
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Improved cycling stability in high-capacity Li-rich vanadium containing disordered rock salt oxyfluoride cathodes†
Christian
Baur
 *a,
Ida
Källquist
b,
Johann
Chable
a,
Jin Hyun
Chang
c,
Rune E.
Johnsen
c,
Francisco
Ruiz-Zepeda
d,
Jean-Marcel
Ateba Mba
d,
Andrew J.
Naylor
e,
Juan Maria
Garcia-Lastra
c,
Tejs
Vegge
c,
Franziska
Klein
a,
Annika R.
Schür
a,
Poul
Norby
c,
Kristina
Edström
e,
Maria
Hahlin
b and
Maximilian
Fichtner
af
*a,
Ida
Källquist
b,
Johann
Chable
a,
Jin Hyun
Chang
c,
Rune E.
Johnsen
c,
Francisco
Ruiz-Zepeda
d,
Jean-Marcel
Ateba Mba
d,
Andrew J.
Naylor
e,
Juan Maria
Garcia-Lastra
c,
Tejs
Vegge
c,
Franziska
Klein
a,
Annika R.
Schür
a,
Poul
Norby
c,
Kristina
Edström
e,
Maria
Hahlin
b and
Maximilian
Fichtner
af
aHelmholtz Institute Ulm for Electrochemical Energy Storage, Helmholtzstraße 11, 89081 Ulm, Germany. E-mail: christian.baur@kit.edu
bUppsala University, Department of Physics and Astronomy, P. O. box 256, SE-751 05 Uppsala, Sweden
cTechnical University of Denmark, Department of Energy Conversion and Storage, 2800 Kgs. Lyngby, Denmark
dNational Institute of Chemistry, Hajdrihova 19, P. O. box 660, SI-1000 Ljubljana, Slovenia
eDepartment of Chemistry, Ångström Laboratory, Uppsala University, Box 538, 75121 Uppsala, Sweden
fInstitute of Nanotechnology, Karlsruhe Institute for Technology, P. O. box 3640, 76021 Karlsruhe, Germany
First published on 26th August 2019
Abstract
Lithium-rich transition metal disordered rock salt (DRS) oxyfluorides have the potential to lessen one large bottleneck for lithium ion batteries by improving the cathode capacity. However, irreversible reactions at the electrode/electrolyte interface have so far led to fast capacity fading during electrochemical cycling. Here, we report the synthesis of two new Li-rich transition metal oxyfluorides Li2V0.5Ti0.5O2F and Li2V0.5Fe0.5O2F using the mechanochemical ball milling procedure. Both materials show substantially improved cycling stability compared to Li2VO2F. Rietveld refinements of synchrotron X-ray diffraction patterns reveal the DRS structure of the materials. Based on density functional theory (DFT) calculations, we demonstrate that substitution of V3+ with Ti3+ and Fe3+ favors disordering of the mixed metastable DRS oxyfluoride phase. Hard X-ray photoelectron spectroscopy shows that the substitution stabilizes the active material electrode particle surface and increases the reversibility of the V3+/V5+ redox couple. This work presents a strategy for stabilization of the DRS structure leading to improved electrochemical cyclability of the materials.
Introduction
Lithium-ion batteries (LIBs) are the most widely used energy storage systems for applications in electric transportation and portable electronic devices.1,2 Cathode materials for LIBs need to meet high energy and power density criteria for several applications and are the main focus of current research activities.1,3,4 In 2015, the lithium-rich disordered rock salt (DRS) Li2VO2F was identified as a promising cathode material with a high theoretical capacity of 462 mA h g−1 by Chen et al.5 This compound exhibits a DRS structure where Li and V ions appear to be randomly distributed at the same crystallographic site in the crystal structure.6 The lithium-excess (Li![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :TM-ratio (transition metal) > 1) in DRS structures enables percolating pathways for Li-ions to diffuse through the structure, facilitating their use as battery materials with relatively high capacities.7,8 Li2VO2F was the first material reported in the class of Li-rich DRS materials and the first material to incorporate F− by partly substituting O2− to lower the oxidation state of the transition metal cation and to increase the average discharge potential.9 The capacity is expected to originate from the two-electron redox couple, V3+/V5+, leading to practical capacities over 300 mA h g−1, considerably higher than what is observed for conventional cathode materials such as LiCoO2 and LiFePO4 (140 mA h g−1 and 170 mA h g−1).1–3
:TM-ratio (transition metal) > 1) in DRS structures enables percolating pathways for Li-ions to diffuse through the structure, facilitating their use as battery materials with relatively high capacities.7,8 Li2VO2F was the first material reported in the class of Li-rich DRS materials and the first material to incorporate F− by partly substituting O2− to lower the oxidation state of the transition metal cation and to increase the average discharge potential.9 The capacity is expected to originate from the two-electron redox couple, V3+/V5+, leading to practical capacities over 300 mA h g−1, considerably higher than what is observed for conventional cathode materials such as LiCoO2 and LiFePO4 (140 mA h g−1 and 170 mA h g−1).1–3
Many different compounds with the Li-rich DRS phase have been reported since the original study. Materials like Li1.3Nb0.3TM0.4O2 (TM = Fe3+, Mn3+, V3+) and Li1.2Ni1/3Ti1/3Mo2/15O2 have been synthesized using a classical high-temperature annealing solid-state approach.10–12 These materials exhibit a relatively stable electrochemical behavior for more than 20 cycles. Fluoride-containing Li-rich DRS materials, such as Li2VO2F, Li2Cr1−xVxO2F (x = 0–1), Li2MnO2F, Li2MoO2F and Li2.1Ti0.2Mo0.7O2F, have on the other hand only been synthesized by high-energy ball milling, leading to nano-sized particles with crystallographic defects.5,13–17 Ball milled DRS oxyfluorides generally suffer from fast capacity fading and the reason for the capacity fading is still not fully understood.5,15,16,18 Recent publications indicate that such DRS phases might be metastable.10,14,19 A recent study of Källquist et al. identified irreversible reactions at the surface of Li2VO2F with the electrolyte as the possible cause of the capacity fading.20 Herein, we propose that the key to understand the degradation mechanism of Li2VO2F and other disordered rock salt oxyfluorides is linked to the metastable structure and surface stability of the DRS phase. Even though Li2VO2F has a lower average discharge voltage compared to other DRS materials, its high theoretical and practical discharge capacities make it an attractive material to study. We report the synthesis of two new Li-rich DRS oxyfluoride phases, Li2V0.5Ti0.5O2F and Li2V0.5Fe0.5O2F. We also elucidate alternative synthesis approaches towards a conventional solid-state synthesis of Li2VO2F and substituted Li-rich DRS oxyfluorides (see ESI†). LiF cannot be incorporated stoichiometrically into the structure under conventional solid-state synthesis conditions. Thus, the isovalent substituted Li2V0.5Ti0.5O2F and Li2V0.5Fe0.5O2F compounds are synthesized by a high energy ball milling approach and the properties of the compounds are compared with those of Li2VO2F. The structural stability of the materials is investigated by density functional theory (DFT) calculations. Further, we analyze the electrochemical performance by galvanostatic charge–discharge experiments and atomic level information from the material surface is obtained from ex situ hard X-ray photoelectron spectroscopy (HAXPES).
Experimental
Synthesis procedures
The compounds were prepared by using a dry ball milling procedure in two steps (each step involves milling at 600 rpm for 20 h using a Fritsch Pulverisette 6 classic line containing an 80 mL Si3N4 jar and 25 balls of 10 mm diameter under the conditions of a ball to powder ratio of 10:1 and the total amount of powder of approx. 4.5 g). All compounds were added into an air-tight Si3N4 jar under inert conditions in an argon-filled glovebox with water and oxygen levels below 0.1 ppm.
In the first step, Li2O (10% excess to compensate loss during the synthesis,21 Alfa Aesar, 99.7%) and the corresponding metal oxide precursors, V2O3 (Alfa Aesar, 99.7%), and Ti2O3 (Alfa Aesar, 99.8%) or Fe2O3 (Alfa Aesar, 99.9%), were milled to form the intermediate products DRS LiVO2, LiV0.5Ti0.5O2 or LiV0.5Fe0.5O2, respectively. In the second step LiF (Alfa Aesar, 99.9%) was added and subsequently milled to stoichiometrically form Li2VO2F, Li2V0.5Ti0.5O2F or Li2V0.5Fe0.5O2F, respectively. After ball milling, powders were handled in a glovebox and were used without further purification.
Powder X-ray diffraction (PXRD)
Synchrotron PXRD patterns were recorded at beamline 11-ID-B at the APS Argonne National Lab using a PerkinElmer flat panel detector (XRD1621) with a pixel size of 200 × 200 μm. The powdered samples were placed in 1.1 mm borosilicate glass capillaries inside an argon-filled glovebox and sealed before they were measured in transmission geometry. A sample-to-detector distance of 641.010 mm and a wavelength of 0.21280 Å were used for data acquisition with a summed exposure time of 30 s per diffraction pattern. The diffraction data were integrated using the Fit2D.22–24 Rietveld refinements were conducted using TOPAS version 5.25 The instrumental resolution function was determined using the CeO2 standard. For the Rietveld refinement, the b-factors were refined for the 4a and 4b Wyckoff positions and not for each element individually. The occupancies of O and F were fixed to 2/3 for O and 1/3 for F, respectively. The TM and Li occupancies were restrained to result in 1 and refined.Ex situ PXRD patterns of cycled electrodes after 100 cycles were recorded in reflection geometry using a STOE STADI-p diffractometer with Mo Kα1 radiation (0.70932 Å), equipped with a DECTRIS MYTHEN 1K strip detector. The electrodes where covered with Kapton® tape to prevent oxidation in air.
Transmission electron microscopy (TEM)
TEM characterization and energy dispersive X-ray spectroscopy (EDX) mapping were performed using a Cs corrected JEOL ARM CF operating at 200 kV, equipped with an SSD Jeol EDX spectrometer.DFT calculations
DFT calculations were carried out using the Vienna Ab initio Simulation Package (VASP)26–29 using the projector augmented-wave (PAW) method.30 The generalized gradient approximation as parametrized by Perdew, Burke and Ernzerhof31 was used as the exchange–correlation functional. The plane-wave cutoff of 600 eV was used, and both the cell and atomic positions were fully relaxed such that all the forces are smaller than 0.02 eV Å−1. A rotationally invariant Hubbard U correction32,33 was applied to the d orbitals of V, Ti and Fe with the U values of 3.25, 3.50 and 4.30 eV, respectively. Integrations over the Brillouin zone were carried out using the Monkhorst–Pack scheme34 with a grid with a maximal interval of 0.04 Å−1.Electrochemical measurements
Electrodes were prepared by pre-mixing Li2VxTM1−xO2F (TM = Ti, Fe, x = 1 or 0.5) with carbon black (acetylene black, Alfa Aesar) in the ball mill to form a composite (300 rpm for 3 h). The obtained composite was mixed with polyvinylidenedifluoride binder (PVDF) (Solvay 6050) and N-methyl-2-pyrrolidone (NMP, Alfa Aesar, 99.5%) solution to obtain a slurry with a weight ratio of 70/20/10. The slurry was coated on aluminum foil acting as the current collector and subsequently dried under vacuum at stepwise increasing temperatures up to a maximum of 120 °C for 12 h. Afterwards electrodes of 12 mm diameter were punched out. The active material mass loading ranges from 1.4 to 1.9 mg cm−2 with a dry film thickness between 12 and 21 μm.For the electrochemical measurements, 2-electrode Swagelok-type cells were assembled using a lithium metal counter electrode, a Li2VO2F working electrode, 200 μL LP30-electrolyte (1 M LiPF6 in an ethylene carbonate (EC)/dimethyl carbonate (DMC) mixture (1:1 by volume, Sigma Aldrich)) and two Whatman glass fiber separators. These Li half-cells were assembled in a glovebox under an argon atmosphere. Galvanostatic charge–discharge tests were conducted in the 2-electrode setup with an ARBIN BT2000 battery testing system. All electrochemical cycling tests were carried out with a C/5-rate in a voltage range of 1.3 V to 4.1 V vs. Li/Li+. Cycling was performed at room temperature.
HAXPES analysis
For HAXPES analysis, pouch-type cells were prepared in an argon-filled glovebox (O2 < 2 ppm, H2O < 1 ppm). 13 mm diameter electrodes of Li2VO2F, Li2V0.5Fe0.5O2F or Li2V0.5Ti0.5O2F were used as working electrodes with a lithium metal (125 μm thick, Cyprus Foote Material) counter electrode and Solupor separator (20 μm thick) soaked in 50 μL LP30-electrolyte. Galvanostatic charge–discharge was performed using a Digatron BTS 600 galvanostat under the same conditions as describe above.HAXPES was performed on pristine, fully charged and fully discharged electrodes cycled 5 or 50 times. These are abbreviated P, Ch5, DCh5, Ch50, and DCh50 respectively. HAXPES samples were prepared by disassembling the pouch cells, rinsing the working electrode with DMC to remove salt residues, and mounting a piece of the electrode on a sample holder using conductive Cu or carbon tape. All sample transfers were made under an Ar atmosphere. For Li2VO2F and Li2V0.5Fe0.5O2F, measurements were performed at the KMC-1 beamline at BESSY II, Germany35 using photon energies of 2005 eV. X-rays were monochromatized using a Si(111) double-crystal monochromator and a Gammadata Scienta R-4000 hemispherical analyzer was used to record the photoelectron spectra. Li2V0.5Ti0.5O2F samples were measured at the I09 beamline at Diamond light source, UK36 using photon energies of 2350 eV. X-rays were monochromatized using a Si(111) double-crystal monochromator and a Scienta EW4000 high-voltage electron analyzer was used to record the photoelectron spectra. The probing depth was estimated to be approximately 10 nm using three times the inelastic mean free path (IMFP) for Li2VO2F derived from the NIST database37 using material parameters from The Materials Project.38 HAXPES data analysis and curve-fitting were performed using Igor Pro 6.37 software. All spectra were intensity normalized to the highest intensity peak and binding energy calibrated by shifting the carbon black peak to 284.4 eV. To curve fit the transition metal spectra, the spin orbit splitting of the 2p3/2 and 2p1/2 doublet peaks was locked to established values (7.3 eV for V, 5.7 eV for Ti, and 13.6 for Fe) while the absolute binding energy values were allowed to vary. In the case of vanadium, only one spin orbit split doublet peak was used to fit the spectra, and its binding energy was used to determine the oxidation state. For V 2p and Ti 2p peaks, parameters from the work by Biesinger et al.39 were used for the fitting. The Fe 2p peaks partially overlap with the plasmon of the F 1s peak.40 By comparing the survey measurements of Li2V0.5Fe0.5O2F and Li2VO2F (ESI, Fig. S11†) it was determined that the Fe 2p1/2 peak lies above the fluorine plasmon and can thus be used to evaluate iron. The Fe 2p1/2 was fitted using established peak parameters.41,42 Further details on the fitting can be found in the ESI.† To evaluate the composition of the samples, peak areas are calculated and normalized by the photoionization cross section. Ratios between different elements are calculated according to eqn (1):
 | (1) |
Results
We synthesized the target compounds Li2V0.5Ti0.5O2F and Li2V0.5Fe0.5O2F by a high energy ball milling approach, similar to the reported synthesis of Li2VO2F.5 High energy ball milling is often used to synthesize metastable phases or to stabilize phases that normally exist at higher temperatures or pressures.44 The synthesis procedures reported in the literature so far synthesize the DRS compounds in a one-step process, which in some cases leads to impurities of unreacted precursor compounds.5,15 In the case of Li2VO2F, a total ball milling time of 40 hours is suggested.5 Here, we introduce a two-step approach. In the first step, we synthesized a LiTMO2 species by using Li2O and the corresponding TM2O3 oxide already exhibits the DRS structure.45 In a second step, LiF was introduced into the structure as a solid solution by ball milling. This approach has the advantage of significantly reducing the transition metal oxide precursor impurities in the product. Additionally, we changed the ball mill jar and ball material from tungsten carbide (WC) to less abrasive silicon nitride (Si3N4), which reduces the amount of impurities in the synthesized compound. Following this approach, we successfully synthesized two new Li-rich DRS materials, Li2V0.5Ti0.5O2F and Li2V0.5Fe0.5O2F, and compared them with the original Li2VO2F compound.Rietveld refinements based on the synchrotron PXRD of Li2V0.5Ti0.5O2F and Li2V0.5Fe0.5O2F, both mixed with carbon black, are presented in Fig. 1. The broad diffraction peaks of both compounds indicate the nanocrystalline nature of the material, as known for Li2VO2F (ESI, Fig. S2†) and similar ball milled materials.5,9,15,21,45 Rietveld refinements are based on the DRS phase (Fm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m) for both Li2V0.5Ti0.5O2F and Li2V0.5Fe0.5O2F and yield in good fits. The Rietveld refinements show that the synthesized products have high purity (the products contain below 1 wt% of Si3N4 impurity). No transition metal precursors (TM2O3) were found. The refinements yield lattice parameters of a = 4.1342(6) Å for Li2V0.5Ti0.5O2F and a = 4.1388(6) Å for Li2V0.5Fe0.5O2F, which are slightly larger compared to that of the unsubstituted compound Li2VO2F (a = 4.1169(4) Å) due to the larger ionic radii of Ti3+ and Fe3+.46 The refined occupancies of the transition metal positions differ only marginally from the ideal stoichiometry for the Ti and Fe containing phases. Detailed structural parameters obtained from the refinement are given in ESI Table S1.† The crystallite size (between 12 and 14 nm) and strain (below 0.6%) were determined from Williamson–Hall plots (ESI, Fig. S3†).47
m) for both Li2V0.5Ti0.5O2F and Li2V0.5Fe0.5O2F and yield in good fits. The Rietveld refinements show that the synthesized products have high purity (the products contain below 1 wt% of Si3N4 impurity). No transition metal precursors (TM2O3) were found. The refinements yield lattice parameters of a = 4.1342(6) Å for Li2V0.5Ti0.5O2F and a = 4.1388(6) Å for Li2V0.5Fe0.5O2F, which are slightly larger compared to that of the unsubstituted compound Li2VO2F (a = 4.1169(4) Å) due to the larger ionic radii of Ti3+ and Fe3+.46 The refined occupancies of the transition metal positions differ only marginally from the ideal stoichiometry for the Ti and Fe containing phases. Detailed structural parameters obtained from the refinement are given in ESI Table S1.† The crystallite size (between 12 and 14 nm) and strain (below 0.6%) were determined from Williamson–Hall plots (ESI, Fig. S3†).47
 | ||
| Fig. 1 (a) PXRD pattern of Li2V0.5Ti0.5O2F mixed with carbon black with Rietveld refinement and the Bragg position of Li2V0.5Ti0.5O2F. (b) PXRD pattern of Li2V0.5Fe0.5O2F mixed with carbon black with Rietveld refinement and the Bragg position of Li2V0.5Fe0.5O2F. λ = 0.21280 Å. Inset: schematic illustration of the DRS crystal structure. | ||
The particle size and morphology of Li2V0.5Ti0.5O2F, Li2V0.5Fe0.5O2F and Li2VO2F were investigated by TEM. The results for Li2V0.5Ti0.5O2F are presented in Fig. 2. The results for Li2V0.5Fe0.5O2F and Li2VO2F are given in the ESI (ESI, Fig. S4–S6†). The sample is composed of agglomerated nanocrystalline particles (Fig. 2a). Similar microstructures have been found for Li2V0.5Fe0.5O2F and Li2VO2F. The lattice d-values obtained by selected area electron diffraction (SAED) (Fig. 2b) correspond to the metrics of the DRS structure (Fmm) and confirm the results of the Rietveld refinement. The high-resolution scanning transmission electron microscopy annular dark field image (STEM-ADF) in Fig. 2c reveals several nanocrystalline (5–10 nm) and some amorphous domains. The corresponding fast Fourier transformation (FFT) of one nanocrystallite is shown in Fig. 2d, matching the orientation along the [101] zone axis of the disordered structure. Despite the local non-uniform mass-thickness contrast in the image, it is possible to identify some differences in the intensity of the atomic columns, indicating variations of the transition metal atomic content. Similar results have been obtained for Li2VO2F and Li2V0.5Fe0.5O2F. Energy dispersive X-ray spectroscopy mapping (EDX) of the materials shows a uniform distribution of elements. However, in the case of Li2V0.5Fe0.5O2F the EDX map reveals a small fraction of V-enriched areas of 40–80 nm size, which are not present in the X-ray diffraction pattern (ESI, Fig. S6†).
 | ||
| Fig. 2 TEM analysis of Li2V0.5Ti0.5O2F. (a) TEM image, (b) SAED pattern, and (c) high-resolution STEM ADF image with corresponding FFT (d). | ||
To shed light on the structural properties of Li2VO2F, Li2V0.5Ti0.5O2F and Li2V0.5Fe0.5O2F we investigated the relative structural stability of DRS oxyfluoride phases using DFT calculations by comparing the energies of Special Quasi-random Structures (SQS) and ordered prototype structures. These are derived from known ordered oxide structures such as α-NaFeO2 and γ-LiFeO2, which are known to be the ground state structure of many lithium transition metal oxides (ESI, Fig. S7 and S8†).48,49 The relative structural stability of DRS oxyfluoride phases is determined via the energy difference between the SQS and the most stable ordered structure, ΔE, defined as
| ΔE = ESQS − min(Eordered) | (2) |
The ordered phase is expected to be more structurally stable than the disordered phase for all considered compounds, because the DRS oxyfluorides are in the metastable phase achieved using a mechanochemical ball milling procedure. Furthermore, the decomposition of Li2VO2F into LiVO2 and LiF upon heating50 indicates that the considered compounds may be metastable in general, irrespective of the ordered or disordered phase. Such metastability of the compounds makes it difficult to investigate their relative structural stabilities. However, the ordering propensity, the extent to which the ordered phase is preferred compared to the disordered phase, can be used to assess the relative stability of the compared DRS oxyfluoride compounds.49 In other words, the relative values of ΔE in eqn (2) are used to assess how stable the compounds are in the disordered phase compared to the ordered phase. The value of ΔE is 179, 144 and 147 meV per atom for Li2VO2F, Li2V0.5Ti0.5O2F and Li2V0.5Fe0.5O2F, respectively. The ordered phase is energetically preferred for all three compounds, and the disordered phase is metastable as expected. More interestingly, the ΔE values of Li2V0.5Ti0.5O2F and Li2V0.5Fe0.5O2F are lower than those of Li2VO2F by more than 30 meV per atom, indicating enhanced structural stability of the disordered phase. Consequently, Li2V0.5Ti0.5O2F and Li2V0.5Fe0.5O2F are more likely to retain the disordered phase upon cycling than Li2VO2F. It should be noted that the relative energies of the decomposed products of the compounds (LiF + LiTMO2 for Li2TMO2F and LiF + 0.5LiTM1O2 + 0.5LiTM2O2 for Li2TM10.5TM20.5O2F, where more stable α-NaFeO2 and γ-LiFeO2 type oxide structures are considered) are also compared (ESI, Table S2†). All of the compounds are found to be most stable in the decomposed state, which aligns with the challenges faced in synthesizing the compounds in conventional techniques.
In addition to the relative stabilities of the disordered and ordered phases, DFT calculations reveal that the disorder leads to a distribution of oxidation states of the transition metal ions in the compounds. The oxidation states of TM in Li2TMO2F and TM1 and TM2 in Li2TM10.5TM20.5O2F (TM, TM1 and TM2 = V, Ti, Fe, respectively) are always 3+ for all of the ordered structures. The oxidation states of V ions are distributed between 2+, 3+ and 4+ in the SQS of Li2TMO2F (the distribution of oxidation states of the transition metals of the SQS is shown in Table S3 of ESI†). Furthermore, it is observed that the substitution of V with Ti leads to a downward shift in the oxidation state distribution of V ions (between 2+ and 3+) while Ti ions have oxidation states of 3+ and 4+. The opposite happens when V ions are substituted with Fe ions; oxidation states of V ions are distributed between 3+ and 4+ while they are distributed between 2+ and 3+ for Fe ions. A constant value of oxidation states in the ordered phase and its distribution pattern in the disordered phase can be used to determine the extent to which the material is disordered, albeit to a first order approximation.
The electrochemical performance of the new DRS oxy-fluoride compounds Li2V0.5Ti0.5O2F and Li2V0.5Fe0.5O2F was investigated. The proposed theoretical capacity of Li2VO2F is 462 mA h g−1 based on a 2 e− redox process of the V3+/V5+ couple. Li2V0.5Ti0.5O2F has a theoretical capacity of 350 mA h g−1 based on a 1.5 e− redox process assuming additional redox activity of Ti3+/Ti4+ in the low voltage range between 1.5 and 2.0 V.51 Li2V0.5Fe0.5O2F has a theoretical capacity of 226 mA h g−1 assuming electrochemical inactivity of Fe3+ (2 e− redox process of 50% V3+/V5+). Galvanostatic charge–discharge tests of Li2VO2F, Li2V0.5Ti0.5O2F and Li2V0.5Fe0.5O2F half cells have been conducted. The cycling performance is shown in Fig. 3. The materials have been cycled within a potential range of 1.3 and 4.1 V vs. Li/Li+ with a C/5-rate. Li2VO2F shows the highest first discharge capacity of all three compounds (Fig. 3a) of around 330 mA h g−1, which is in good agreement with the literature, accompanied by rapid capacity fading known from previous reports.5,14 45% of the initial discharge capacity is lost after 25 cycles. After 50 cycles the discharge capacity is already below 150 mA h g−1, which corresponds to less than 40% capacity retention (Fig. 3b). Both substituted compounds, Li2V0.5Ti0.5O2F and Li2V0.5Fe0.5O2F, exhibit a lower discharge capacity of 285 mA h g−1 and 218 mA h g−1 in the first cycle, respectively. The discharge capacity of Li2V0.5Ti0.5O2F is 67 mA h g−1 higher compared to Li2V0.5Fe0.5O2F, which may be explained by additional contribution to the capacity of the Ti3+/Ti4+ redox couple. The capacity fading is significantly reduced for both substituted compounds; Li2V0.5Ti0.5O2F and Li2V0.5Fe0.5O2F retain 81% and 83% of the initial discharge capacity after 25 cycles and 66% and 73% after 50 cycles, respectively. The coulombic efficiency (Fig. 3a) is improved for both new materials (around 97% for 50 cycles) compared to Li2VO2F (around 93%). Altogether, the substitution of V with 50% Ti or Fe clearly improves the cycling performance compared to Li2VO2F. Li2V0.5Fe0.5O2F shows the best cycling stability over 50 cycles, whereas Li2V0.5Ti0.5O2F exhibits the highest overall discharge capacities. The corresponding voltage profiles of Li2VO2F, Li2V0.5Ti0.5O2F and Li2V0.5Fe0.5O2F exhibit a steep and sloping profile shape enhanced by the disorder suggesting a single-phase insertion process for Li+ (ESI, Fig. S9†).5,6,14 No voltage plateaus are observed. The average discharge voltage of Li2VO2F is about 2.53 V with a voltage hysteresis of 0.51 V. Both substituted compounds exhibit a slightly lower average discharge voltage, 2.31 V for Li2V0.5Ti0.5O2F and 2.45 V for Li2V0.5Fe0.5O2F. The voltage hysteresis for Li2V0.5Ti0.5O2F is 0.59 V, which is the highest of all three compounds. Li2V0.5Fe0.5O2F shows the smallest voltage hysteresis of 0.43 V.
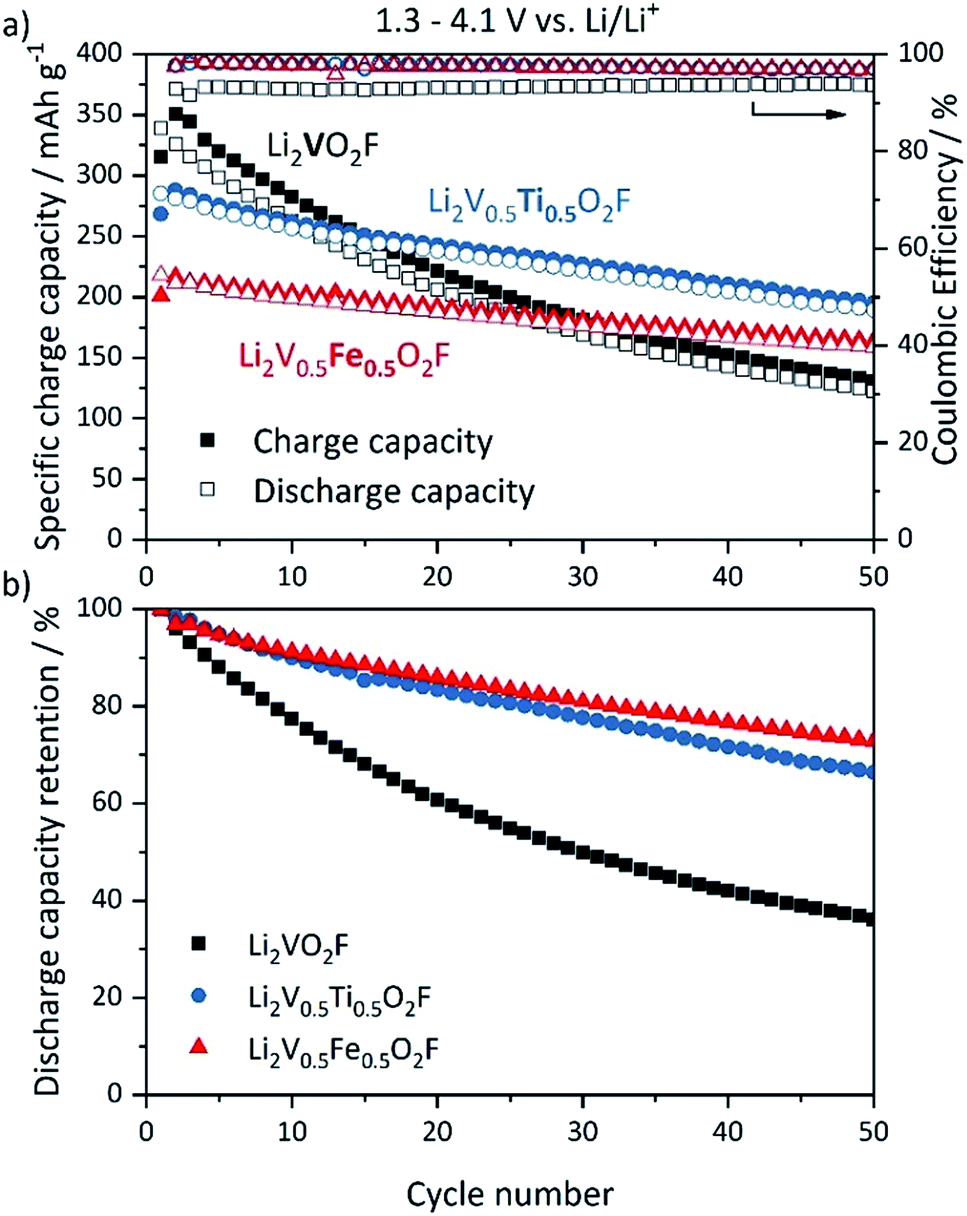 | ||
| Fig. 3 (a) Cycling performance (filled symbols: charge capacity, hollow symbols: discharge capacity) and coulombic efficiency of Li2VO2F (black), Li2V0.5Ti0.5O2F (blue) and Li2V0.5Fe0.5O2F (red) as a function of cycle number for half-cells cycled within a potential range of 1.3–4.1 V vs. Li/Li+ with C/5-rate at 25 °C. (b) Corresponding discharge capacity retention of the compounds. | ||
To understand the redox processes occurring during electrochemical cycling the differential capacity dQ/dV plots are shown in Fig. 4. In Li2VO2F, the oxidation of V3+ to V4+ is located in the area of 2.6 V and that of V4+ to V5+ is located above 3.5 V (indicated with dashed lines).14,15 For Li2V0.5Ti0.5O2F, the assumed redox peaks of vanadium (dashed lines) are slightly shifted to higher voltages and exhibit the highest overpotentials, which may be related to kinetic effects. Furthermore, additional peaks in the charge and discharge directions are observed at 2.2 V and 1.8 V, respectively (dotted lines). These peaks are not present in the samples that do not contain Ti (Li2VO2F and Li2V0.5Fe0.5O2F) and thus are expected to originate from the Ti3+/Ti4+ redox couple leading to an additional discharge capacity. Li2V0.5Fe0.5O2F shows the smallest overpotential and behaves like Li2VO2F in the low voltage regime indicating a similar redox behavior, but differs in the voltage region above 3.5 V during charging. This deviation may be related to processes at high voltages associated with irreversible reactions affecting the cycling stability. Upon extended cycling the dQ/dV plot of Li2VO2F tends to a flattening differential capacity peak response, indicating a loss of V-redox activity.20 In contrast, Li2V0.5Ti0.5O2F and Li2V0.5Fe0.5O2F preserve the characteristic redox peaks for a longer cycling period. This suggests that the maintained electrochemical activity of the TM is related to the improved cycling stability of the materials. Like for Li2VO2F, the ex situ PXRD pattern of Li2V0.5Ti0.5O2F and Li2V0.5Fe0.5O2F after extended cycling does not exhibit any development of new crystalline phases (ESI, Fig. S10†).5
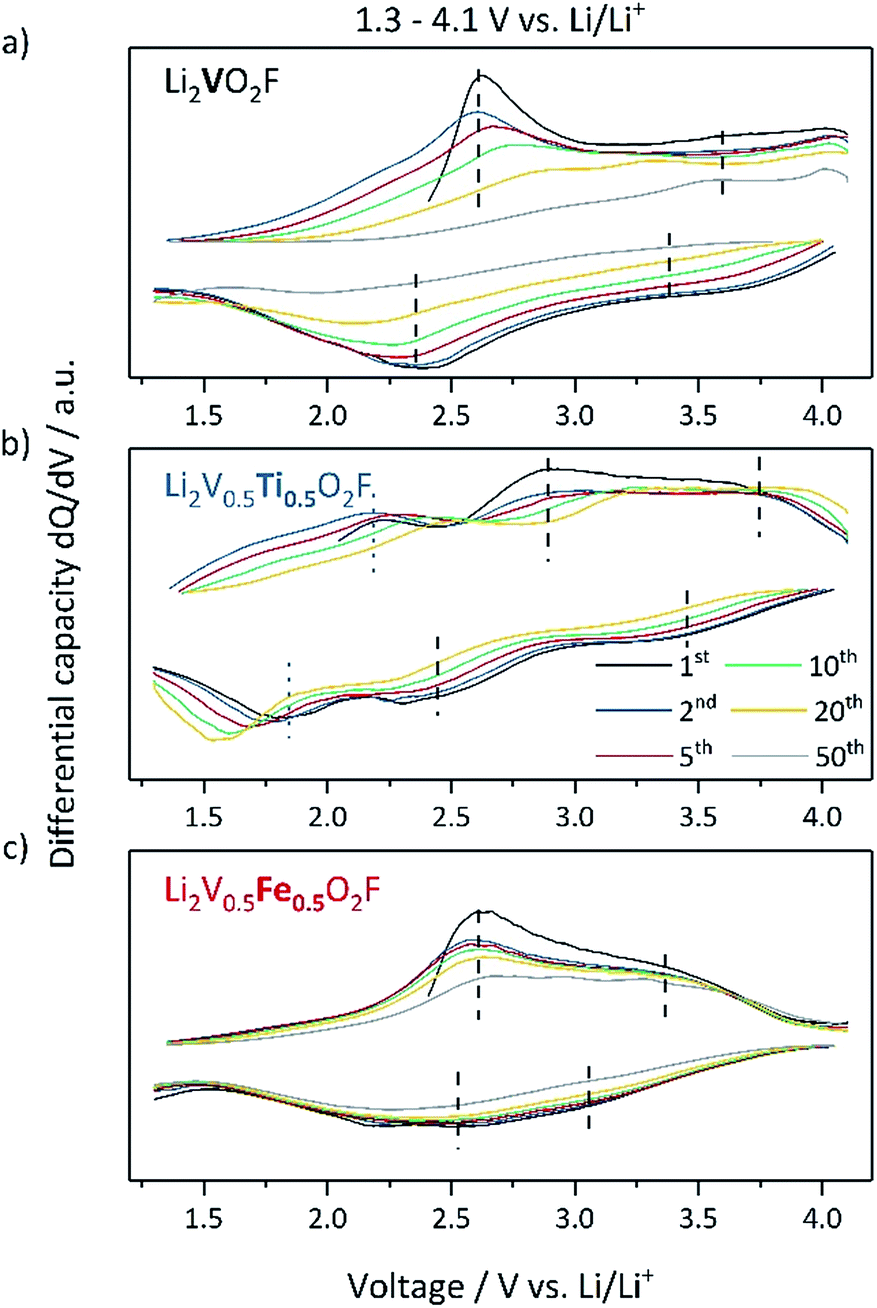 | ||
| Fig. 4 Differential capacity curves of (a) Li2VO2F, (b) Li2V0.5Ti0.5O2F and (c) Li2V0.5Fe0.5O2F half-cells cycled within a potential range of 1.3–4.1 V vs. Li/Li+ with C/5-rate at 25 °C. | ||
To further analyze the materials' surface stability, HAXPES was used to probe the redox activity of the transition metals and the surface layer evolution. Since HAXPES is a surface sensitive technique it has commonly been used to study the surface layers built up on the active material, known to be crucial for the cycling performance.52,53 Thus, to understand the improved capacity retention of the substituted materials the O 1s, V 2p and C 1s spectra are analyzed for pristine (P) and samples cycled 5 or 50 times in both charged (Ch5 and Ch50, respectively) and discharged (DCh5 and DCh50, respectively) states. The photon energy for the measurements is chosen so that both the outer layers of the active material and the surface layer can be probed.
The O 1s and V 2p spectra are shown in Fig. 5. Five different peaks are used to fit the data, from left to right corresponding to carboxyl/hydroxyl compounds (∼534 eV), carbonates (∼532 eV), metal oxide (530 eV) and vanadium that is detected with a spin orbit splitting of 7.33 eV at ∼517 and 524 eV according to peak parameters summarized by Biesinger et al.39 The energy difference of the O 1s metal oxide peak and V 2p3/2 can be used to determine the oxidation state of V, where a larger value corresponds to a lower oxidation state. The values obtained from the fitting are presented in Table 1. The pristine samples show differences in the average vanadium oxidation state depending on the material. Li2VO2F and Li2V0.5Ti0.5O2F show vanadium in a mix of V3+ and V4+, while Li2V0.5Fe0.5O2F contains a mix of V4+ and V5+ in the region probed. The deviation from an average oxidation state of 3+ might be related to surface oxidation, as seen also for other vanadium oxides.39 The mix of oxidation states is seen from the rather large FWHM of the V 2p peaks. This can be expected due to the disordered structure, where vanadium can be found with a different coordination of oxygen and fluorine, affecting the binding energy. Upon fifth charge all samples are as expected close to a fully oxidized V5+ state. After the following fifth discharge Li2V0.5Fe0.5O2F and Li2V0.5Ti0.5O2F return close to their respective pristine state, while the vanadium in Li2VO2F is no longer redox active and stays in a highly oxidized state. For the Li2VO2F material this reduced redox activity of vanadium has previously been suggested to be linked to a partial oxidation of oxygen forming reactive compounds that leads to a breakdown of the active material, starting at the surface.20 In this context both the Li2V0.5Fe0.5O2F and Li2V0.5Ti0.5O2F materials clearly show improved reversibility of the vanadium redox behavior. Still, after 50 cycles the materials show less to no redox activity of vanadium in the depth region probed by HAXPES. This trend can also be followed in the FWHM, which changes upon cycling. Especially on fifth discharge a broadening can be seen, indicating that some of the material can no longer return to its original state. This broadening is most significant for the unsubstituted material. The smaller FWHM after 50 cycles can be explained by a more uniform V5+ state of the probed material, in combination with that V5+ exhibits narrower peaks than V4+ and V3+, since V5+ does not have any unpaired valence electrons.39
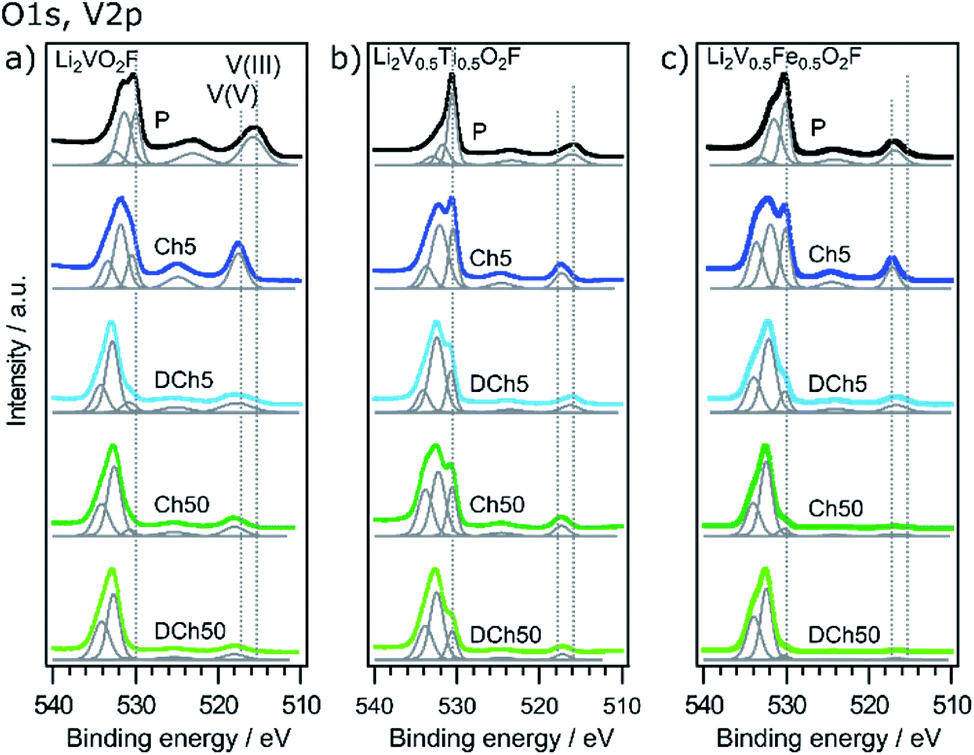 | ||
| Fig. 5 O 1s and V 2p photoelectron spectra of the three different materials in pristine, charged and discharged states after 5 and 50 cycles, obtained using a photon energy of 2005 eV (a and c) or 2350 eV (b). Dotted lines indicate reference values for metal oxide (530 eV) and different oxidation states of vanadium.39 | ||
| Material | Li2VO2F | Li2V0.5Ti0.5O2F | Li2V0.5Fe0.5O2F | |||
|---|---|---|---|---|---|---|
| ΔBE | FWHM | ΔBE | FWHM | ΔBE | FWHM | |
| a Corresponding ΔBE values for V3+, V4+ and V5+ in vanadium oxide references are 14.7, 14.2 and 12.8 eV, respectively.39 | ||||||
| P | 14.2 | 3.3 | 14.5 | 2.7 | 13.3 | 2.5 |
| Ch5 | 12.9 | 2.3 | 13.2 | 2.0 | 12.9 | 1.8 |
| DCh5 | 13.0 | 4.3 | 14.4 | 2.6 | 13.5 | 3.2 |
| Ch50 | 12.7 | 3.0 | 13.3 | 2.0 | 13.5 | 3.3 |
| DCh50 | 12.7 | 3.2 | 13.4 | 2.0 | 13.5 | 3.3 |
To gain a deeper insight into the improved redox activity of the substituted materials it is interesting to look at the intensity ratio between vanadium and the metal oxide (MO) peak.20 In Table 2 the V:MO ratios are presented, with the oxygen content normalized to two for easy comparison. According to the structural formulas, the V content should be 1 for Li2VO2F and 0.5 for Li2V0.5Fe0.5O2F and Li2V0.5Ti0.5O2F. For the pristine materials the ratio is slightly higher than expected for the Li2VO2F material, while for the Li2V0.5Fe0.5O2F and Li2V0.5Ti0.5O2F materials the ratio is slightly lower. Comparing charged and discharged samples, it is seen that the relative ratio is higher in the discharged samples for Li2VO2F and Li2V0.5Fe0.5O2F, while the Li2V0.5Ti0.5O2F material shows a small but opposite trend. For Li2VO2F the V:MO ratio increases significantly during cycling together with a binding energy shift of the MO peak. As discussed in detail in another study,20 the relative increase and decrease of vanadium compared to oxygen can be coupled both to possible oxygen redox processes as well as the formation and dissolution of a surface layer containing vanadium. The same trend cannot be seen for the Li2V0.5Fe0.5O2F and Li2V0.5Ti0.5O2F materials. Only a slight increase of the V:MO ratio is seen for Li2V0.5Fe0.5O2F, while for Li2V0.5Ti0.5O2F a close to constant ratio is obtained up to 50 cycles. Additionally, only small binding energy shifts of the MO peak are seen (<0.2 eV). These results clearly indicate that substitution with iron and titanium mitigates the detrimental reactions causing vanadium dissolution and incorporation in the surface layer and thus improves the chemical stability of the materials.
| Material | V:MO |
||
|---|---|---|---|
| Li2VO2F | Li2V0.5Ti0.5O2F | Li2V0.5Fe0.5O2F | |
| a To facilitate comparison the oxygen content is normalized to two according to the structural formula. | |||
| Ratio | 1.0:2 |
0.50:2 |
0.50:2 |
| P | 1.2:2 |
0.33:2 |
0.42:2 |
| Ch5 | 1.5:2 |
0.41:2 |
0.43:2 |
| DCh5 | 1.9:2 |
0.35:2 |
0.74:2 |
| Ch50 | 2.6:2 |
0.31:2 |
0.53:2 |
| DCh50 | 5.2:2 |
0.28:2 |
0.75:2 |
Looking further at the other transition metals (ESI, Fig. S12†), both iron and titanium are found to be partially redox active at the surface. Iron is found in a mix of Fe3+ and Fe2+ in the pristine material and upon cycling the Fe3+ content increases after fifth charge while more Fe2+ is found after subsequent discharge. Titanium is predominately found in the Ti4+ state for the pristine material, with some amount of Ti3+ upon discharge. As already mentioned for vanadium, the deviation from the 3+ oxidation state indicates that an oxidized surface layer is present already after the synthesis of the materials. This kind of passivating surface film is often seen on cathode materials.54,55
The surface layer evolution during cycling is evaluated using the carbon spectra, as shown in Fig. 6. Here it is particularly interesting to look at the relative intensities between the carbon black (CB) bulk peak (shaded in red) and the hydrocarbon (C–H) surface peak. A relatively lower CB peak intensity signifies a thicker surface layer. Starting with the unsubstituted material (Fig. 6a), a buildup of a surface layer is seen upon charge, followed by its partial dissolution upon discharge. This is consistent with previous results for Li2VO2F.20 The substituted samples on the other hand show a stabilized surface after 50 cycles (similar Ch50/DCh50 spectra). Especially Li2V0.5Ti0.5O2F (Fig. 6b) shows a rather thin and stable surface layer with similar spectra for all samples. For Li2V0.5Fe0.5O2F the surface layer is of similar thickness compared to the Li2VO2F sample, but the layer is more stable and no dissolution is observed after 50 cycles.
 | ||
| Fig. 6 C 1s photoelectron spectra of the three different materials in pristine, charged and discharged states after 5 and 50 cycles, obtained using a photon energy of 2005 eV (a and c) or 2350 eV (b). Dotted lines are a guide to the eye to track shifts in the hydrocarbon (C–H) peak relative to the carbon black (CB) peak. | ||
The other peaks in the C 1s spectra stem from the PVDF binder (two peaks at ∼286 and ∼290 eV) and different carbon oxygen compounds (at ∼286.5, 288 and 290 eV), typically stemming from electrolyte degradation. The surface layers are seen to consist of mostly hydrocarbons and some C–O compounds. On Li2V0.5Fe0.5O2F (Fig. 6c) the surface layer is built up with a relatively larger amount of C–O compounds, probably stemming from electrolyte breakdown. The presence of a surface layer indicates that some side reactions occur for all materials, but a more stable layer, as found on the substituted materials, can limit the extent of these reactions by passivating the surface.
Discussion
Combining the results of the synthesis, the DFT calculations, the electrochemical experiments and the HAXPES analysis, the results can be discussed related to disorder of the structure and related to the electrochemical cycling behavior of the materials. The disorder in the crystal structure of the pristine TM-oxyfluorides leads to a distribution of oxidation states deviating from the original 3+ state in the ordered structures for the presented compounds as has been revealed by the DFT calculations. The computed mixed 2+, 3+, and 4+ oxidation states for vanadium in Li2VO2F are consistent with the HAXPES results, considering additional contribution of Li-deficiency and oxidation at the surface.14,20 The DFT and HAXPES results further agree with the mixed 2+ and 3+ oxidation states of iron in Li2V0.5Fe0.5O2F, which lead to a shift for vanadium to 4+ and 5+. For Li2V0.5Ti0.5O2F, where the titanium oxidation state is 3+ and 4+, the substitution instead lowers the oxidation state of vanadium to 3+. In addition, the DFT calculations showed that the metastable disordered rock salt structure was stabilized when partially substituting vanadium with iron or titanium.The electrochemical cycling behavior of the substituted materials differs from that of Li2VO2F. Both substituted materials exhibit a significantly more stable cycling behavior but a lower discharge capacity. The differential capacity analysis reveals additional capacity contributions of Li2V0.5Ti0.5O2F compared to Li2V0.5Fe0.5O2F and Li2VO2F indicating redox activity of titanium in the bulk. Whittingham et al. determined the oxidation state of vanadium in Li2VO2F in the charged state by X-ray absorption spectroscopy. They observed an average oxidation state of only 4.2+ when charged to 4.1 V vs. Li/Li+.14 Complete oxidation to V5+ could not be achieved. The HAXPES results of Li2V0.5Ti0.5O2F and Li2V0.5Fe0.5O2F confirm a partial redox activity of iron and titanium at the surface (ESI, Fig. S12†). Furthermore, we believe that some of the additional capacity of Li2V0.5Ti0.5O2F compared to Li2V0.5Fe0.5O2F stems from the fact that substitution with titanium promotes a complete use of the V3+/V5+ redox couple. Further analysis by X-ray absorption techniques may shed light on the different redox reactions occurring in the bulk material.
In recent reports, DRS materials are cycled to high potentials up to 4.8 V vs. Li/Li+, which facilitates anionic redox activity of oxygen in the lattice that leads to additional capacity contribution.11,14–16,21 Materials containing Ti, Li1.2Ti0.4Mn0.4O2 for instance, experimentally sustain a stable oxygen-redox reaction above 4.1 vs. Li/Li+.10 The observed shift in binding energy of the MO peak together with the relative changes in intensity between the MO peak and vanadium indicates that such anionic redox activity occurs in the surface region of the Li2VO2F material already when cycling to 4.1 V vs. Li/Li+.56 In Li2VO2F this is believed to create highly reactive oxygen atoms in the lattice leading to the instability of the surface, as discussed in more detail in the work of Källquist et al.20 The reaction between the oxidized lattice oxygen and the electrolyte creates an interfacial layer rich in vanadium in oxidation state 5+. Li2V0.5Ti0.5O2F and Li2V0.5Fe0.5O2F show a higher reversibility of the V redox reaction according to the differential capacity analysis and the HAXPES results. In the dQ/dV plot, the substituted materials show a reduced irreversible capacity contribution above 3.5 V up to. 4.1 V vs. Li/Li+. At the same time, the HAXPES data only show small changes of the V:MO-ratio and the binding energy of the MO peak after 50 cycles for Li2V0.5Ti0.5O2F and Li2V0.5Fe0.5O2F. Together this suggests a reduced reactivity of the lattice oxygen species at the surface in these new materials when cycled to 4.1 V vs. Li/Li+. We propose this as an explanation for the improved cycling stability. This is further supported by the cycling performance when the materials are cycled up to 4.5 V vs. Li/Li+. These results (see ESI, Fig. S13†) show additional contribution to the capacity for all three compounds, possibly originating from anionic redox activity, but are also accompanied by a reduced cycling stability.
Additional contribution to the cycling stability likely comes from mitigation of the dissolution and rebuilding of the surface layer for the substituted materials that otherwise is seen on Li2VO2F. Although the thickness of the surface layers on Li2V0.5Ti0.5O2F and Li2V0.5Fe0.5O2F varies, both are to a large part preserved during cycling. This is in agreement with a recent study of Takeda et al. on Li2.1Ti0.2Mo0.7O2F showing a reduced dissolution of Mo when Ti is in the structure.17
Coupling these results to the electrochemical performance the improved cycling behavior of the Fe- or Ti-substituted materials can be summarized by a mitigated reactivity of the surface that previously has been perceived as one of the main reasons for capacity fading in Li-rich DRS materials.10,20,57
Conclusions
We have demonstrated the successful approach of improving the cycling stability of Li-rich vanadium disordered rock salt oxyfluoride cathodes by stabilization of the structure with Ti3+ or Fe3+. Two new disordered rock salt compounds, Li2V0.5Fe0.5O2F and Li2V0.5Ti0.5O2F, have been successfully synthesized by mechanochemical ball milling. The materials were characterized by synchrotron X-ray diffraction followed by an extensive analysis of the crystal structure by Rietveld refinement and transmission electron microscopy. By using the special quasi-random structure approach in density functional theory calculations, a distribution of oxidation states of the transition metal in the structure was revealed. The theoretical model further predicted an improvement of the structural stability for disordered rock salt Li2V0.5Fe0.5O2F and Li2V0.5Ti0.5O2F compared to unsubstituted Li2VO2F. The stabilizing effect on the structure further manifests in a significantly improved cycling stability over 50 cycles. HAXPES analysis of cycled electrodes supports our hypothesis and shows improved surface stability of the substituted materials. The cycling stability of the substituted compounds is significantly improved and supports our hypothesis of stabilizing the disordered rock salt structure of oxyfluorides in the bulk and at the surface with elements promoting the DRS structure (Fe3+ and Ti3+). Optimizing the TM stoichiometry by reducing the amount of stabilizing elements (e.g. Ti3+ or Fe3+) will help to increase discharge capacities. This work is one step further in understanding the structural and electrochemical cycling stability of DRS materials as a promising class of Co-free Li-ion insertion cathode materials.Author contributions
CB and JC planned the project. JC proposed the idea of DRS structure and capacity stabilization through substitution. CB, JC and AS synthesized the materials. CB performed the electrochemical measurements with the help of FK. The PXRD synchrotron data were acquired by REJ with the help of CB and PN. The PXRD data were analysed by CB with the help of REJ and JC. HAXPES samples were prepared by IK and measurements were performed by IK, AN and MH. HAXPES analysis was conducted by IK. Computational investigations were performed by JHC. TEM data were acquired by FRZ with the help of JMA. The manuscript was written by CB with the help of IK, JHC and MH. The manuscript was revised with the help of all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project has received funding from the European Union's Horizon 2020 research and innovation program (FET-OPEN project “LiRichFCC”) under grant agreement No. 711792. This research used resources of the Advanced Photon Source, an Office of Science User Facility operated for the U.S. Department of Energy (DOE) office of Science by Argonne National Laboratory and was supported by the U.S. DOE under contract No. DE-AC02-06CH11357. DANSCATT is acknowledged for covering travel expenses in relation to the synchrotron experiment. The research leading to the HAXPES result has been supported by the project CALIPSOplus under the Grant Agreement 730872 from the EU Framework Programme for Research and Innovation HORIZON 2020. We acknowledge Diamond Light Source for time on Beamline I09 under Proposal 19602 and HZB for allocated synchrotron radiation beamtime on beamline KMC-1 at BESSY II. The work is part of the platform CELEST (Center for Electrochemical Storage Ulm-Karlsruhe).Notes and references
- J. B. Goodenough and K.-S. Park, J. Am. Chem. Soc., 2013, 135, 1167–1176 CrossRef CAS.
- M. Armand and J.-M. Tarascon, Nature, 2008, 451, 652–657 CrossRef CAS.
- M. Winter, J. O. Besenhard, M. E. Spahr and P. Novák, Adv. Mater., 1998, 10, 725–763 CrossRef CAS.
- J. B. Goodenough and Y. Kim, Chem. Mater., 2010, 22, 587–603 CrossRef CAS.
- R. Chen, S. Ren, M. Knapp, D. Wang, R. Witter, M. Fichtner and H. Hahn, Adv. Energy Mater., 2015, 5, 1401814 CrossRef.
- A. Abdellahi, A. Urban, S. Dacek and G. Ceder, Chem. Mater., 2016, 28, 5373–5383 CrossRef CAS.
- J. Lee, A. Urban, X. Li, D. Su, G. Hautier and G. Ceder, Sci. 80, 2014, 343, 519–522 CrossRef CAS.
- A. Urban, J. Lee and G. Ceder, Adv. Energy Mater., 2014, 4, 1400478 CrossRef.
- R. Chen, S. Ren, M. Yavuz, A. A. Guda, V. Shapovalov, R. Witter, M. Fichtner and H. Hahn, Phys. Chem. Chem. Phys., 2015, 17, 17288–17295 RSC.
- N. Yabuuchi, M. Nakayama, M. Takeuchi, S. Komaba, Y. Hashimoto, T. Mukai, H. Shiiba, K. Sato, Y. Kobayashi, A. Nakao, M. Yonemura, K. Yamanaka, K. Mitsuhara and T. Ohta, Nat. Commun., 2016, 7, 13814 CrossRef CAS.
- N. Yabuuchi, M. Takeuchi, M. Nakayama, H. Shiiba, M. Ogawa, K. Nakayama, T. Ohta, D. Endo, T. Ozaki, T. Inamasu, K. Sato and S. Komaba, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 7650–7655 CrossRef CAS.
- J. Lee, D. H. Seo, M. Balasubramanian, N. Twu, X. Li and G. Ceder, Energy Environ. Sci., 2015, 8, 3255–3265 RSC.
- N. Takeda, S. Hoshino, L. Xie, S. Chen, I. Ikeuchi, R. Natsui, K. Nakura and N. Yabuuchi, J. Power Sources, 2017, 367, 122–129 CrossRef CAS.
- X. Wang, Y. Huang, D. Ji, F. Omenya, K. Karki, S. Sallis, L. F. J. Piper, K. M. Wiaderek, K. W. Chapman, N. A. Chernova and M. S. Whittingham, J. Electrochem. Soc., 2017, 164, A1552–A1558 CrossRef CAS.
- S. Ren, R. Chen, E. Maawad, O. Dolotko, A. A. Guda, V. Shapovalov, D. Wang, H. Hahn and M. Fichtner, Adv. Sci., 2015, 2, 1500128 CrossRef.
- R. A. House, L. Jin, U. Maitra, K. Tsuruta, J. W. Somerville, D. P. Förstermann, F. Massel, L. Duda, M. R. Roberts and P. G. Bruce, Energy Environ. Sci., 2018, 11, 926–932 RSC.
- N. Takeda, I. Ikeuchi, R. Natsui, K. Nakura and N. Yabuuchi, ACS Appl. Energy Mater., 2019, 2, 1629–1633 CrossRef CAS.
- T. Matsuhura, Y. Tsuchiya, K. Yamanaka, K. Mitsuhura, T. Ohta and N. Yabuuchi, Electrochemistry, 2016, 84, 797–801 CrossRef.
- M. Freire, N. V. Kosova, C. Jordy, D. Chateigner, O. I. Lebedev, A. Maignan and V. Pralong, Nat. Mater., 2016, 15, 173–177 CrossRef CAS.
- I. Källquist, A. J. Naylor, C. Baur, J. Chable, J. Kullgren, M. Fichtner, K. Edstrom, D. Brandell and M. Hahlin, Chem. Mater., 2019, 31, 6084–6096 CrossRef.
- J. Lee, D. A. Kitchaev, D.-H. Kwon, C.-W. Lee, J. K. Papp, Y.-S. Liu, Z. Lun, R. J. Clément, T. Shi, B. D. McCloskey, J. Guo, M. Balasubramanian and G. Ceder, Nature, 2018, 556, 185–190 CrossRef CAS.
- A. P. Hammersley, ESRF Intern. Rep Search PubMed.
- A. P. Hammersley, S. O. Svensson, M. Hanfland, A. N. Fitch and D. Hausermann, High Pressure Res., 1996, 14, 235–248 CrossRef.
- A. P. Hammersley, S. O. Svensson and A. Thompson, Nucl. Instrum. Methods Phys. Res., Sect. A, 1994, 346, 312–321 CrossRef CAS.
- A. A. Coelho, J. Appl. Crystallogr., 2018, 51, 210–218 CrossRef CAS.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558–561 CrossRef CAS.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 49, 14251–14269 CrossRef CAS.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS.
- V. I. Anisimov, J. Zaanen and O. K. Andersen, Phys. Rev. B: Condens. Matter Mater. Phys., 1991, 44, 943–954 CrossRef CAS.
- M. Cococcioni and S. de Gironcoli, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 71, 035105 CrossRef.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Solid State, 1976, 13, 5188–5192 CrossRef.
- F. Schaefers, M. Mertin and M. Gorgoi, Rev. Sci. Instrum., 2007, 78, 123102 CrossRef CAS.
- T.-L. Lee and D. A. Duncan, Synchrotron Radiation News, 2018, 31, 16–22 CrossRef.
- U. Guide, C. J. Powell, M. S. Division and A. Jablonski, Nat'l Std. Ref. Data Ser. (NIST NSRDS) Search PubMed.
- K. A. Persson, D. Skinner, W. Chen, S. Cholia, G. Hautier, D. Gunter, S. P. Ong, S. Dacek, W. D. Richards, A. Jain and G. Ceder, APL Mater., 2013, 1, 011002 CrossRef.
- M. C. Biesinger, L. W. M. Lau, A. R. Gerson and R. S. C. Smart, Appl. Surf. Sci., 2010, 257, 887–898 CrossRef CAS.
- J. F. Moulder, W. F. Stickle, P. E. Sobol, K. D. Bomben and J. Chastain, Handbook of X-Ray Photoelectron Spectroscopy A Reference Book of Standard Spectra for Identification and Interpretation of XPS Data, Perkin-Elmer Corporation, 1992 Search PubMed.
- T. C. Lin, G. Seshadri and J. A. Kelber, Appl. Surf. Sci., 1997, 119, 83–92 CrossRef CAS.
- M. C. Biesinger, B. P. Payne, A. P. Grosvenor, L. W. M. Lau, A. R. Gerson and R. S. C. Smart, Appl. Surf. Sci., 2011, 257, 2717–2730 CrossRef CAS.
- J. H. Scofield, Theoretical Photoionization Cross Sections from 1 to 1500 keV, U.S. Atomic Energy Commission, 1973 Search PubMed.
- V. Šepelák, A. Düvel, M. Wilkening, K. D. Becker and P. Heitjans, Chem. Soc. Rev., 2013, 42, 7507–7520 RSC.
- C. Baur, J. Chable, F. Klein, V. S. K. Chakravadhanula and M. Fichtner, ChemElectroChem, 2018, 5, 1484–1490 CrossRef CAS.
- R. D. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751–767 CrossRef.
- G. Williamson and W. Hall, Acta Metall., 1953, 1, 22–31 CrossRef CAS.
- A. Urban, A. Abdellahi, S. Dacek, N. Artrith and G. Ceder, Phys. Rev. Lett., 2017, 119, 1–6 CrossRef.
- A. Urban, I. Matts, A. Abdellahi and G. Ceder, Adv. Energy Mater., 2016, 6, 1600488 CrossRef.
- X. Wang, Y. Huang, D. Ji, F. Omenya, K. Karki, S. Sallis, L. F. J. Piper, K. M. Wiaderek, K. W. Chapman, N. A. Chernova and M. S. Whittingham, J. Electrochem. Soc., 2017, 164, A1552–A1558 CrossRef CAS.
- R. Chen, R. Witte, R. Heinzmann, S. Ren, S. Mangold, H. Hahn, R. Hempelmann, H. Ehrenberg and S. Indris, Phys. Chem. Chem. Phys., 2016, 18, 7695–7701 RSC.
- E. Peled and S. Menkin, J. Electrochem. Soc., 2017, 164, A1703–A1719 CrossRef CAS.
- B. Philippe, M. Hahlin, K. Edström, T. Gustafsson, H. Siegbahn and H. Rensmo, J. Electrochem. Soc., 2015, 163, A178–A191 CrossRef.
- K. Xu, Chem. Rev., 2014, 114, 11503–11618 CrossRef CAS.
- D. Aurbach, B. Markovsky, G. Salitra, E. Markevich, Y. Talyossef, M. Koltypin, L. Nazar, B. Ellis and D. Kovacheva, J. Power Sources, 2007, 165, 491–499 CrossRef CAS.
- G. Assat, D. Foix, C. Delacourt, A. Iadecola, R. Dedryvère and J.-M. Tarascon, Nat. Commun., 2017, 8, 2219 CrossRef.
- J. Lee, J. K. Papp, R. J. Clément, S. Sallis, D.-H. Kwon, T. Shi, W. Yang, B. D. McCloskey and G. Ceder, Nat. Commun., 2017, 8, 981 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: PXRD pattern of ceramic synthesis attempts; structural parameters of the Rietveld refinements; PXRD pattern of Li2VO2F with Rietveld refinement; Williamson–Hall-plots; TEM and EDX analysis; SQS of Li2TMO2F and Li2TM10.5TM20.5O2F; ordered structures of Li2TM10.5TM20.5O2F; table of energy difference between the ordered/decomposed state and disordered state; table of oxidation states of TMs; voltage profiles of Li2VO2F, Li2V0.5Ti0.5O2F and Li2V0.5Fe0.5O2F half-cells cycled up to 4.1 V; PXRD pattern of cycled electrodes; HAXPES Fe 2p peak fitting; HAXPES survey of Li2V0.5Fe0.5O2F and Li2VO2F and fluorine plasmon overlaps with the Fe 2p3/2 peak; core level photoelectron spectra of Fe 2p and Ti 2p; cycling performance of Li2VO2F, Li2V0.5Ti0.5O2F and Li2V0.5Fe0.5O2F half-cells cycled up to 4.5 V. See DOI: 10.1039/c9ta06291b |
| This journal is © The Royal Society of Chemistry 2019 |