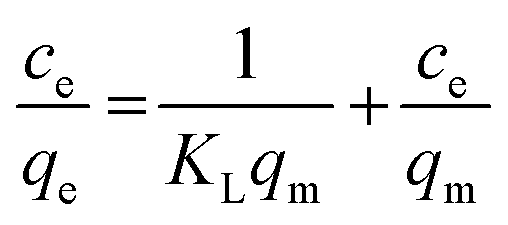DOI:
10.1039/C9TA06735C
(Paper)
J. Mater. Chem. A, 2019,
7, 19081-19086
Core–shell TiO2@C ultralong nanotubes with enhanced adsorption of antibiotics†
Received
22nd June 2019
, Accepted 18th July 2019
First published on 24th July 2019
Abstract
As materials capable of adsorbing antibiotics continue to be developed, composite adsorbents have been shown to offer advantages over mono-material adsorbents. In this work, ultralong titanium dioxide@carbon nanotubes were prepared by a simple hydrothermal treatment, followed by carbonization. The composite material is able to adsorb three different categories of antibiotics, including tetracycline (TC), ofloxacin (OFO) and norfloxacin (NFO). The adsorption results show that the adsorption properties of composite materials have been greatly improved compared with single inorganic adsorbent materials, for which the adsorption capacities are 240 mg g−1 (TC), 232 mg g−1 (OFO), and 190 mg g−1 (NFO), respectively. The adsorption mechanism is consistent with a Langmuir pseudo-first-order kinetic model.
Introduction
With the discovery of antibiotics, such as penicillin, for human beings to fight infectious diseases, there is a common phenomenon of abusing antibiotics to cope with various illnesses.1 However, antibiotics are difficult to metabolize in a short period of time in animals, plants and humans. Simultaneously, emissions from pharmaceutical companies have also caused enormous environmental pollution.2,3 Thus, a large amount of used antibiotics went to the soils, rivers, animals, etc. The accumulation of these residual antibiotics not only pollutes the environment, but also causes harm to human health.4 With the emergence of super bacteria, there is serious concern that society will lack the required antibiotics in future.5 The warnings of scientists have attracted the attention of the public and governments. Therefore, searching for effective and non-toxic methods to remove antibiotic pollutants has become a hot research topic.
There are a number of methods which aim to decrease the amount of antibiotics in the environment, including adsorption,6 photocatalysis,7 electrochemical oxidation,8 advanced oxidation9 and biodegradation.10 Among these methods, adsorption is a simple and cheap way to solve the problem of antibiotic pollution and can be divided into two categories: chemisorption and physisorption. Chemisorption requires high activation energy and usually higher temperatures, while physisorption is usually caused by molecular force and can occur at lower temperature. Furthermore, the chemisorption reaction may be undetectable at low temperature or pressure.11 As characteristic of physisorption, for gases at temperatures above their critical temperature, adsorption is confined to a monolayer (unimolecular).12 Therefore, the adsorption capacity is related to the specific surface area and structures of the adsorbents. The effects of the specific surface area and structure of different types of single adsorbent materials and composite adsorbent materials on the adsorption capacity have been studied in depth.13 Promising materials, such as zeolites for adsorption of tetracycline,14 attapulgite/carbon composites for adsorption of tetracycline,15 and oil shale powders16 and functional MOFs for adsorption of norfloxacin,17 were reported. A large number of experimental results show that the specific surface area and the structure of composites are the key factors that make adsorption more effective than with a single material. Therefore, there is an increasing focus on developing composite adsorbent materials with an optimized structure and high specific surface area via simple preparation processes amenable to mass production.
In this work, ultralong TiO2 nanotubes were prepared using a simple hydrothermal treatment. In order to sustain the ultralong structure and increase the specific surface area, the ultralong TiO2 nanotubes were covered with an ultrathin carbon layer by carbonizing ethanol, and thus a core–shell TiO2@C ultralong nanotube composite was formed. Three common antibiotics, i.e., tetracycline (TC), ofloxacin (OFO) and norfloxacin (NFO), were adsorbed by the TiO2@C composite, and the adsorption capacities were measured to be 240 mg g−1, 232 mg g−1, 190 mg g−1, respectively, which are higher than those of many of the reported single and composite adsorbent materials. Therefore, the core–shell TiO2@C nanotubes are promising adsorbent materials for antibiotic pollutants. This study sheds light on the mechanism of metal oxide/carbon hybrids for adsorbing antibiotics and identifies routes to improve performance.
Experimental section
Preparation of ultralong titanium dioxide nanotubes
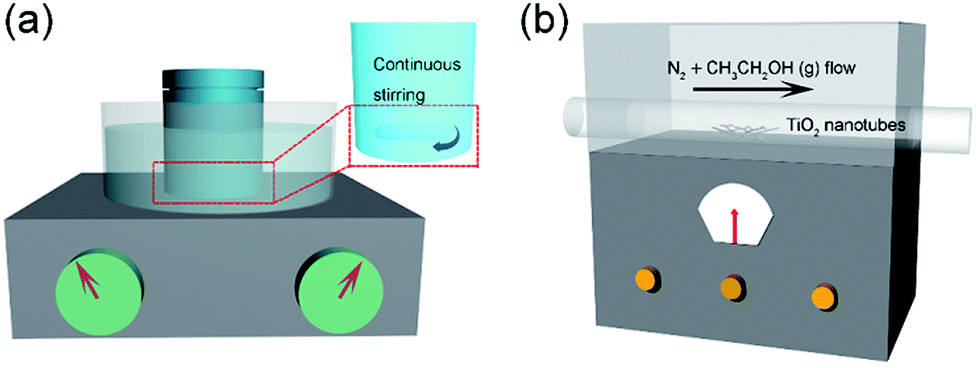
Firstly, 0.5 g P25 (a commercial titanium dioxide which is an anatase and rutile two-phase mixture nanopowder with an average particle diameter of 25 nm) was added into 40 mL of 10 mol L−1 NaOH solution and dispersed in a 50 mL reaction kettle, which was ultrasonicated for 5 min to obtain a homogeneous solution. The reaction kettle was sealed and put into an oil bath at 120 °C for 48 h with magnetic stirring (Fig. 1a). When the reaction finished, the reactor was taken out and cooled to room temperature. The obtained white products were washed with 0.1 mol L−1 HNO3 and centrifuged several times until the pH reached 7. The final products were washed with deionized water and placed in a vacuum drying oven at 60 °C for 8 h.
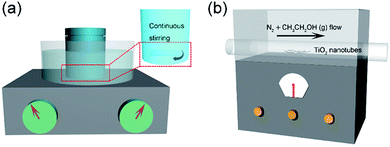 |
| | Fig. 1 Schematic of the preparation process for TiO2@C. | |
Preparation of TiO2@C core–shell structures
The as-prepared products were placed in the middle of a tube furnace, N2 gas was purged through anhydrous ethanol and then into the tube furnace, and the reaction temperature was set at 400 °C for 10 h under a nitrogen atmosphere. Black products (TiO2@C composite) were obtained as the reaction finished. The experimental schematic diagram is shown in Fig. 1b.
Characterization
The prepared TiO2@C composites were characterized by scanning electron microscopy (SEM, S-4800, Japan) and transmission electron microscopy (TEM, JEM-2100F, Japan). The crystal structure and components of the material were determined using an X-ray diffractometer (XRD, Rigaku, Cu-Kα radiation, Japan). The specific surface area and the pore size distribution of the material were determined from the Brunauer–Emmett–Teller (BET) test (ASAP 2020, Micromeritics, America). The surface chemical structure was determined by Fourier transform infrared spectroscopy (FT-IR) (6700, Nicolet, USA) and Raman spectroscopy (Lab RAM, HR800) with 532 nm laser light. The absorbance of the TiO2@C composite materials was measured using a UV spectrophotometer (G-9, Rangqi, China). The functional groups and the elemental states on the surface of the adsorbents were examined by X-ray photoelectron spectroscopy (XPS) analysis, which was conducted on a Kratos Axis Ultra DLD spectrometer, using monochromated Al Kα X-rays at a base pressure of 1 × 10−9 Torr.
Results and discussion
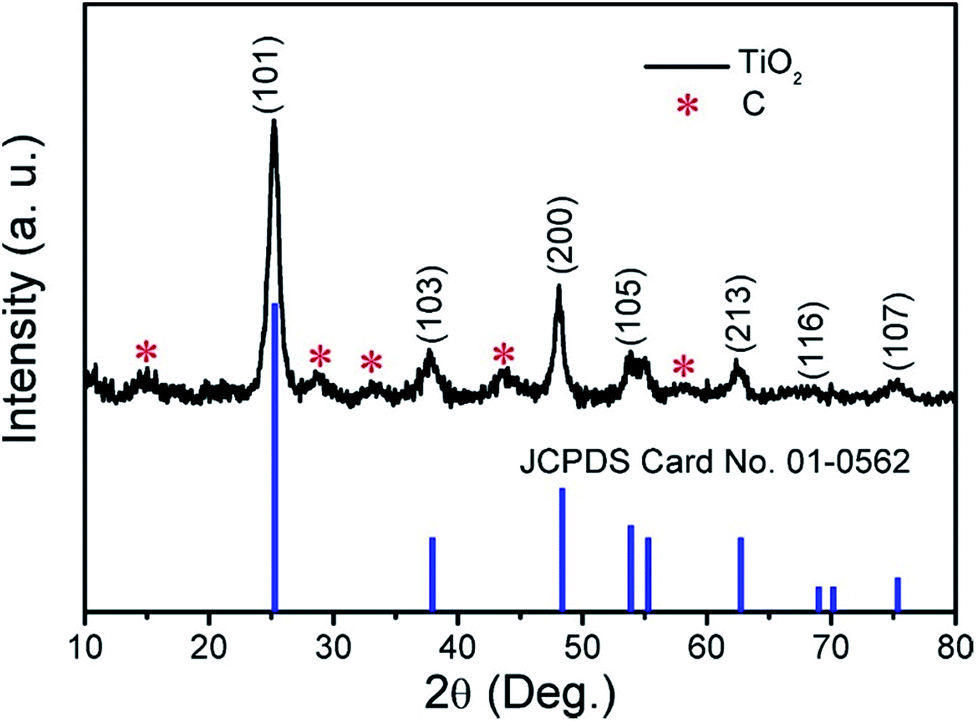
The result of XRD analysis is shown in Fig. 2; the main diffraction peaks could be indexed to the (101), (103), (200), (105), (213), (116) and (107) reflections, corresponding to anatase phase TiO2 with the JCPDS card no. 01-0562. The result also confirms that C exists in the TiO2 products (marked with red asterisks). The C species were further tested by Raman spectroscopy and the result is shown in Fig. S1.† The peak intensity ratio between the G band and D band (IG/ID) is 1.1796, indicating that the proportion of carbon defect structures in the carbon structure is relatively small and the dominant component in the carbon structures is the sp2.
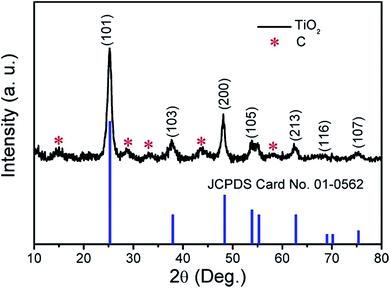 |
| | Fig. 2 XRD pattern of the obtained products. | |
The low- and high-magnification SEM images of the TiO2@C products are shown in Fig. 3a and b. From the SEM images, it can be seen that the length of the TiO2@C nanotubes can be up to several micrometers, representing an ‘ultralong’ structure. From the TEM image of Fig. 3c, it can be confirmed that the composite material is a tubular nanostructure. More detailed information on the material can be obtained from Fig. 3d; the outer diameter of the nanotubes is ∼35 nm, and the inner diameter is ∼15 nm, and the thickness of the carbon layer is 5–6 nm (Fig. 3e). This shows that the carbon layer was successfully coated on the outside of the TiO2 nanotubes, and the interplanar spacing of 0.35 nm indicates the (101) lattice plane of the TiO2 crystal.
 |
| | Fig. 3 Microstructure characterization of the core–shell TiO2@C nanotubes: (a and b) low and high-magnification SEM images, and (c) TEM and (d and e) HRTEM images. | |
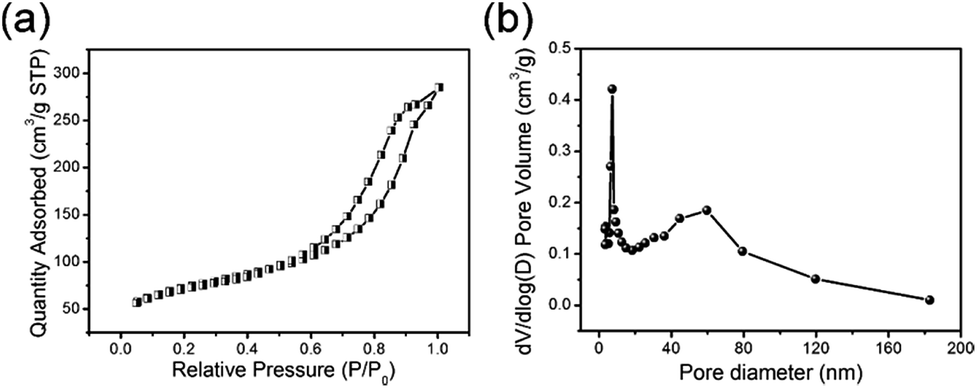
The detailed specific surface area of the TiO2@C core–shell nanotubes can be obtained from the N2 adsorption–desorption curve (Fig. 4a). The specific surface area of TiO2@C can be calculated from the N2 adsorption–desorption isotherm, and the BET value is 346.26 m2 g−1, which is higher than that of the single TiO2 (Fig. S2 in the ESI†). Fig. 4b shows the pore size distribution of the TiO2@C nanotubes, with a main diameter of around 15 nm, which corresponds to the diameter of the nanotubes. The diameter of ∼60 nm can be attributed to the gaps between the nanotubes.
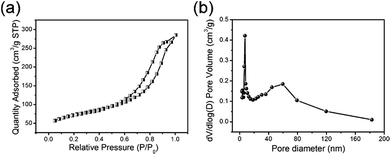 |
| | Fig. 4 (a) The N2 adsorption–desorption isotherm and (b) pore diameter distribution of the TiO2@C nanotubes. | |
The adsorption of the three kinds of antibiotics is based on the classical monolayer Langmuir model and the multilayer Freundlich adsorption model. The Langmuir equation depicts the process of randomly adsorbing species onto the surface of the adsorbent. The linearization equation of the Langmuir adsorption model is shown as follows:18
| | 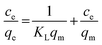 | (1) |
where
Ce is the balance concentration (mg L
−1),
KL is the adsorption constant (L mg
−1),
qm is the maximum adsorption capacity (mg g
−1), and
qe represents the quantity (mg g
−1) of antibiotics adsorbed at equilibrium.
The Freundlich adsorption model is an empirical equation that is simple and widely used. It can simulate a multi-layer adsorption process, which means that the adsorption sites on the adsorption surface are not uniform. The linearized form of the Freundlich adsorption model equation is as follows:19
| | 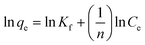 | (2) |
where
KF and
n represent the Freundlich constant [(mg g
−1) (L mg
−1)
1/n] and adsorption intensity of the adsorbents, respectively.
20–22 The mass of the TiO
2@C materials used in this study was 5 mg, for adsorption of 5–40 mg L
−1 tetracycline (TC), 5–40 mL ofloxacin (OFO) and 2–20 mg L
−1 norfloxacin (NFO). Adsorption equilibrium was achieved after magnetic stirring at room temperature.
Fig. 4 shows the experimental data of the TiO
2@C nanotubes adsorbing the three kinds of antibiotics: OFO, TC and NFO.
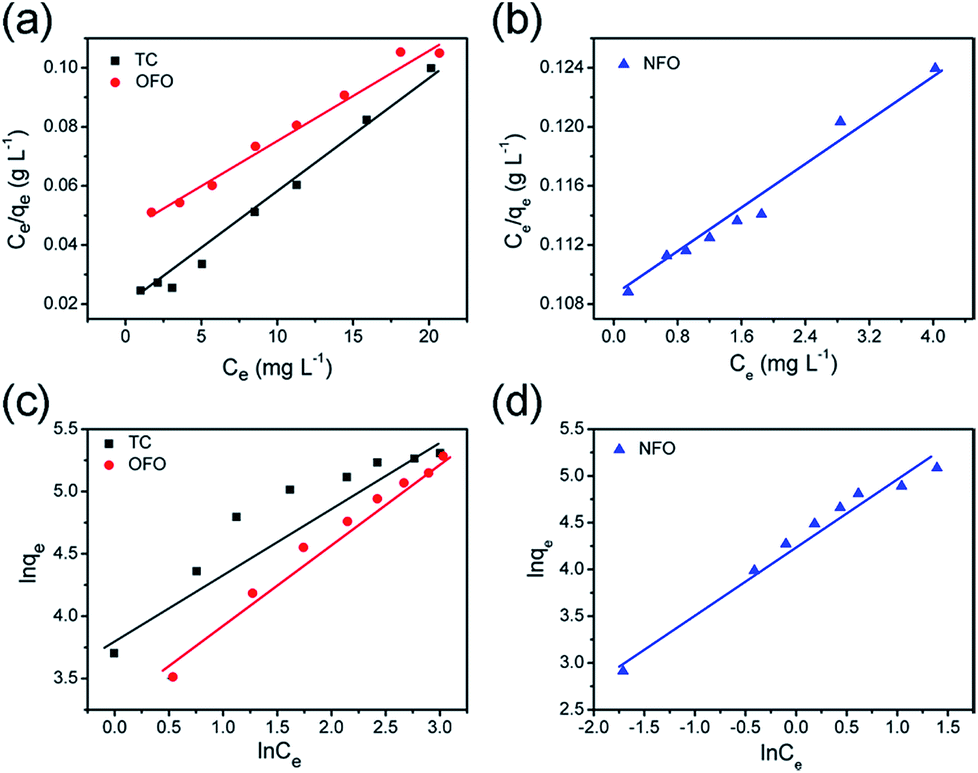
Fig. 5a and b are the Langmuir model fitting charts, and
Fig. 5c and d show the corresponding Freundlich model fitting charts. The fitting data are shown in
Table 1. From the correlation coefficient (
R2) value of the two models, it can be concluded that the three kinds of antibiotics adsorbed by the material are more consistent with the Langmuir model. The maximum adsorption capacities of OFO, TC and NFO, calculated using the Langmuir model, are 232 mg g
−1, 240 mg g
−1, and 190 mg g
−1, respectively. The experimental results show that the adsorption amount of this material is greatly improved compared with other inorganic materials, as shown in
Table 2.
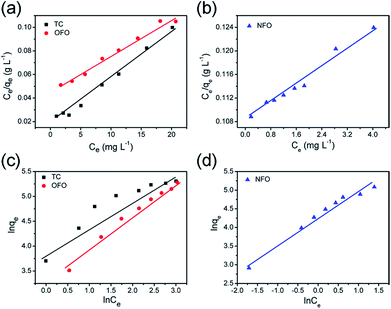 |
| | Fig. 5 The liner fitting curves of the Langmuir (a and b) and Freundlich (c and d) models for TiO2@C towards NFO, TC and OFO. | |
Table 1 Langmuir and Freundlich regression data from the adsorption isotherms of NFO, TC and OFO for the TiO2@C
| Pollutant |
K
L (L mg−1) |
Langmuir qm (mg g−1) |
R
2
|
k
f
|
Freundlich n |
R
2
|
| NFO |
0.0488 |
190 |
0.979 |
70.8014 |
1.4105 |
0.940 |
| TC |
0.2562 |
240 |
0.989 |
53.4171 |
1.9861 |
0.874 |
| OFO |
0.0963 |
232 |
0.980 |
26.0860 |
1.4714 |
0.960 |
Table 2 The adsorption capacity comparison of the adsorbent materials
| Antibiotics |
Adsorbents |
q
m (mg g−1) |
Conditions |
Ref. |
| Tetracycline |
Zeolite |
130 |
pH = 7 |
14
|
| GSO |
137 |
pH = 7 |
23
|
| PS/GO |
197.9 |
pH = 6 |
24
|
| PC@CMCS |
136.9 |
pH = 7 |
25
|
| Ultralong hydrogen titanate nanobelts |
151.51 |
PH = 7 |
26
|
| TiO2@C |
240 |
pH = 7 |
This work |
| Norfloxacin |
AMPH |
1.42 |
pH = 7 |
27
|
| MIP |
29.35 |
pH = 8 |
28
|
| Ultralong hydrogen titanate nanobelts |
111.73 |
pH = 7 |
26
|
| TiO2@C |
190 |
pH = 7 |
This work |
| Ofloxacin |
Oil shale powders |
15.4 |
pH = 7.5 |
16
|
| Ultralong hydrogen titanate nanobelts |
148.14 |
pH = 7 |
26
|
| TiO2@C |
232 |
pH = 7 |
This work |
Adsorption kinetic model
The adsorption kinetics of the three kinds of antibiotics were studied. The pseudo-first-order model was based on the assumption that the adsorption was controlled by diffusion.29 The pseudo-first-order dynamic model is as follows:30| | ln(qe − qt) = ln![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) qe − K1t qe − K1t | (3) |
The pseudo-second-order kinetic model assumes that the adsorption rate is determined by the square value of the number of adsorbed vacancies on the surface of the adsorbent. The pseudo-second-order dynamic model is as follows:31
| | 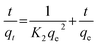 | (4) |
where
qe is the concentration of adsorption at equilibrium (mg g
−1),
qt is the amount of adsorption (mg g
−1) at time
t,
K1 is the pseudo-first-order adsorption rate constant, and
K2 is the pseudo-second-order adsorption rate constant.
17 The fitting data of the dynamics of the three kinds of antibiotics are given in
Table 3.
Table 3 OFO, TC and NFO dynamic correlation fitting data
| Model |
C
0
|
Pseudo-first-order, K1 |
q
e,cal.
|
R
2
|
Pseudo-second-order, K2 |
q
e,cal.
|
R
2
|
| OFO |
40 |
0.00647 |
230.2 |
0.95 |
7.32 × 10−6 |
769.2 |
0.52 |
| TC |
40 |
0.00909 |
252.1 |
0.97 |
2.84 × 10−6 |
454.5 |
0.50 |
| NFO |
20 |
0.00874 |
191.2 |
0.97 |
2.99 × 10−5 |
248.7 |
0.47 |
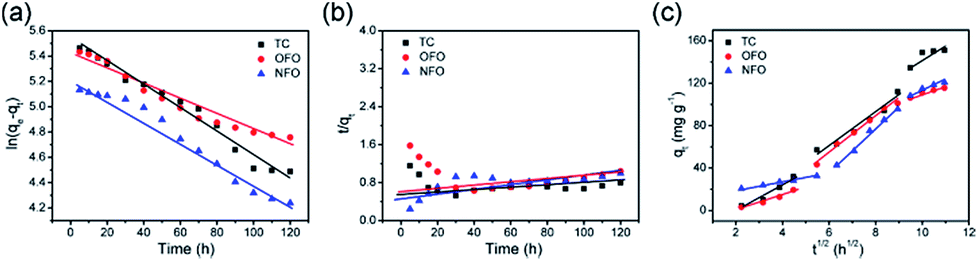
Fig. 6 shows the pseudo-first-order and pseudo-second-order kinetic models of the three antibiotics, and the intra-particle-diffusion model. As Table 3 shows, the pseudo-first-order model best describes the adsorption mechanism. Thus, the adsorption process of TiO2@C adsorbing OFO, TC and NFO is mainly controlled by diffusion.32 In order to obtain more information on the adsorption process, data are fitted to the intra-particle-diffusion model, as follows:33
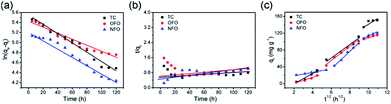 |
| | Fig. 6 (a) The pseudo-first-order model of OFO, TC, and NFO, (b) the pseudo-second-order model of OFO, TC, and NFO, and (c) the intra-particle-diffusion model of OFO, TC, and NFO. | |
Adsorption can be divided into two processes: adsorbent surface adsorption and slow pore diffusion. The particle diffusion model is the most suitable for describing the dynamics of particles in the particle diffusion process.34 The linear fitting of qt and t1/2 gives a straight line that passes through the origin and shows that particle diffusion is the rate determining step.35 From Fig. 6c, it can be seen that the TiO2@C particle diffusion model is divided into three stages; the first phase is the diffusion of the boundary layer, which is mainly the external mass transfer of the adsorbent. The second phase refers to the internal diffusion of particles, which is mainly the transport of microstructures of adsorbents, such as holes. These two stages make up the intra-particle diffusion model. Because the contact angle between the material itself and the solution is different, the time needed for different materials to reach adsorption equilibrium is different. The third phase is usually not considered as a rate-controlling step, generally referring to the procession of adsorption being close to adsorption equilibrium, which means that the active sites of the internal and external surfaces are mostly occupied. Compared with single titanium dioxide nanotube materials, the contact angle of the composites changed due to the synergistic effect of titanium dioxide nanotubes and the carbon layer, which greatly improved the adsorption performance of the material36 (Fig. S3†).
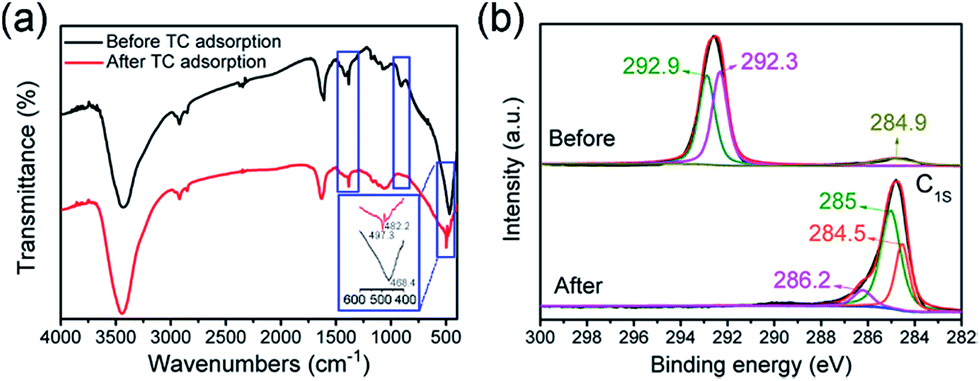
In order to understand the change that has occurred on the surface of the TiO2 nanotubes, FTIR and XPS tests were carried out and TC was selected as an example. Fig. 7a shows the FTIR comparison, before and after the adsorption of TC. The absorption peak at 2920 cm−1 is attributed to the C–H bond. The hydrogen ion was introduced into the soaking TiO2 nanotubes from dilute nitric acid, combined with a small amount of carbon into the C–H bond. The absorption peak at 1384 cm−1 is the result of the N–O bond introduced by TiO2 nanotubes with dilute nitric acid.5 The smaller absorption peaks at 1066 cm−1 and 903 cm−1 are due to the telescopic vibration of the C–O bond. The absorption peak of TiO2@C at 1609 cm−1 is the characteristic peak of the amide group. After adsorption of TC, the peak shifted to 1630 cm−1, indicating that TC was successfully adsorbed on TiO2@C. The sharp peak at 468 cm−1 is the result of the vibration of the Ti–O bond of titanium dioxide nanotubes. In addition, the carbonyl group is formed by the combination of some oxygen elements, which makes the Ti–O bond peak of the TiO2@C composite blunt and the peak shifts to 497 cm−1. The analysis of FT-IR results showed that the adsorption of TiO2@C on TC was mainly physical adsorption, which was in accordance with the pseudo-first-order kinetic model. To further prove that TC has been adsorbed by TiO2@C, XPS spectra (Fig. S4†) of the C 1s peaks of adsorbents before and after adsorption are compared. After deconvolution, the C 1s signal can be separated into a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C/C–C peak at 284.9 eV, a C–O peak at 292.3 eV and a CO peak at 292.9 eV (Fig. 8b). It is noted that the content of CC/C–C is much lower than that of CO. However, after the TC antibiotic was adsorbed by TiO2@C, the adsorbed C 1s signal is deconvoluted to a CC/C–C peak at 284.5 eV, a C–O peak at 285 eV and a CO peak at 286.2 eV. Simultaneously, the content of C–C/CC increased sharply. In contrast, the content of CO is the lowest among the three kinds of chemical bonds. In summary, the increase of CC/C–C is due to the benzene ring of TC. When the TC antibiotic was adsorbed by TiO2@C, the CO would be replaced by CC/C–C. Thus, it is noted from the XPS spectra that the TC antibiotic can be adsorbed by TiO2@C effectively.
C/C–C peak at 284.9 eV, a C–O peak at 292.3 eV and a CO peak at 292.9 eV (Fig. 8b). It is noted that the content of CC/C–C is much lower than that of CO. However, after the TC antibiotic was adsorbed by TiO2@C, the adsorbed C 1s signal is deconvoluted to a CC/C–C peak at 284.5 eV, a C–O peak at 285 eV and a CO peak at 286.2 eV. Simultaneously, the content of C–C/CC increased sharply. In contrast, the content of CO is the lowest among the three kinds of chemical bonds. In summary, the increase of CC/C–C is due to the benzene ring of TC. When the TC antibiotic was adsorbed by TiO2@C, the CO would be replaced by CC/C–C. Thus, it is noted from the XPS spectra that the TC antibiotic can be adsorbed by TiO2@C effectively.
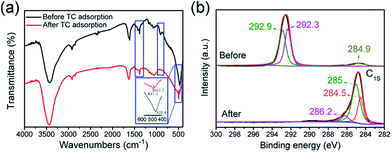 |
| | Fig. 7 (a) The FTIR spectra of TiO2@C before and after adsorbing TC, (b) high-resolution XPS spectra of the C 1s peaks before and after adsorption of the TC antibiotic by TiO2@C. | |
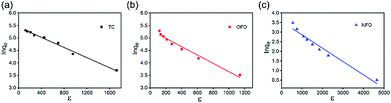 |
| | Fig. 8 (a–c) D–R isotherm regression fitting lines from the adsorption isotherms of TC, OFO and NFO for the TiO2@C. | |
The D–R isotherm model can be applied to identify the type of adsorption, i.e., physical or chemical adsorption process. The linear D–R isotherm37 models are as follow and the results are shown in Fig. 8:
| | | Inqe = Inqm − BDε | (6) |
| | | ε = RTIn(1 + 1/Ce) | (7) |
where
qe and
qm (mmol g
−1) are the equilibrium antibiotics solution concentration on the adsorbent and maximum adsorption capacity, respectively,
BD is a constant related to the adsorption energy (mol
2 J
−2),
ε is the Polanyi potential,
Ce is the equilibrium concentration (mol L
−1),
R is the gas constant and
T is the absolute temperature (K).
The adsorption free energy (E) can be calculated according to Polanyi potential from the following equation:
Compared with the value of R2, it is obvious that the Langmuir model is more fit for the experimental data. As for the D–R equations, if the value of E is less than 8 kJ mol−1, physical sorption is considered as the main mechanism, while chemical sorption will be expected if the value of E is in the range of 8–16 kJ mol−1. As shown in Table 4, the three values of E are all less than 8 kJ mol−1. This proves that physisorption is the main sorption mechanism. The results of this analysis yielded consistent pseudo-first-order adsorption kinetics.
Table 4 D–R equation regression data from the adsorption isotherms of NFO, TC and OFO for the TiO2@C
| Pollutant |
B
D (mol2 J−2) |
D–R equation qm(mmol g−1) |
R
2
|
E (KJ mol−1) |
| TC |
1.01 × 10−3 |
225.88 |
0.983 |
0.700 |
| OFO |
1.67 × 10−3 |
202.35 |
0.966 |
0.423 |
| NFO |
6.90 × 10−4 |
35.59 |
0.953 |
1.025 |
The thermodynamics parameters regarding antibiotic adsorption onto TiO@C, such as Gibbs free energy (ΔG), and the solid and liquid phases at equilibrium (Kd), can be calculated using the following equations:38
| | | ΔG = −RIn(Kd) | (10) |
Generally, if −20 < ΔG < 0 kJ mol−1, physical adsorption is dominant and it changes to more negative values than −40 kJ mol−1 for chemical adsorption. As shown in Table 5, the calculated result is consistent with D–R equations and pseudo-first-order adsorption kinetics.
Table 5 The values of ΔG calculated for adsorption of the three antibiotics by TiO2@C
| Pollutant (40 mg L−1) |
TC |
OFO |
NFO |
| ΔG (kJ mol−1) |
−5.71 |
−5.58 |
−5.172 |
Conclusions
Core–shell TiO2@C ultralong nanotubes were prepared using a solvothermal process followed by anhydrous ethanol carbonization. The TiO2@C exhibits a mesoporous structure and large specific surface area. The adsorption experiment proves that the TiO2@C composite adsorbent can adsorb three kinds of typical antibiotics (TC, OFO, and NFO) effectively. Moreover, the main determinant of the adsorption process is proved to be physical adsorption rather than chemical adsorption, and the adsorption process fits the Langmuir and pseudo-first-order models. Therefore, the work provides a practical case for the future development of more composite adsorbents and lays a foundation for the industrialization of composite materials.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The work has been funded by the National Natural Science Foundation of China (51602193, 21601122, and 51701022), the Shanghai “Chen Guang” Project (16CG63), the Fundamental Research Funds for the Central Universities (WD1817002), and the Talent Program of Shanghai University of Engineering Science, Science and Technology Facilities Council for STFC/MDC Futures Early Career Award.
References
- E. A. Abdullah, A. H. Abdullah, Z. Zainal, M. Z. Hussein and T. K. Ban, J. Environ. Sci., 2012, 24(10), 1876–1884 CrossRef CAS.
- A. K. Sarmah, M. T. Meyer and A. B. Boxall, Chemosphere, 2006, 65, 725–759 CrossRef CAS PubMed.
- M. Saravanana, J. H. Hur, N. Arul and M. Ramesh, Environ. Toxicol. Pharmacol., 2014, 38, 948–958 CrossRef PubMed.
- H. A. Al-Aoh, R. Yahya, M. Jamil Maah and B. A. M. Radzi, Desalin. Water Treat., 2014, 52(34–36), 6720–6732 CrossRef CAS.
- S. Rodriguez-Mozaz, S. Chamorro, E. Marti, B. Huerta, M. Gros, A. Sànchez-Melsió, C. M. Borrego, D. Barcelo and J. L. Balcazar, Water Res., 2015, 69, 234–242 CrossRef CAS PubMed.
- T. T. Lu, Y. F. Zhu, Y. X. Qi, W. B. Wang and A. Q. Wang, Int. J. Biol. Macromol., 2018, 106, 870–877 CrossRef CAS PubMed.
- F. Bianculloa, N. F. F. Moreiraa, A. R. Ribeiroa, C. M. Manaia, J. L. Faria, O. C. Nunes, S. M. Castro-Silva and A. M. R. Silva, J. Chem. Eng., 2019, 367, 304–313 CrossRef.
- H. Yu, B. X. Zhang, M. Zhao, L. Zhang, H. Dong and H. B. Yu, Catal. Today, 2019, 327, 308–314 CrossRef CAS.
- J. C. Espíndola, K. Szymanski, R. O. Cristovao, A. Mendes, V. J. P. Vilara and S. Moziab, Catal. Today, 2019, 328, 274–280 CrossRef.
- C. L. Amorim, I. S. Moreira, A. S. Maia, M. E. Tiritan and P. M. Castro, Appl. Microbiol. Biotechnol., 2014, 98, 3181–3190 CrossRef CAS PubMed.
-
R. Françoise, R. Jean and S. Kenneth, Adsorption by Powders and Porous Solids, 1999, pp. 1–26 Search PubMed.
-
E. Can, Supercritical Fluid Science & Technology, 2011, vol. 1, pp. 41–77 Search PubMed.
- D. D. Wang, Y. L. Shen, Y. L. Chen, L. L. Liu and Y. F. Zhao, J. Chem. Eng., 2019, 367, 260–268 CrossRef CAS.
- H. Huang, Y. L. Zou and Y. N. Li, Adv. Mater. Res., 2012, 512–515, 2355–2360 CAS.
- J. Tang, L. Zong, B. Mu, Y. R. Kang and A. Q. Wang, Korean J. Chem. Eng., 2018, 35, 1650–1661 CrossRef CAS.
- A. Gouza, S. Saoiabi, M. El Karbane, S. Masse, G. Laurent, A. Rami, A. Saoiabi, A. Laghzizil and T. Coradin, Environ. Sci. Pollut. Res., 2017, 24(33), 25977–25985 CrossRef CAS PubMed.
- Y. Zhuang, F. Yu, J. Ma and J. H. Chen, Desalin. Water Treat., 2016, 57(20), 9510–9519 CrossRef CAS.
- P. K. Baskaran, B. R. Venkatraman and S. Arivoli, Eur. J. Chem., 2011, 8(1), 9–18 CAS.
- A. H. Ali, Desalin. Water Treat., 2013, 51(28–30), 5547–5558 CrossRef CAS.
- L. Yu, X. Liu, W. Yuan, L. J. Brown and D. Wang, Langmuir, 2015, 31, 6351–6366 CrossRef CAS PubMed.
- S. K. Singh, T. G. Townsend, D. Mazyck and T. H. Boyer, Water Res., 2012, 46, 491–499 CrossRef CAS PubMed.
- M. Iram, C. Guo, Y. Guan, A. Ishfaq and H. Z. Liu, J. Hazard. Mater., 2010, 181, 1039–1050 CrossRef CAS PubMed.
- Y. Zhuang, F. Yu, J. Ma and J. H. Chen, Desalin. Water Treat., 2016, 57(20), 9510–9519 CrossRef CAS.
- L. C. Chen, S. Lei, M. Z. Wang, J. Yang and X. W. Ge, Chin. Chem. Lett., 2016, 27(4), 511–517 CrossRef CAS.
- J. S. He, J. D. Dai, A. T. Xie, S. J. Tian, Z. S. Chang, Y. S. Yan and P. W. Huo, RSC Adv., 2016, 6(87), 84536–84546 RSC.
- W. Y. Li, J. R. Wang, G. J. He, L. Yu, N. Noor, Y. G. Sun, X. Y. Zhou, J. Q. Hu and I. P. Parkin, J. Mater. Chem. A, 2017, 5(9), 4352–4358 RSC.
- R. A. Wuana, R. Sha'Ato and S. Iorhen, Desalin. Water Treat., 2016, 57(25), 11904–11916 CrossRef CAS.
- Y. Kong, N. W. Wang, X. N. Ni, Q. Y. Yu, H. Liu, W. H. Huang and W. Z. Xu, J. Appl. Polym. Sci., 2016, 133(1), 42817 CrossRef.
- L. Zhang, H. A. Loaiciga, M. Xu, C. Du and Y. Du, Int. J. Environ. Res. Public Health, 2015, 12(11), 14312–14326 CrossRef CAS PubMed.
- D. Feng, H. M. Yu, H. Deng, F. Z. Li and C. J. Ge, BioResources, 2015, 10(4), 6751–6768 CAS.
- M. A. Al-Anber, Desalin. Water Treat., 2014, 52(13–15), 2560–2571 CrossRef CAS.
- N. A. Rashidi, S. Yusup, A. Borhan and L. H. Loong, Clean Technol. Environ. Policy, 2014, 16(7), 1353–1361 CrossRef CAS.
- X. Y. Cao, H. L. Pang and G. P. Yang, J. Soils Sediments, 2015, 15(7), 1635–1643 CrossRef CAS.
- A. R. Kul and H. Koyuncu, J. Hazard. Mater., 2010, 179(1–3), 332–339 CrossRef CAS PubMed.
- Q. Liu, Q. Z. Liu, W. Ma, W. L. Liu, X. X. Cai and J. S. Yao, Colloids Surf., A, 2016, 511, 8–16 CrossRef CAS.
- L. L. Jia, X. Y. Huang, H. E. Liang and Q. Tao, Int. J. Biol. Macromol., 2019, 132, 1039–1043 CrossRef CAS PubMed.
- M. A. Barakat and R. Kumar, J. Ind. Eng. Chem., 2015, 23, 93–99 CrossRef CAS.
- O. Pezoti, A. L. Cazetta, K. C. Bedin, L. S. Souza, A. C. Martins, T. L. Silva, O. O. Santos, J. V. Visentainer and V. C. Almeida, Chem. Eng. J., 2016, 288, 778–788 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ta06735c |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *ab,
Jianwei
Li
c,
Ruoyu
Xu
b,
Kenan
Zhang
a,
Guanjie
He
*ab,
Jianwei
Li
c,
Ruoyu
Xu
b,
Kenan
Zhang
a,
Guanjie
He