
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLayered cobalt hydrotalcite as an advanced lithium-ion anode material with high capacity and rate capability†
Long
Pan
 *,
Haijian
Huang
and
Markus
Niederberger
*,
Haijian
Huang
and
Markus
Niederberger
Department of Materials, Laboratory for Multifunctional Materials, ETH Zurich, Vladimir-Prelog-Weg 5, 8093, Zurich, Switzerland. E-mail: long.pan@mat.ethz.ch
First published on 26th August 2019
Abstract
Lithium-ion anode materials with high energy density and high power density are growing in demand for practical applications. However, conventional electrode materials such as graphite and lithium titanium oxide (LTO) cannot meet these requirements. Here, we report layered cobalt hydrotalcite (LCH) as an advanced anode material for lithium-ion batteries with high capacity and rate capability. The large interlayer distance of the LCH structure offers sufficient space for lithium-ion insertion, which is demonstrated using various electrochemical analyses, operando X-ray diffraction, and ex situ X-ray photoelectron spectroscopy. Owing to the fast lithium-ion insertion kinetics and the unique 2D morphology, the LCH exhibits a high reversible capacity of 743.9 and 643.4 mA h g−1 at 500 and 2000 mA g−1 over 600 and 2000 cycles, respectively, and an outstanding rate capability (490.0 mA h g−1 at 5000 mA g−1) in lithium-ion half cells. This excellent electrochemical performance makes LCH a promising alternative to the currently existing pseudocapacitive materials as an anode for lithium-ion batteries.
Introduction
Currently, lithium-ion batteries (LIBs) are widely used in portable electronic devices and electric vehicles. With the ever-growing demand for energy and power density, LIBs with the conventional anode material, i.e., graphite (theoretical capacity = 372 mA h g−1), have reached their limit.1 Therefore, great efforts are being made to discover alternative anode materials with higher capacity and better rate capability. Transition metal oxides/sulfides (e.g., Fe3O4, Mn3O4, and MoS2) with a conversion mechanism and elementary substances (e.g., Si, P, Sn, and Ge) with an alloying mechanism have been proposed due to their high theoretical capacities.2–12 However, these materials always suffer from sluggish rate capabilities because of their low conductivity.6,8 Recently, new types of anode materials such as Nb2O5 and TiO2(B) have been explored as high-rate anode materials, despite their low electrical conductivity.11,13–15 These hosts exhibit a unique Li+ storage mechanism, i.e., pseudocapacitance, thereby offering fast Li+ insertion/extraction.15,16 Unfortunately, such materials deliver relatively low theoretical capacity (e.g., Nb2O5vs. graphite, 200 vs. 372 mA h g−1), which hinders their practical application in next-generation, high-energy LIBs.15,16 Therefore, it is particularly urgent to pursue an advanced host with high capacity and high rate Li+ storage.Hydrotalcite is a layered material with anions (e.g., NO3− and CO32−) located in the interlayer voids.17 It has attracted much interest in the field of supercapacitors and aqueous alkaline batteries because of the possibility of adjusting the metal cations and the intercalating anions.18–20 Additionally, hydrotalcite exhibits flexible and large interlayer spacing, which is supposed to be a favorable structure for alkali-ion intercalation electrode materials.21 In spite of these promising characteristics, however, neither the Li+ storage property nor its mechanism has been investigated for hydrotalcite.
Here, we demonstrate the suitability of layered cobalt hydrotalcite (LCH) as a conversion-type Li+ storage anode material with high capacity and high rate capability via various electrochemical analyses, operando powder X-ray diffraction (XRD), and ex situ X-ray photoelectron spectroscopy (XPS). It is confirmed that the layered structure with large interlayer spacing offers unique diffusion pathways for fast Li+ storage. These merits, together with the small surface area, endow the LCH with a high reversible capacity of 1369.5 mA h g−1 at 100 mA g−1 in the first cycle. In addition, the LCH exhibits a good cycling performance, e.g., 743.9 mA h g−1 at 500 mA g−1 after 600 cycles and 643.4 mA h g−1 at 2000 mA g−1 over 2000 cycles, and an outstanding rate capability (490.0 mA h g−1 at 5000 mA g−1).
Results and discussion
The LCH nanosheets were synthesized from Co(NO3)2·6H2O with the assistance of microwave radiation. LCH has been widely studied over the last several decades.17,22 The general chemical formula of LCH is proposed to be Co(OH)1.5(NO3)0.5,22 where the –OH groups are covalently bound to Co2+, forming octahedral Co(OH)1.5 units, and where the NO3− anions are intercalated in between the layers, as shown in Fig. 1a and S1 (ESI†). The interlayer spacing of LCHs is about 6.96 Å, which is much larger than the radius of Li+ (0.76 Å), enabling fast Li+ diffusion.22,23 The nanosheet morphology of the LCH is revealed by scanning electron microscopy (SEM) and transmission electron microscopy (TEM), as shown in Fig. 1b, c and S2 (ESI†). The LCH nanosheets are of hexagonal shape and their lateral sizes are in the range of several microns. According to the high-resolution TEM (HRTEM) image (Fig. 1d), the LCH nanosheets are composed of a layered structure with an interlayer distance of 7.0 Å. Apart from the high crystallinity (Fig. S3, ESI†), the individual LCH nanosheets are single crystalline as revealed by the well-developed spots in the selected area electron diffraction (SAED) pattern (Fig. 1e). The scanning TEM (STEM) together with the element mapping analysis (Fig. 1f) shows the homogeneous distribution of Co, O, and N throughout the LCH nanosheets, confirming the presence of NO3− in the LCH structure.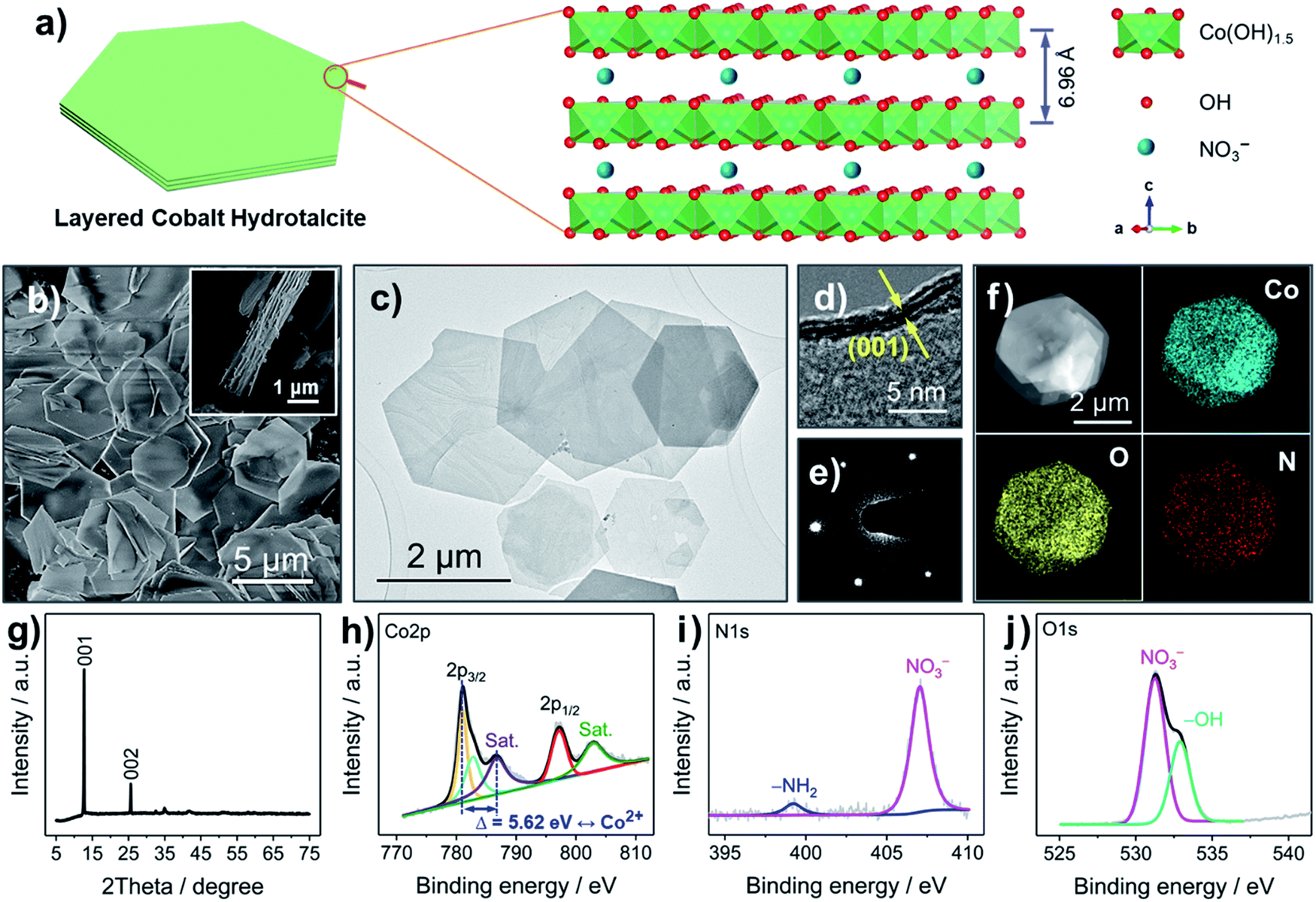 | ||
| Fig. 1 Characterization of the LCH nanosheets. (a) Schematic illustration of the LCH nanosheets and the crystal structure (hexagonal symmetry). (b) SEM, (c) TEM, (d) HRTEM, (e) SAED, and (f) STEM with elemental mapping images of the LCH nanosheets. (g) XRD pattern, and (h) Co 2p, (i) N 1s, and (j) O 1s XPS spectra of the LCH nanosheets. | ||
The crystal structure and the phase of the LCH nanosheets are disclosed by XRD, as depicted in Fig. 1g. The XRD pattern exhibits two intensive peaks corresponding to the (001) and (002) reflections of a typical layered structure.22 The calculated interlayer spacing from the (001) plane is 6.97 Å, which is in good agreement with the HRTEM result (Fig. 1d) and the crystal structure in Fig. 1a. Fig. S4 (ESI†) displays the XPS survey spectrum, in which well-resolved Co 2p, N 1s, and O 1s peaks are observed. Besides, the atomic ratio of Co, N, and O obtained from XPS analysis (Table S1, ESI†) agrees well with the theoretical ratio, indicating that the chemical formula of LCH is indeed Co(OH)1.5(NO3)0.5. The high-resolution Co 2p spectrum in Fig. 1h shows two well-resolved peaks at 781.06 and 797.28 eV, corresponding to the 2p3/2 and 2p1/2 orbitals of Co2+. Two additional broad peaks at 786.68 and 803.08 eV can be assigned to satellite features of Co2+.24 These results together with the asymmetrical shape of the Co 2p peaks imply that the Co cations in the LCH nanosheets are divalent (Co2+). The prominent peak at 407.04 eV in the N 1s spectrum (Fig. 1i) fits to NO3−, once again indicating the presence of NO3− in the LCH nanosheets.22 The small, broad peak located at 399.22 eV can be related to –NH2, presumably as a result of oleylamine residues.25 The very small amount of –NH2 groups, therefore, has little influence on the compositional analysis of the XPS results. Besides, the presence of –OH and NO3− is further confirmed by the O 1s spectrum, as shown in Fig. 1j.
To explore the Li+ storage properties of the LCH nanosheets, galvanostatic tests using a half cell with lithium foil as the counter electrode were performed. Fig. 2a shows the first discharging–charging curves of the LCH anode at a specific current of 100 mA g−1. During the first discharge, an obvious plateau around 1.0 V is observed, which may be attributed to the Li+ reaction with the LCH. After discharging to 0.8 V, the solid–electrolyte interphase (SEI) starts to form and electrochemically activated electrolyte decomposition occurs.26 In total, the specific capacity of the first discharge is 2296.7 mA h g−1. During the charging process, Li+ is extracted from the LCH, delivering a reversible capacity of 1369.5 mA h g−1 (3.7 times that of graphite). Therefore, the coulombic efficiency of the first cycle is about 60%. This value is comparable to or even higher than that of Co(OH)2-based electrode materials, indicating the favorable kinetics of Li+ storage in LCH.27,28 Another reason for this high value is that the small specific surface area of the LCH (6.7 m2 g−1 shown in Fig. S5, ESI†) brings along fewer irreversible side reactions from the electrolyte decomposition compared to those having large surface area.29 Thanks to the SEI layer, the coulombic efficiency increases to 90% in the second cycle and further rises to 97% in the fifth cycle, as displayed in Fig. 2b. The Li+ storage behavior is also examined by cyclic voltammetry (CV) (Fig. S6, ESI†), which confirms the results from the galvanostatic tests.
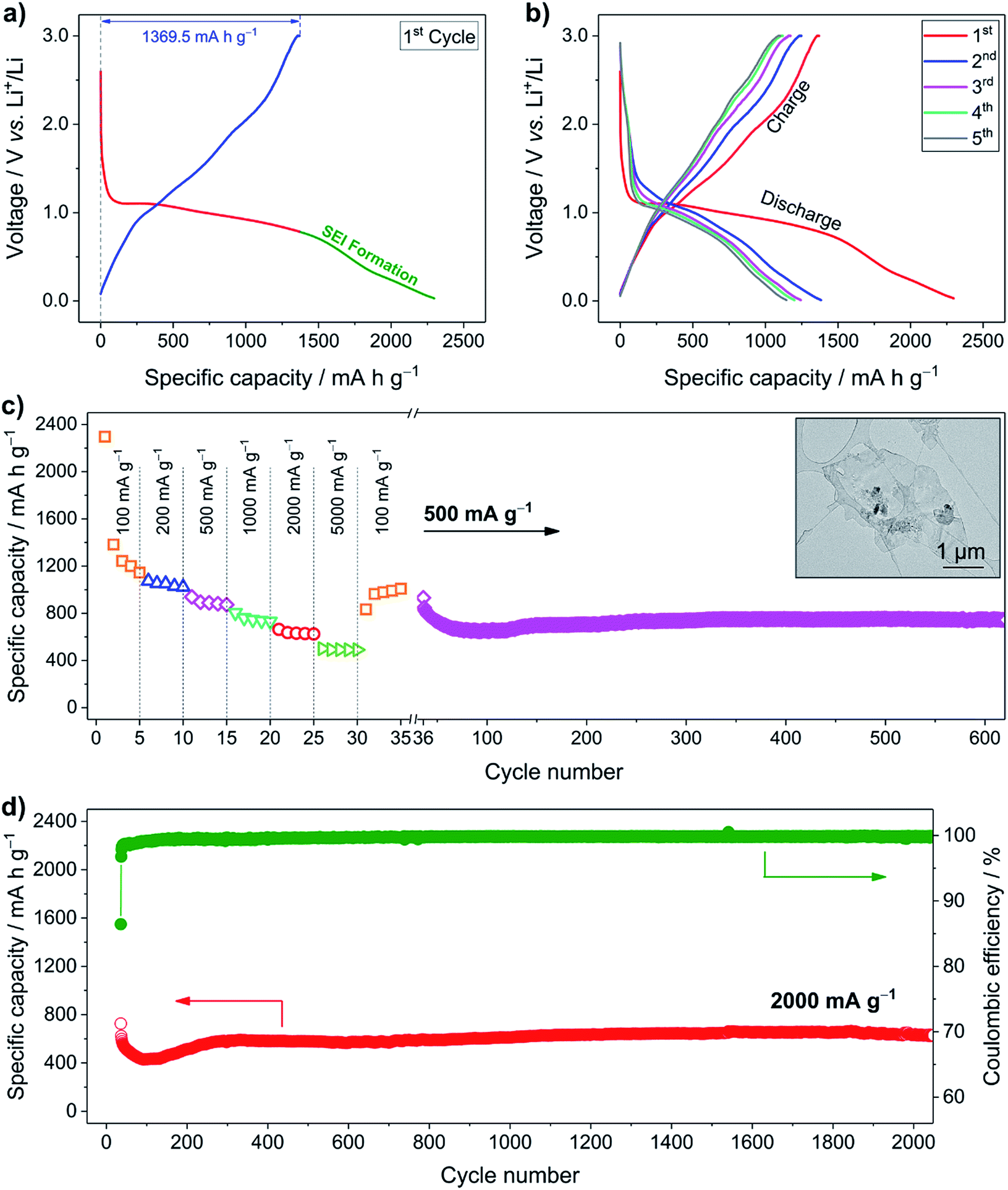 | ||
| Fig. 2 Electrochemical performance of the LCH nanosheets. (a) First and (b) first five discharging–charging curves of the LCH nanosheets (specific current = 100 mA g−1). (c) Rate capability and cycling performance of the LCH nanosheets. (d) Long-term cyclability of the LCH nanosheets (specific current = 2000 mA g−1). The inset in (c) shows a TEM image of the LCH nanosheets after 600 cycles at 500 mA g−1. | ||
The good rate capability of the LCH anode material is presented in Fig. 2c. In the first five cycles, a high reversible capacity of 1144.5 mA h g−1 at a specific current of 100 mA g−1 is delivered. Then, the specific current is gradually increased to 200, 500, 1000, and 2000 mA g−1 with five cycles for each rate, resulting in a reversible capacity of 1022.0, 872.4, 730.5, and 624.4 mA h g−1, respectively. Even at a specific current as high as 5000 mA g−1, a relatively high reversible capacity of 490.0 mA h g−1 is delivered. The LCH anode material is further tested at 500 mA g−1 over 600 cycles to analyze its cycling performance. It delivers a reversible capacity of 743.9 mA h g−1, which is almost two times that of graphite. After 600 cycles, the nanosheet-like morphology of the LCH is mainly retained, despite the partial fracture of the pristine hexagonal nanosheets, as displayed in the inset of Fig. 2c. The long-term cycling performance is further investigated at a specific current of 2000 mA g−1 over 2000 cycles (Fig. 2d). A reversible capacity as high as 643.4 mA h g−1 is delivered, indicating outstanding cyclability of the LCH anode material. Even compared to the typical conversion-type anode materials such as Co3O4 and Fe3O4 (Table S2 and Fig. S7, ESI†), the LCH still exhibits comparable or even better cyclability and rate capability, indicating its great potential for high-performance Li+ storage applications. As seen from the cycling curves in both Fig. 2c and d, the capacities gradually increase in the beginning and then become stable, which is common in cobalt-containing electrode materials.30–34 Several reasons have been proposed to explain this phenomenon, including a reversible growth of a polymer gel-like layer and gradual wetting of electrode materials by the electrolyte.33,35 Besides these reasons, in our case, another possible explanation is the gradual Li+ reaction with electrode materials at high specific current, as discussed later.
To uncover the Li+ storage mechanism of the LCH, operando XRD and ex situ high-resolution XPS were performed. Generally, when Li+ is inserted in conversion type (e.g., Co3O4) or alloying type (e.g., Sn) anode materials, they transform to materials with crystal structures that are totally different from the original ones (e.g., Co3O4 + 8Li+ + 8e− → 3Co + 4Li2O, Sn + xLi+ + xe− → LixSn).30,36 Therefore, new characteristic peaks corresponding to these phases are expected to appear during the operando XRD measurements.37 In some cases, however, the newly formed products are amorphous, meaning that all peaks disappear.38Fig. 3a–c present the operando XRD patterns and the corresponding discharging curve of the first discharging of the LCH. During the Li+ reaction at a relatively low specific current of 50 mA g−1 (Fig. 3a), all peaks of the LCH structure gradually disappear, and no new peaks except a broad one located at 25° are observed in all patterns (Fig. 3b and c). We stress that the 25° peak comes from the background of the operando cell, which is proved in Fig. S8 (ESI†). Besides, the characteristic reflections of LCH do not appear after charging to 3.0 V (Fig. S9, ESI†), which is similar to other conversion type anode materials.38 These results confirm a conversion or alloying type mechanism for the LCH.38 Generally, conversion mechanisms occur in compounds, whereas alloying reactions take place in elementary substances.2–12 Therefore, it can be inferred that the LCH goes through a conversion reaction with Li+. Also, we recorded the operando XRD patterns at a high discharging/charging specific current (200 mA g−1), as shown in Fig. S10 (ESI†). Similarly, the intensities of the (00l) reflections gradually decrease and no new peaks appear during the discharge. Note that the (00l) reflections are still observable after several cycles, indicating that the LCH does not fully react with Li+ in the first few cycles because of the high specific current. As the cycling goes on, more and more LCH is converted, resulting in a steady decrease of reflection intensities and a gradual increase of specific capacity. After complete conversion of LCH, the specific capacity becomes stable (Fig. 2c and d). These results demonstrate that the specific current does not change the Li+ storage mechanism but affects the reaction degree of Li+ with electrode materials.
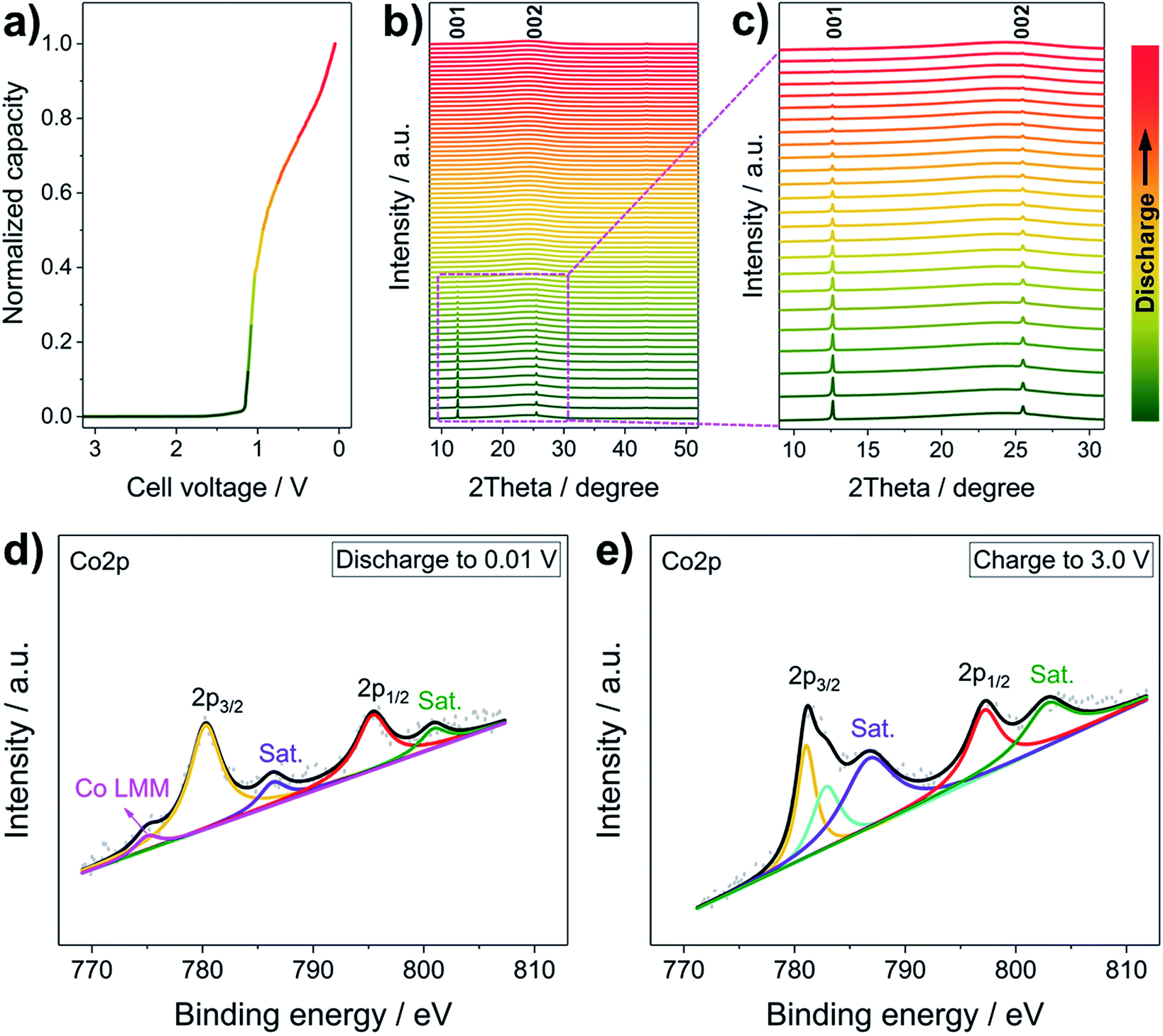 | ||
| Fig. 3 Investigation of the Li+ storage mechanism of the LCH nanosheets. (a) The first discharging profile (50 mA g−1) and (b and c) corresponding operando XRD patterns. High-resolution Co 2p XPS spectra of the LCH nanosheets (d) discharged to 0.01 V and (e) charged to 3.0 V. | ||
Further confirmation of the conversion mechanism is revealed by ex situ XPS characterization. The high-resolution Co 2p XPS spectra at different discharging and charging states are presented in Fig. 3d and e, respectively. Generally, the Co 2p3/2 orbital of metallic Co exhibits an asymmetric peak at ∼778.1 eV with a loss satellite feature and a weak Co LMM Auger peak.39,40 In this case, however, the asymmetric Co 2p3/2 peak of pristine LCH (Fig. 1h) turns to be semi-asymmetric (Fig. 3d) after discharging to 0.01 V. The Co 2p3/2 binding energy of the discharged LCH is 780.42 eV, which is higher than that of metallic Co but lower than that of Co2+. Besides, two weak satellite peaks and a small Co LMM Auger peak are witnessed, indicating that the LCH is reduced to metallic Co during Li+ uptake. Note that the semi-asymmetric peak, the higher binding energy and the weak satellite peaks may come from a small amount of unreacted LCH at a relatively high specific current, which is in good agreement with the operando results (Fig. S10, ESI†). After charging to 3.0 V, the Co 2p spectrum of the LCH (Fig. 3e) is almost the same as that of the pristine LCH (Fig. 1h), implying a highly reversible Li+ storage behavior of LCH. On the basis of the operando XRD and ex situ XPS results, the Li+ storage mechanism in LCH is demonstrated to follow a conversion process, in which LCH is converted to metallic Co. It should be pointed out that the role of NO3− anions in the Li+ storage property is not clear yet, which will be systematically investigated in the future work.
Conclusions
In this work we show that LCH is a promising conversion-type Li+ anode material with high capacity and high rate capability. Various electrochemical analyses were performed, including operando XRD measurement and ex situ XPS characterization. These results demonstrate the conversion mechanism of the LCH. Owing to the unique 2D morphology with large interlayer spacing and fast ion-storage kinetics, the LCH exhibits excellent cycling stability at high specific current and good rate performance, satisfying the high requirements of next-generation, high-performance LIBs. Note that there are still some problems remaining for conversion-type anode materials such as voltage drops arising from the multiphase nature of the reaction, which should be addressed toward practical applications. Our work introduces a new class of potential electrode materials, paving the way for discovering other layered hydrotalcites for high energy and high power metal-ion batteries.Experimental
Materials
Co(NO3)2·6H2O (≥99.0%), oleic acid (technical grade, 90%), benzyl alcohol (99–100.5%), and chloroform (≥99.8%) were purchased from Sigma-Aldrich. Tetrahydrofuran and acetone were purchased from VWR Chemicals. Oleylamine (C18 content 80–90%) and carboxymethylcellulose sodium salt (CMC, DS = 0.9, M.W. = 700![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1) were purchased from Acros Organics. All chemicals were used as received.
000 g mol−1) were purchased from Acros Organics. All chemicals were used as received.
Synthesis of LCH nanosheets
Typically, 5 mmol of Co(NO3)2·6H2O, 25 mmol of oleylamine, 25 mmol of oleic acid, and 42.6 mmol of benzyl alcohol were mixed in a 35 mL microwave tube. The mixture was subjected to a microwave reactor (Discover SP, CEM) at 180 °C for 20 min. After cooling down, the product was washed with acetone (2 times), chloroform (2 times), tetrahydrofuran (2 times), and acetone (2 times). Finally, the LCH nanosheets were obtained by vacuum drying at 50 °C overnight.Electrochemical tests
LCH nanosheets (active materials), carbon black (conductive additive), and CMC (binder) were mixed with deionized water at a ratio of 60:30:10 wt%. After mixing in a ball mill (Mixer Mill MM 400, Retsch) at 20 Hz for 20 min, the homogeneous slurry was coated on a titanium holder (current collector) and vacuum-dried. Half-cells with lithium foil as the counter electrodes were assembled. 1 M LiPF6 solution in dimethyl carbonate/ethylene carbonate (volume ratio, 1/1) was used as the electrolyte. Glass fiber membranes (GF/D, Whatman) were used as separators. The mass loading of the active material is about 1 mg cm−2. Note that the relatively high content of carbon black was added to ensure sufficient conductivity of LCH. Besides, the capacity contribution of carbon black to the total capacity is small (∼8%), as shown in Fig. S11 (ESI†).
Characterization
TEM and SAED measurements were performed on a Hitachi HT7700 microscope at an accelerating voltage of 100 kV or on an FEI Tecnai F30 FEG microscope at an accelerating voltage of 300 kV. For STEM and HRTEM measurements we used an FEI Talos F 200X microscope at an accelerating voltage of 200 kV. SEM data were acquired on a ZEISS LEO 1530 microscope at an accelerating voltage of 5 kV. XPS experiments were carried out on an Escalab 250 Xi spectrometer or a Thermo scientific Sigma 2 spectrometer. XRD measurements were performed on a PANalytical Empyrean X-ray diffractometer with Cu Kα radiation (λ = 0.154 nm). The specific surface area was calculated according to the Brunauer–Emmett–Teller (BET) method based on nitrogen adsorption data from a Quantachrome Autosorb iQ analyzer.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Dr E. Tervoort and Dr F. Gramm for TEM characterization and discussions, Prof. A. Studart for providing the SEM equipment, Prof. A. Rossi and G. Cossu for providing the XPS equipment, the Scientific Center for Optical and Electron Microscopy (ScopeM) of ETH Zürich for access to TEM facilities, and Dr D. Kundu for helpful discussions. The authors acknowledge financial support from ETH Zürich. L. P. is grateful for the fellowship from the Office of China Postdoctoral Council (No. 32 Document of OCPC, 2017). H. H. acknowledges the financial support from the China Scholarship Council.Notes and references
- N. Mahmood, T. Tang and Y. Hou, Adv. Energy Mater., 2016, 6, 1600374 CrossRef.
- L. Pan, Y.-T. Liu, X.-M. Xie and X.-D. Zhu, Chem.–Asian J., 2014, 9, 1519 CrossRef CAS.
- L. Pan, K.-X. Wang, X.-D. Zhu, X.-M. Xie and Y.-T. Liu, J. Mater. Chem. A, 2015, 3, 6477 RSC.
- Y. M. Chen, X. Y. Yu, Z. Li, U. Paik and X. W. Lou, Sci. Adv., 2016, 2, e1600021 CrossRef.
- L. Pan, X.-D. Zhu, X.-M. Xie and Y.-T. Liu, J. Mater. Chem. A, 2015, 3, 2726 RSC.
- D. Bresser, S. Passerini and B. Scrosati, Energy Environ. Sci., 2016, 9, 3348 RSC.
- L. Pan, X.-D. Zhu, K.-N. Sun, Y.-T. Liu, X.-M. Xie and X.-Y. Ye, Nano Energy, 2016, 30, 347 CrossRef CAS.
- Y. Lu, L. Yu and X. W. Lou, Chem, 2018, 4, 972 CAS.
- L. Pan, Y.-T. Liu, X.-M. Xie and X.-Y. Ye, Small, 2016, 12, 6703 CrossRef CAS.
- L. Pan, X.-D. Zhu, X.-M. Xie and Y.-T. Liu, Adv. Funct. Mater., 2015, 25, 3341 CrossRef CAS.
- B. Y. Guan, L. Yu, J. Li and X. W. Lou, Sci. Adv., 2016, 2, e1501554 CrossRef.
- L. Pan, H. Huang, M. Zhong and M. Niederberger, Energy Storage Materials, 2019, 16, 519 CrossRef.
- L. Pan, Z.-W. Zhou, Y.-T. Liu and X.-M. Xie, J. Mater. Chem. A, 2018, 6, 7070 RSC.
- L. Pan, Y.-T. Liu, X.-M. Xie, X.-Y. Ye and X.-D. Zhu, Nano Res., 2016, 9, 2057 CrossRef CAS.
- V. Augustyn, J. Come, M. A. Lowe, J. W. Kim, P.-L. Taberna, S. H. Tolbert, H. D. Abruña, P. Simon and B. Dunn, Nat. Mater., 2013, 12, 518 CrossRef CAS.
- A. G. Dylla, G. Henkelman and K. J. Stevenson, Acc. Chem. Res., 2013, 46, 1104 CrossRef CAS.
- S. Nishimura, A. Takagaki and K. Ebitani, Green Chem., 2013, 15, 2026 RSC.
- J. Yu, Q. Wang, D. O'Hare and L. Sun, Chem. Soc. Rev., 2017, 46, 5950 RSC.
- J. H. Lee, H. J. Lee, S. H. Choi, J. Shin, S.-Y. Chung and J. W. Choi, Adv. Energy Mater., 2018, 8, 1703572 CrossRef.
- R. Zhao, M. Wang, D. Zhao, H. Li, C. Wang and L. Yin, ACS Energy Lett., 2018, 3, 132 CrossRef CAS.
- S. Kajiyama, L. Szabova, K. Sodeyama, H. Iinuma, R. Morita, K. Gotoh, Y. Tateyama, M. Okubo and A. Yamada, ACS Nano, 2016, 10, 3334 CrossRef CAS.
- R. Xu and H. C. Zeng, Chem. Mater., 2003, 15, 2040 CrossRef CAS.
- M. D. Slater, D. Kim, E. Lee and C. S. Johnson, Adv. Funct. Mater., 2013, 23, 947 CrossRef CAS.
- D. Lee, Q. X. Xia, J. M. Yun and K. H. Kim, Appl. Surf. Sci., 2018, 433, 16 CrossRef CAS.
- L. Lai, L. Chen, D. Zhan, L. Sun, J. Liu, S. H. Lim, C. K. Poh, Z. Shen and J. Lin, Carbon, 2011, 49, 3250 CrossRef CAS.
- P. Verma, P. Maire and P. Novák, Electrochim. Acta, 2010, 55, 6332 CrossRef CAS.
- X.-l. Huang, X. Zhao, Z.-l. Wang, L.-m. Wang and X.-b. Zhang, J. Mater. Chem., 2012, 22, 3764 RSC.
- Y.-S. He, D.-W. Bai, X. Yang, J. Chen, X.-Z. Liao and Z.-F. Ma, Electrochem. Commun., 2010, 12, 570 CrossRef CAS.
- M. Okubo, A. Sugahara, S. Kajiyama and A. Yamada, Acc. Chem. Res., 2018, 51, 591 CrossRef CAS.
- Z.-S. Wu, W. Ren, L. Wen, L. Gao, J. Zhao, Z. Chen, G. Zhou, F. Li and H.-M. Cheng, ACS Nano, 2010, 4, 3187 CrossRef CAS.
- G. Huang, F. Zhang, X. Du, Y. Qin, D. Yin and L. Wang, ACS Nano, 2015, 9, 1592 CrossRef CAS.
- C. Yan, G. Chen, X. Zhou, J. Sun and C. Lv, Adv. Funct. Mater., 2016, 26, 1428 CrossRef CAS.
- Z. Li, X.-Y. Yu and U. Paik, J. Power Sources, 2016, 310, 41 CrossRef CAS.
- Y. Ma, J. He, Z. Kou, A. M. Elshahawy, Y. Hu, C. Guan, X. Li and J. Wang, Adv. Mater. Interfaces, 2018, 1800222 CrossRef.
- Z. Le, F. Liu, P. Nie, X. Li, X. Liu, Z. Bian, G. Chen, H. B. Wu and Y. Lu, ACS Nano, 2017, 11, 2952 CrossRef CAS.
- Y. Liu, N. Zhang, L. Jiao and J. Chen, Adv. Mater., 2015, 27, 6702 CrossRef CAS.
- D. Larcher, G. Sudant, J.-B. Leriche, Y. Chabre and J.-M. Tarascon, J. Electrochem. Soc., 2002, 149, A234 CrossRef CAS.
- G. X. Wang, Y. Chen, K. Konstantinov, M. Lindsay, H. K. Liu and S. X. Dou, J. Power Sources, 2002, 109, 142 CrossRef CAS.
- M. C. Biesinger, B. P. Payne, A. P. Grosvenor, L. W. M. Lau, A. R. Gerson and R. S. C. Smart, Appl. Surf. Sci., 2011, 257, 2717 CrossRef CAS.
- XPS Reference Table of Elements, https://xpssimplified.com/elements/cobalt.php.
Footnote |
| † Electronic supplementary information (ESI) available: Supplementary figures and tables. See DOI: 10.1039/c9ta06593h |
| This journal is © The Royal Society of Chemistry 2019 |