of both the electronic properties of the buried TiO2/bimetallic sulfide HER interfaces and the steric effects by TiO2-coating confinement. The optimally designed 5 nm TiO2/NiFeS heterostructures showed the highest HER activity, displaying a current density of −50 mA cm−2 at a small overpotential of −107 mV, a Tafel slope as small as 69 mV dec−1, and 30 hour stability in alkaline media. We achieved small overpotentials and long-term stability for 10 mA cm−2, which is an important benchmark for light-driven water splitting. This enhanced performance for bare NiFeS nanosheets can be explained by the favorable electronic structure at the buried interface: the charge-transfer driving force from NiFeS band edges to HER intermediates was increased. The ALD TiO2 coating is considered an electron donor and proton sponge. Observing a significant reduction of the Tafel slope from 127 mV dec−1 for bare NiFeS to 69 mV dec−1 for TiO2/NiFeS, we proposed that the rate of the alkaline HER was improved by facilitating the Heyrovsky step at the buried interface, and optimizing the transport of hydrogen intermediates through the TiO2 membrane. The results of the X-ray absorption near-edge structure (XANES) further verified electronic interactions between NiFeS and the TiO2 coating. This study leads to a set of strategies including fine tuning the surface electronic properties of semi-metallic or semiconductive catalysts and confining charges and intermediates at a buried interface formed by a permeable coating. This approach has potential for general applicability in improving the performance of practical water splitting and other electrocatalytic reactions.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the financial support of the University of Chinese Academy of Sciences (UCAS[2015]37) Joint PhD Training Program, the National Natural Science Foundation of China (No. 51774261), and the Petroleum Research Fund (ND55524). This research used resources of the Center for Functional Nanomaterials, and beamline 8-ID ISS (Inner Shell Spectroscopy) at the National Synchrotron Light Source II; both are DOE Office of Science User Facilities operated for the DOE Office of Science by Brookhaven National Laboratory under Contract No. DE-SC00122704. The authors would like to thank Dr Min Li of Yale's Materials Characterization Core (MCC) and Dr Michael Rooks of the Yale Institute for Nanoscience and Quantum Engineering (YINQE) for their invaluable help.References
- E. J. Popczun, C. G. Read, C. W. Roske, N. S. Lewis and R. E. Schaak, Angew. Chem., Int. Ed., 2014, 53, 5427–5430 CrossRef CAS PubMed.
- C. C. McCrory, S. Jung, I. M. Ferrer, S. M. Chatman, J. C. Peters and T. F. Jaramillo, J. Am. Chem. Soc., 2015, 137, 4347–4357 CrossRef CAS PubMed.
- E. J. Popczun, J. R. McKone, C. G. Read, A. J. Biacchi, A. M. Wiltrout, N. S. Lewis and R. E. Schaak, J. Am. Chem. Soc., 2013, 135, 9267–9270 CrossRef CAS PubMed.
- C. Xia, H. Liang, J. Zhu, U. Schwingenschlögl and H. N. Alshareef, Adv. Energy Mater., 2017, 7, 1602089 CrossRef.
- I. Dincer and C. Acar, Int. J. Hydrogen Energy, 2015, 40, 11094–11111 CrossRef CAS.
- M. Xiao, B. Luo, M. Lyu, S. Wang and L. Wang, Adv. Energy Mater., 2018, 8, 1701605 CrossRef.
- J. Chen, X. J. Wu, L. Yin, B. Li, X. Hong, Z. Fan, B. Chen, C. Xue and H. Zhang, Angew. Chem., Int. Ed., 2015, 54, 1210–1214 CrossRef CAS PubMed.
- P. Zhang, L. Yu and X. W. D. Lou, Angew. Chem., Int. Ed., 2018, 57, 15076–15080 CrossRef CAS PubMed.
- G. Zhang, G. Wang, Y. Liu, H. Liu, J. Qu and J. Li, J. Am. Chem. Soc., 2016, 138, 14686–14693 CrossRef CAS PubMed.
- T. Zheng, W. Sang, Z. He, Q. Wei, B. Chen, H. Li, C. Cao, R. Huang, X. Yan, B. Pan, S. Zhou and J. Zeng, Nano Lett., 2017, 17, 7968–7973 CrossRef CAS PubMed.
- R. Miao, B. Dutta, S. Sahoo, J. He, W. Zhong, S. A. Cetegen, T. Jiang, S. P. Alpay and S. L. Suib, J. Am. Chem. Soc., 2017, 139, 13604–13607 CrossRef CAS PubMed.
- X. Wang, W. Ma, Z. Xu, H. Wang, W. Fan, X. Zong and C. Li, Nano Energy, 2018, 48, 500–509 CrossRef CAS.
- Y. Qiu, Z. Wen, C. Jiang, X. Wu, R. Si, J. Bao, Q. Zhang, L. Gu, J. Tang and X. Guo, Small, 2019, 15, 1900014 CrossRef PubMed.
- X. Du, W. Lian and X. Zhang, Int. J. Hydrogen Energy, 2018, 43, 20627–20635 CrossRef CAS.
- X. Du, Q. Wang, Y. Li and X. Zhang, Dalton Trans., 2018, 47, 10273–10280 RSC.
- X. Du, X. Zhang, Y. Li and M. Zhao, Int. J. Hydrogen Energy, 2018, 43, 19955–19964 CrossRef CAS.
- N. Mahmood, Y. Yao, J.-W. Zhang, L. Pan, X. Zhang and J.-J. Zou, Adv. Sci., 2018, 5, 1700464 CrossRef PubMed.
- X. P. Yin, H. J. Wang, S. F. Tang, X. L. Lu, M. Shu, R. Si and T. B. Lu, Angew. Chem., Int. Ed., 2018, 57, 9382–9386 CrossRef CAS PubMed.
- J. N. Tiwari, S. Sultan, C. W. Myung, T. Yoon, N. Li, M. Ha, A. M. Harzandi, H. J. Park, D. Y. Kim, S. S. Chandrasekaran, W. G. Lee, V. Vij, H. Kang, T. J. Shin, H. S. Shin, G. Lee, Z. Lee and K. S. Kim, Nat. Energy, 2018, 3, 773–782 CrossRef CAS.
- X. Du, Z. Yang, Y. Li, Y. Gong and M. Zhao, J. Mater. Chem. A, 2018, 6, 6938–6946 RSC.
- X. Li, W. Liu, M. Zhang, Y. Zhong, Z. Weng, Y. Mi, Y. Zhou, M. Li, J. J. Cha, Z. Tang, H. Jiang, X. Li and H. Wang, Nano Lett., 2017, 17, 2057–2063 CrossRef CAS PubMed.
- Z. W. Seh, K. D. Fredrickson, B. Anasori, J. Kibsgaard, A. L. Strickler, M. R. Lukatskaya, Y. Gogotsi, T. F. Jaramillo and A. Vojvodic, ACS Energy Lett., 2016, 1, 589–594 CrossRef CAS.
- L.-A. Stern, L. Feng, F. Song and X. Hu, Energy Environ. Sci., 2015, 8, 2347–2351 RSC.
- Y. Zhou, J. V. Pondick, J. L. Silva, J. M. Woods, D. J. Hynek, G. Matthews, X. Shen, Q. Feng, W. Liu, Z. Lu, Z. Liang, B. Brena, Z. Cai, M. Wu, L. Jiao, S. Hu, H. Wang, C. M. Araujo and J. J. Cha, Small, 2019, 15, e1900078 CrossRef PubMed.
- H. Zhou, F. Yu, Y. Huang, J. Sun, Z. Zhu, R. J. Nielsen, R. He, J. Bao, W. A. Goddard III, S. Chen and Z. Ren, Nat. Commun., 2016, 7, 12765 CrossRef CAS PubMed.
- Y. Li, H. Zhang, M. Jiang, Q. Zhang, P. He and X. Sun, Adv. Funct. Mater., 2017, 27, 1702513 CrossRef.
- X. Y. Yu and X. W. Lou, Adv. Energy Mater., 2018, 8, 1701592 CrossRef.
- S.-Y. Huang, D. Sodano, T. Leonard, S. Luiso and P. S. Fedkiw, J. Electrochem. Soc., 2017, 164, F276–F282 CrossRef CAS.
- C. Bae, T. A. Ho, H. Kim, S. Lee, S. Lim, M. Kim, H. Yoo, J. M. Montero-Moreno, J. H. Park and H. Shin, Sci. Adv., 2017, 3, e1602215 CrossRef PubMed.
- Y. Wu, X. Liu, D. Han, X. Song, L. Shi, Y. Song, S. Niu, Y. Xie, J. Cai, S. Wu, J. Kang, J. Zhou, Z. Chen, X. Zheng, X. Xiao and G. Wang, Nat. Commun., 2018, 9, 1425 CrossRef PubMed.
- X. Long, G. Li, Z. Wang, H. Zhu, T. Zhang, S. Xiao, W. Guo and S. Yang, J. Am. Chem. Soc., 2015, 137, 11900–11903 CrossRef CAS PubMed.
- C. Chu, D. Huang, Q. Zhu, E. Stavitski, J. A. Spies, Z. Pan, J. Mao, H. L. Xin, C. A. Schmuttenmaer, S. Hu and J.-H. Kim, ACS Catal., 2018, 9, 626–631 CrossRef.
- Y. Huang, J. Hu, H. Xu, W. Bian, J. Ge, D. Zang, D. Cheng, Y. Lv, C. Zhang, J. Gu and Y. Wei, Adv. Energy Mater., 2018, 8, 1800789 CrossRef.
- Y. Xue, Z. S. Fishman, J. A. Röhr, Z. Pan, Y. Wang, C. Zhang, S. Zheng, Y. Zhang and S. Hu, J. Mater. Chem. A, 2018, 6, 21918–21926 RSC.
- C. E. Finke, S. T. Omelchenko, J. T. Jasper, M. F. Lichterman, C. G. Read, N. S. Lewis and M. R. Hoffmann, Energy Environ. Sci., 2019, 12, 358–365 RSC.
- N. Cheng, Y. Shao, J. Liu and X. Sun, Nano Energy, 2016, 29, 220–242 CrossRef CAS.
- S. Hu, M. R. Shaner, J. A. Beardslee, M. Lichterman, B. S. Brunschwig and N. S. Lewis, Science, 2014, 344, 1005–1009 CrossRef CAS PubMed.
- D. V. Esposito, ACS Catal., 2017, 8, 457–465 CrossRef.
- N. Y. Labrador, E. L. Songcuan, C. De Silva, H. Chen, S. J. Kurdziel, R. K. Ramachandran, C. Detavernier and D. V. Esposito, ACS Catal., 2018, 8, 1767–1778 CrossRef CAS.
- J. G. Vos, T. A. Wezendonk, A. W. Jeremiasse and M. T. M. Koper, J. Am. Chem. Soc., 2018, 140, 10270–10281 CrossRef CAS PubMed.
- T. Moehl, J. Suh, L. Sévery, R. Wick-Joliat and S. D. Tilley, ACS Appl. Mater. Interfaces, 2017, 9, 43614–43622 CrossRef CAS PubMed.
- P. Poodt, D. C. Cameron, E. Dickey, S. M. George, V. Kuznetsov, G. N. Parsons, F. Roozeboom, G. Sundaram and A. Vermeer, J. Vac. Sci. Technol., A, 2012, 30, 010802 CrossRef.
- J. Divisek, H. Schmitz and B. Steffen, Electrochim. Acta, 1994, 39, 1723–1731 CrossRef CAS.
- M. Grdeń, M. Alsabet and G. Jerkiewicz, ACS Appl. Mater. Interfaces, 2012, 4, 3012–3021 CrossRef PubMed.
- G. Zhang, Y.-S. Feng, W.-T. Lu, D. He, C.-Y. Wang, Y.-K. Li, X.-Y. Wang and F.-F. Cao, ACS Catal., 2018, 8, 5431–5441 CrossRef CAS.
- T. A. Ho, C. Bae, H. Nam, E. Kim, S. Y. Lee, J. H. Park and H. Shin, ACS Appl. Mater. Interfaces, 2018, 10, 12807–12815 CrossRef CAS PubMed.
- L. X. Chen, Z. W. Chen, Y. Wang, C. C. Yang and Q. Jiang, ACS Catal., 2018, 8, 8107–8114 CrossRef CAS.
- F. Lyu, Y. Bai, Z. Li, W. Xu, Q. Wang, J. Mao, L. Wang, X. Zhang and Y. Yin, Adv. Funct. Mater., 2017, 27, 1702324 CrossRef.
- J. Luo, J. H. Im, M. T. Mayer, M. Schreier, M. K. Nazeeruddin, N. G. Park, S. D. Tilley, H. J. Fan and M. Gratzel, Science, 2014, 345, 1593–1596 CrossRef CAS PubMed.
- R. Frydendal, E. A. Paoli, I. Chorkendorff, J. Rossmeisl and I. E. L. Stephens, Adv. Energy Mater., 2015, 5, 1500991 CrossRef.
- S. Hu, M. H. Richter, M. F. Lichterman, J. Beardslee, T. Mayer, B. S. Brunschwig and N. S. Lewis, J. Phys. Chem. C, 2016, 120, 3117–3129 CrossRef CAS.
- T. Sun, J. Wang, X. Chi, Y. Lin, Z. Chen, X. Ling, C. Qiu, Y. Xu, L. Song, W. Chen and C. Su, ACS Catal., 2018, 8, 7585–7592 CrossRef CAS.
- S. Liu, M. Li, C. Wang, P. Jiang, L. Hu and Q. Chen, ACS Sustainable Chem. Eng., 2018, 6, 9137–9144 CrossRef CAS.
- G. V. Gibbs, R. T. Downs, C. T. Prewitt, K. M. Rosso, N. L. Ross and D. F. Cox, J. Phys. Chem. B, 2005, 109, 21788–21795 CrossRef CAS PubMed.
- L. L. Feng, G. Yu, Y. Wu, G. D. Li, H. Li, Y. Sun, T. Asefa, W. Chen and X. Zou, J. Am. Chem. Soc., 2015, 137, 14023–14026 CrossRef CAS PubMed.
- J. H. Wang, Z. Cheng, J. L. Bredas and M. Liu, J. Chem. Phys., 2007, 127, 214705 CrossRef PubMed.
- G. A. Saunders, Contemp. Phys., 1973, 14, 149–166 CrossRef CAS.
- S. M. Rommel and R. Weihrich, Chem.–Eur. J., 2015, 21, 9863–9867 CrossRef CAS PubMed.
- J. N. Schrauben, R. Hayoun, C. N. Valdez, M. Braten, L. Fridley and J. M. Mayer, Science, 2012, 336, 1298–1301 CrossRef CAS PubMed.
- C. N. Valdez, M. Braten, A. Soria, D. R. Gamelin and J. M. Mayer, J. Am. Chem. Soc., 2013, 135, 8492–8495 CrossRef CAS PubMed.
- S. Intikhab, J. D. Snyder and M. H. Tang, ACS Catal., 2017, 7, 8314–8319 CrossRef CAS.
- Y. R. Zheng, P. Wu, M. R. Gao, X. L. Zhang, F. Y. Gao, H. X. Ju, R. Wu, Q. Gao, R. You, W. X. Huang, S. J. Liu, S. W. Hu, J. Zhu, Z. Li and S.-H. Yu, Nat. Commun., 2018, 9, 2533 CrossRef PubMed.
- H. Feng, Z. Xu, L. Ren, C. Liu, J. Zhuang, Z. Hu, X. Xu, J. Chen, J. Wang, W. Hao, Y. Du and S. X. Dou, ACS Catal., 2018, 8, 4288–4293 CrossRef CAS.
- F. Farges, G. E. Brown and J. J. Rehr, Phys. Rev. B: Condens. Matter Mater. Phys., 1997, 56, 1809–1819 CrossRef CAS.
- Y. Li, K. A. Kuttiyiel, L. Wu, Y. Zhu, E. Fujita, R. R. Adzic and K. Sasaki, ChemSusChem, 2017, 10, 68–73 CrossRef CAS PubMed.
- I. Harada, T. Suzuki and A. Kotani, J. Phys. Soc. Jpn., 1996, 65, 3016–3020 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ta07123g |
| This journal is © The Royal Society of Chemistry 2019 |